

Abstract
Atopic dermatitis (AD) results from gene and environment interactions that lead to a range of immunological abnormalities and breakdown of the skin barrier. Protease-activated receptor 2 (PAR2) belongs to a family of G-protein coupled receptors and is expressed in suprabasal layers of the epidermis. PAR2 is activated by both trypsin and a specific agonist peptide, SLIGKV-NH2 and is involved in both epidermal permeability barrier homeostasis and epithelial inflammation. In this study, we investigated the effect of lobaric acid on inflammation, keratinocyte differentiation, and recovery of the skin barrier in hairless mice. Lobaric acid blocked trypsin-induced and SLIGKV-NH2-induced PAR2 activation resulting in decreased mobilization of intracellular Ca2+ in HaCaT keratinocytes. Lobaric acid reduced expression of interleukin-8 induced by SLIGKV-NH2 and thymus and activation regulated chemokine (TARC) induced by tumor necrosis factor-a (TNF-α) and IFN-γ in HaCaT keratinocytes. Lobaric acid also blocked SLIGKV-NH2-induced activation of ERK, which is a downstream signal of PAR2 in normal human keratinocytes (NHEKs). Treatment with SLIGKV-NH2 downregulated expression of involucrin, a differentiation marker protein in HaCaT keratinocytes, and upregulated expression of involucrin, transglutamase1 and filaggrin in NHEKs. However, lobaric acid antagonized the effect of SLIGKV-NH2 in HaCaT keratinocytes and NHEKs. Topical application of lobaric acid accelerated barrier recovery kinetics in a SKH-1 hairless mouse model. These results suggested that lobaric acid is a PAR2 antagonist and could be a possible therapeutic agent for atopic dermatitis.
Keywords: PAR2, Skin barrier, Atopic dermatitis, Lobaric acid
INTRODUCTION
Atopic dermatitis (AD) is a chronic inflammatory skin disease associated with cutaneous hyperreactivity to environmental triggers. AD is the product of interaction between susceptibility genes, the environment, defective skin barrier function, and immunologic responses. AD is characterized by dry skin and increased transepidermal water loss (TEWL). Impairment of the skin barrier function in AD leads to increased antigen absorption contributing to cutaneous hyperreactivity (Leung et al., 2004; Lee et al., 2010).
Protease-activated receptor 2 (PAR2) is a G protein-coupled receptor activated by proteolytic cleavage of its amino terminal domain by trypsin-like serine proteases (SPs). Cleavage of PAR2 exposes the tethered ligand neoepitope that binds intramolecularly within the receptor to stimulate signal transduction via coupled G proteins. PAR2-mediated signal transduction is also experimentally stimulated by hexapeptide agonist that are homologous to the tethered ligand sequence. The irreversible nature of PAR2 proteolysis means that downstream signal transduction is tightly regulated (Bohm et al., 1996; Oldham and Hamm, 2008). In skin, PAR2 is associated with regulation of keratinocyte proliferation, differentiation, epidermal barrier recovery and inflammation (Derian et al., 1997; Macfarlane and Plevin, 2003; Frateschi et al., 2011).
The epidermis has protective and defensive functions that are mediated by its unique differentiation end product, the stratum corneum. The stratum corneum comprises a unique, two-compartment system: protein-enriched corneocyte “bricks” embedded in a lipid-enriched, extracellular matrix “mortar” (Elias, 1983; Elias, 2005; Elias and Choi, 2005; Elias and Choi, 2008). Both compartments are required to form an effective permeability barrier (Madison, 2003). Inhibition of serine protease-mediated signaling stimulates barrier recovery responses by both differentiating keratinocytes and the LB secretory machinery (Hachem et al., 2006). For example, recent studies demonstrated that proteolytically active allergens, house dust mites and cockroaches activated PAR2 and delayed barrier recovery by inhibiting LB secretion (Jeong et al., 2008).
PAR2 activation correlates with inflammatory responses. Thymus and activation-regulated chemokine (TARC, CCL-17) is overexpressed in LEKTI-deficient epidermis that has increased amounts of SP (Briot et al., 2009; Morita et al., 2010). PAR2 activation also increase secretion of IL-8, promoting granulocyte and T-cell recruitment (Hou et al., 1998) via extracellular signal-regulated kinase 1 and 2 (ERK1/2) and nuclear factor kappaB (NF-κB) signaling pathways (Guo et al., 2011). PAR2-deficient mice have diminished ear swelling and inflammatory infiltrates in a model of contact hypersensitivity, indicating that PAR2 mediates inflammation in allergic dermatitis (Kawagoe et al., 2002). In addition, increased PAR2 is observed in the lesional skin of patients with AD, suggesting that PAR2 is involved in inflammatory dermatosis (Komatsu et al., 2007).
Lobaric acid, a phenolic compound isolated from lichen Stereocaulon aplinum, has been reported to show antitumor and antimitotic effects (Gissurarson et al., 1997, Morita et al., 2009). However, there is no report showing how lobaric acid affects skin barrier function.
This study investigated, the effects of lobaric acid on PAR2 activation, the inflammation response, keratinocyte differentiation, and permeability barrier recovery after acute barrier abrogation in mouse skin.
MATERIALS AND METHODS
Materials
Lobaric acid was from Greenpharma (Orléans, France). Peptides corresponding to the tethered ligand of human PAR2 (activating peptide SLIGKV-NH2) were from Bachem (Bubendorf, Switzerland). INF-γ and TNF-α were from ProSpec protein specialists (East Brunswick, NJ, USA). Dexamethasone, pertussis toxin from Bordetella pertussis (PTX) and pertussis toxin B oligomer (PTX-B) were from Sigma (St. Louis, MO, USA).
Cell culture
The immortalized human keratinocyte cell line HaCaT was generally cultured in high glucose dulbecco’s modified eagle’s medium (DMEM) with 10% FBS, 100 units/ml penicillin and 100 mg/ml streptomycin (WelGENE Inc., Gyeongsan, Korea) or in DMEM (Invitrogen, Carlsbad, CA, USA) with 10% chelex-treated FCS, 4.5 g/L D-glucose, L-glutamine, sodium pyruvate, 0.05 mM calcium chloride, 100 units/ml penicillin and 100 mg/ml streptomycin. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Normal human epidermal keratinocytes (NHEKs) of neonatal origin were from Invitrogen. NHEKs were used until passage 5. Cells were cultured in EpiLife medium with 60 μM calcium and human keratinocyte growth supplement (Invitrogen), 100 units/ml penicillin and 100 mg/ml streptomycin and incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Measurement of intracellular Ca2+
HaCaT cells were seeded in 96-well plates (black wells and clear bottoms, Greiner Bio-one, Germany) at 4×104 cells/well. After 24 hours, medium was changed to 2 μM Fluo-4, AM (Invitrogen), 0.02% Pluronic F-127 (Invitrogen) and 2.5 mM Probenecid (Sigma) in Hank’s balanced salt solution (HBSS, Sigma) containing 20 mM HEPES (Sigma) for 30 minutes at 37°C, protected from light. Cells were stabilized for 15 minutes at room temperature during shaking. Cells were pretreated with lobaric acid before PAR2 activation. After 30 minutes, 20 μM PAR2-activating peptide SLIGKV-NH2 or 2 unit/ml trypsin (Sigma) was added and intracellular Ca2+ was simultaneously measured using Flexstation 3 (Molecular Devices, Sunnyvale, CA, USA), 485 nm excitation and 525 nm emission at room temperature. Data were analyzed with SoftMax Pro (Molecular Devices), and mobilization of intracellular Ca2+ was calculated as the minimum value subtracted from the maximum value.
Measurement of secreted IL-8
HaCaT cells were seeded in 96-well plates as above. After 24 hours, cells were starved in DMEM without FBS for 12 hours and pretreated with lobaric acid for 30 minutes before adding 100 μM SLIGKV-NH2 and culturing for 24 hours. Supernatants were harvested and analyzed with Human CXCL8/IL-8 DuoSet kits (R&D Systems, Minneapolis, MN, USA). Absorbance was measured at 450 nm and corrected at 540 nm using auto microplate reader (infinite M200, TECAN, Männedorf, Switzerland).
TARC measurement
HaCaT cells were seeded in 96-well plates at 3×104 cells/well. After 24 hours, cells were starved using DMEM without FBS for 12 hours. The inflammatory response was induced by treating with both INF-γ and TNF-α (East Brunswick) at 10 ng/ml for 24 hours. Lobaric acid was added for 72 hours. Dexamethasone (Sigma), the positive control was also treated with 100 μg/ml. Supernatants were harvested and analyzed with Quantikine Human CCL17/TARC immunoassay kits (R&D Systems). Absorbance was measured and corrected as above.
Western blot
Cells were treated with ice-cold RIPA solution (Noble Bio, Hwaseong, Korea), protease inhibitor cocktail (Sigma) and 1 mM phenylmethanesulfonyl fluoride (Sigma) for 60 minutes. Lysates were sonicated on ice and centrifuged at 13,000×g and 4°C for 20 minutes. Supernatants were harvested and protein concentrations were normalized using Pierce Protein Assay kits (Pierce Biotechnology, Inc., Rockford, IL, USA) with bovine serum albumin as the standard. Samples were stored at −20°C. Samples were mixed with LDS sample buffer and sample reducing buffer (Invitrogen) and boiled at 95°C for 5 minutes and loaded on NuPAGE 10% Bis-Tris gels run with MOPS SDS running buffer (Invitrogen). Proteins were transferred to polyvinylidene fluoride transfer membranes (PALL Corporation, Port Washington, NY, USA) that were blocked with 5% skim milk in TBS-T buffer (0.1% Tween 20, 100 mM NaCl and 10 mM Tris-HCl, pH 7.5) and probed with mouse monoclonal anti-involucrin (1/1000, Abcam, Cambridge, UK), rabbit monoclonal anti-transglutamase 1 (1/1000, Santa Cruz, CA, USA), rabbit polyclonal anti-filaggrin (1/1000, Abcam), mouse monoclonal anti-b-actin (1/10,000, Sigma), rabbit monoclonal anti-p42/44 MAPK (Erk1/2) (1/1000, Cell Signaling, Beverly, MA, USA), rabbit monoclonal anti-Phospho-p42/44 MAPK (Erk1/2) (1/2000, Cell Signaling), donkey anti-rabbit IgG (1/5,000, Bethyl Laboratories, Montgomery, TX, USA) or goat anti-mouse IgG (1/10,000, Bio-Rad, Hercules, CA, USA). Bands were detected with WEST-ZOL plus Western Blot Detection systems (iNtRON Biotechnology, Seongnam, Korea) and visualized with ChemiDoc XRS (Bio-Rad). Densitometric analysis was performed using Imaging software (Image J, Bethesda, MD, USA). Band density was normalized to beta-actin intensity.
Animal studies
Female hairless mice (SKH-1), aged 7–12 weeks, were from HOSHINO laboratory animals (Shizuoka, Japan) and fed mouse diet and water ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committees of Gyeonggi Bio Center and performed in accordance with their guidelines. Procedures were as in Hachem et al. (2006). Before acute barrier disruption by sequential cellophane tape stripping, mice were treated with a topical application of lobaric acid (0.2 μM) in propylene glycol (7:3 vol/vol) or vehicle alone. After 2 hours, tape-stripping was performed (TEWL rate ≥50 g/m2h; normal ≤8.5 g/m2h) and samples were applied again. Permeability barrier function was assessed as TEWL with a tewameter (TM300/Courage+Khazaka electronic GmbH, Köln, Germany), before treatment and immediately after treatment and 3 and 24 hours after acute barrier disruption.
Statistical analysis
Data are presented as means ± standard error of the mean (SEM). All statistical analysis was done using SPSS statistical software package Version 17.0, considering below p<0.05 as statistically significant.
RESULTS
Effects of lobaric acid on intracellular Ca2+ uptake in keratinocytes
PAR-2 activation induces intracellular Ca2+ mobilization. HaCaT keratinocytes were treated with SLIGKV or trypsin to induce PAR-2 activation, followed by lobaric acid treatment at several concentrations. Changes in intracellular Ca2+ concentration were observed. Activation of Ca2+ influx by SLIGKV or trypsin (Fig. 1B) was dose-dependently decreased with lobaric acid treatment. Lobaric acid at 2, 10, or 20 μM inhibited Ca2+ uptake by HaCaT keratinocytes by 1%, 34% and 73% after activation by SLIGKV and by 8%, 26% and 54% after activation by trypsin relative to a DMSO control (Fig. 1C).
Fig. 1.
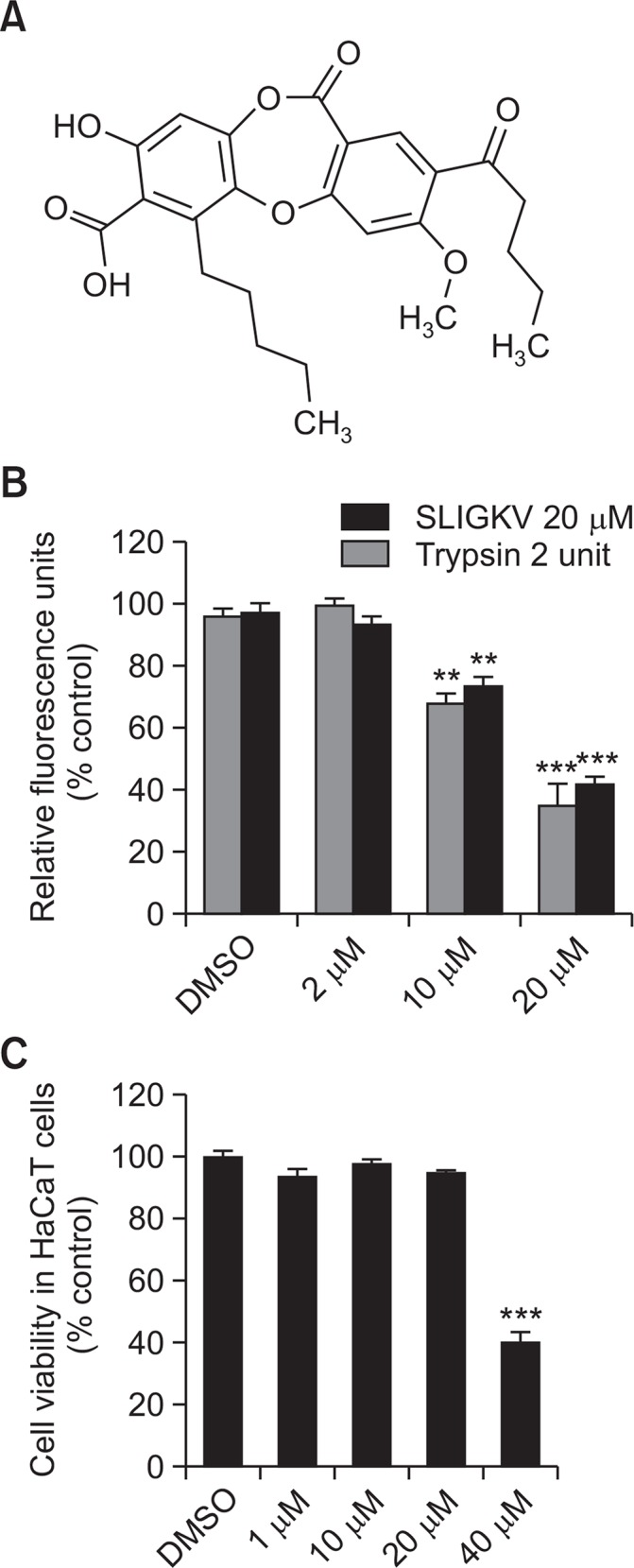
Lobaric acid inhibited PAR2-induced intracellular Ca2+ influx in keratinocytes. Chemical structure of lobaric acid (A). HaCaT keratinocytes were treated with various concentrations of lobaric acid. After 30 minutes, PAR2-activating peptide, SLIGKV-NH2, or trypsin was added. Mobilization of intracellular Ca2+ was dose-dependently inhibited by pretreatment with lobaric acid (B). Cytotoxicity of lobaric acid in HaCaT cells (C). Data are mean ± SEM, n=2. **p<0.01, ***p<0.005 compared with control.
Lobaric acid decreased PAR2-induced inflammatory responses in keratinocytes
HaCaT keratinocytes were treated with SLIGKV-NH2 or INF-γ/TNF-α to induce the inflammatory response and simultaneously exposed to lobaric acid at various concentrations. Treatment with 100 μM SLIGKV-NH2 increased IL-8 secretion but treatment with lobaric acid at 0.3125 μM to 10 μM dose-dependently and significantly decreased IL-8 secretion (Fig. 2A). TARC was induced by exposure to INF-γ and TNF-α at 10 ng/ml. However, treatment with 2.5, 5, or 25 μM lobaric acid decreased TARC secretion significantly and dose dependently (Fig. 2B). The steroid dexamethasone at 100 μg/ml was used as positive control and also significantly decreased TARC. To further assess the pathway of blocking inflammatory response, we investigated whether ERK1/2 activation by SLIGKV-NH2 could be influenced by lobaric acid. Treatment with 100 μM SLIGKV-NH2 induced phosphorylation of ERK1/2 (p-ERK), starting from 4 min after treatment and declining at 12 min. When NHEKs were pretreated with 2 μM lobaric acid for 2 hours, SLIGKV-NH2-induced upregulation of p-ERK was inhibited (Fig. 3C).
Fig. 2.

Lobaric acid decreased PAR2-induced inflammatory response in keratinocytes. After pretreatment with lobaric acid, 100 μM SLIGKV-NH2 was added to HaCaT keratinocytes for 24 hours and immunoassays were performed (A). INF-γ and TNF-α were added to HaCaT keratinocytes followed by lobaric acid or dexamethasone. Supernatants were used in immunoassays (B). ERK phosphorylation in NHEKs induced by 100 μM SLIGKV-NH2 was blocked by lobaric acid. NHEKs were stimulated for indicated times with 100 μM SLIGKV-NH2 and stimulation was blocked by pretreatment with 2 μM lobaric acid (C). Whole cell lysates were used to determine ERK phosphorylation (p-ERK) and total-ERK (t-ERK) by Western blots. Data are mean ± SEM, n=2. *p<0.05, **p<0.01, ***p<0.005, compared with controls.
Fig. 3.

Lobaric acid antagonized SLIGKV-NH2 effects on differentiation protein expression. Effect of SLIGKV-NH2 on involucrin, transglutamase 1 (TGM1), and filaggrin in HaCaT keratinocytes (A–B) and normal human epidermal keratinocytes (NHEKs) (C–D) by Western blot. HaCaT keratinocytes were treated with 100 μM SLIGKV-NH2 at indicated calcium concentration for 72 hrs. SLIGKV-NH2 downregulated involucrin at 0.4 mM calcium (A). Lobaric acid (0.1, 0.5, 2 μM) was used to treat cells before SLIGKV-NH2 treatment. Lobaric acid antagonized SLIGKV-NH2 effects in a non-dose-dependent manner. Positive control was 2.8 mM calcium and G protein inhibitor PTX (0.5 μg/ml) to induce differentiation and inhibit PAR2. PTX negative control was PTX-B (0.5 μg/ml) (B). SLIGKV-NH2 upregulated involucrin, TGM1 and filaggrin and lobaric acid antagonized SLIGKV-NH2 in NHKS (C–D). Positive control was 1.5 mM calcium to induce differentiation (C). Filaggrin was increased by SLIGKV-NH2 treatment and lobaric acid normalized the expression of filaggrin at 72 hours (D).
Lobaric acid antagonized SLIGKV-NH2 effects on differentiation protein expression in keratinocytes
In differentiating keratinocytes, expression increases for the suprabasal makers K1 and K10, filaggrin, involucrin, loricrin, and transglutamase (Eckert et al., 1997). PAR2 activation regulates terminal differentiation of keratinocytes and persistent PAR2 activation might cause abnormal differentiation (Derian et al., 1997; Kim et al., 2012). We investigated whether lobaric acid antagonizes these changes in protein expression induced by PAR2 activation of HaCaT keratinocytes and NHEKs.
In HaCaT keratinocytes, treatment with SLIGKV-NH2 (100 μM) decreased involucrin expression that was induced by calcium treatment for 72 hours. The SLIGKV-NH2 effect was strongest at 0.4 mM calcium, so 0.4 mM was used for experiments (Fig. 3A). SLIGKV-NH2 decreased involucrin expression and this effect was antagonized by lobaric acid treatment (Fig. 3B). PTX (0.5 μg/ml), or a G-protein inhibitor that was used as a positive control also increased expression of involucrin. PTX-B (0.5 μg/ml), which was used as a negative control to PTX, decreased involucrin expression. Treatment with 2.8 mM calcium was used as a positive control to induce involucrin expression. Treatment of SLIGKV-NH2 with PTX reversed the effect of SLIGKV-NH2 alone and high concentrations of calcium (2.8 mM) induced normal keratinocyte differentiation.
In NHEKs, treatment with SLIGKV-NH2 for various times increased expression of the differentiation markers involucrin and transglutamase 1 (TGM1). Filaggrin and lobaric acid antagonized the effect of SLIGKV-NH2 (Fig. 3C, 3D). A positive control of 1.5 mM calcium induced expression of involucrin and TGM1. At 48 hours of lobaric acid treatment, the expression of involucrin, TGM1, and filaggrin decreased (Fig. 3C). At 72 hours of treatment, filaggrin expression was further reduced (Fig. 3D).
Lobaric acid accelerated permeability barrier recovery after mouse skin barrier disruption
To investigate whether lobaric acid aided in recovery of skin barrier functions in vivo, we assessed barrier recovery kinetics in hairless mice. Mice were treated with 0.2 μM of lobaric acid or vehicle (70:30 propylene glycol: ethanol) 2 hours before tape stripping. Acute barrier disruption was induced by sequential tape stripping and TEWL levels measured. TEWL levels were elevated more than six fold compared to the normal conditions and samples were reapplied to acutely disrupted skin. Lobaric acid significantly accelerated the kinetics of barrier recovery at 3 hours following tape-stripping. Skin treated with either lobaric acid or vehicle had recovered by over 80% after 24 hours (Fig. 4).
Fig. 4.

Lobaric acid improved barrier recovery after acute disruption. Acute barrier disruption in hairless mice was by repeated cellophane tape stripping followed by application of lobaric acid (0.2 mM) and vehicle. Lobaric acid before and after tape stripping significantly improved barrier recovery compared to vehicle-treated skin. TEWL was measured at baseline, immediately after, and 3 and 24 h after acute barrier disruption. Results are mean ± SEM, n=9. *p<0.05 compared with control.
DISCUSSION
AD is a chronic inflammatory skin disease characterized by eczema, pruritus, and cutaneous hyperreactivity to environmental factors that are innocuous to normal nonatopic individuals (Briot et al., 2009). Children with poorly controlled AD have a poorer quality of life, and AD results in financial, time, and psychologic burdens (Spergel and Paller, 2003). AD has a complex etiology that results from interactions between environment and several susceptibility genes involved in skin barrier function and systemic and local immunological responses (Cork et al., 2006). AD and PAR2 are closely related. AD-like lesions caused by PAR2 are seen in a serine protease inhibitor knockout mouse model (Briot et al., 2009). In the absence of PAR2, mice have a rescued inflammation response and altered skin barrier permeability (Frateschi et al., 2011). Thus, PAR2 correlates with the inflammatory response and barrier permeability and homeostasis. The proliferation and differentiation of keratinocytes in the epidermis also controls barrier permeability and homeostasis.
The aim of this study was to find new PAR2 antagonists that interfered with PAR2 activation and might halt AD symptoms. We found that lobaric acid decreased intracellular Ca2+ mobilization induced by SLIGKV-NH2 and trypsin in HaCaT keratinocytes (Fig. 1). Our results indirectly indicated that PAR2 activation was specifically inhibited by lobaric acid. We also showed that lobaric acid inhibited PAR2-induced inflammation and keratinocyte differentiation in vitro. PAR2 activation increases secretion of IL-8 via ERK activation (Hou et al., 1998; Guo et al., 2011). Lobaric acid blocked ERK activation and decreased secretion of IL-8 level significantly induced by SLIGKV-NH2 (Fig. 2). TARC was also downregulated significantly by lobaric acid in HaCaT keratinocytes (Fig. 2B).
A number of autocrine and paracrine factors drive keratinocyte differentiation and calcium is crucial in this process (Su et al., 1994). The epidermis has a calcium gradient with the lowest concentrations in the basal layer and the highest in the granulosum layer, where critical proteins for barrier function are produced (Yuspa et al., 1989). PAR2 activation regulates terminal differentiation of keratinocytes; however, the effect of PAR-2 on keratinocyte differentiation is controversial. Some studies found that activation of PAR-2 diminished keratinocytes differentiation (Derian et al., 1997) but other studies did not (Lee at al., 2010). Our study did not find that SLIGKV-NH2 had a consistent effect on keratinocyte differentiation but lobaric acid always antagonized the effect of SLIGKV-NH2. SLIGKV-NH2 decreased involucrin expression and lobaric acid increased expression (Fig. 3A, 3B). SLIGKV-NH2 upregulated expression of the differentiation markers involucrin, TGM1, and filaggrin and lobaric acid downregulated their expression (Fig. 3C, 3D).
To determine the effects of lobaric acid on skin barrier recovery, we assessed permeability barrier homeostasis in mice. SP activity increased after acute perturbation of the permeability barrier and inhibition of SP activation. Topical serine protease inhibitors accelerate barrier recovery in rodents and in humans (Hachem et al., 2006). A tryptic SP signals downstream responses through PAR2 so we measured barrier recovery kinetics of skin after topical lobaric acid application compared to application of vehicle only. Topical treatment with lobaric acid significantly accelerated barrier recovery compared to the control (Fig. 4).
In summary, blocking a PAR2-mediated signal pathway with lobaric acid rescued inflammation and promoted skin barrier recovery. Our results suggested that lobaric acid was a PAR2 antagonist and might be a potential therapeutic agent for AD. Further studies needed include measuring the effects of lobaric acid on AD using NC/Nga mice, a strain that spontaneously develops AD.
Acknowledgments
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (Grant NO: HN15C0102 and HI14C0779).
REFERENCES
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M,, Nelken NA, Coughlin SR, Payan DG, Bunnett NW. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P, Hovnanian A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206:1135–1147. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork MJ, Robinson DA, Vasilopoulos Y, Ferguson A, Moustafa M, MacGowan A, Duff GW, Ward SJ, Tazi-Ahnini R. New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene-environment interactions. J Allergy Clin Immunol. 2006;118:3–21. doi: 10.1016/j.jaci.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Derian CK, Eckardt AJ, Andrade-Gordon P. Differential regulation of human keratinocyte growth and differentiation by a novel family of protease-activated receptors. Cell Growth Differ. 1997;8:743–749. [PubMed] [Google Scholar]
- Eckert RL, Crish JF, Robinson NA. The epidermal keratinocyte as a model for the study of gene regulation and cell differentiation. Physiol Rev. 1997;77:397–424. doi: 10.1152/physrev.1997.77.2.397. [DOI] [PubMed] [Google Scholar]
- Elias PM. Epidermal lipids, barrier function, and desquamation. J Invest Dermatol. 1983;80:44s–49s. doi: 10.1038/jid.1983.12. [DOI] [PubMed] [Google Scholar]
- Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;125:183–200. doi: 10.1111/j.0022-202X.2005.23668.x. [DOI] [PubMed] [Google Scholar]
- Elias PM, Choi EH. Interactions among stratum corneum defensive functions. Exp Dermatol. 2005;14:719–726. doi: 10.1111/j.1600-0625.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121:1337–1343. doi: 10.1016/j.jaci.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frateschi S, Camerer E, Crisante G, Rieser S, Membrez M, Charles RP, Beermann F, Stehle JC, Breiden B, Sandhoff K, Rotman S, Haftek M, Wilson A, Ryser S, Steinhoff M, Coughlin SR, Hummler E. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun. 2011;2:161. doi: 10.1038/ncomms1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissurarson SR, Sigurdsson SB, Wagner H, Ingolfsdottir K. Effect of lobaric acid on cysteinyl-leukotriene formation and contractile activity of guinea pig taenia coli. J Pharmacol Exp Ther. 1997;280:770–773. [PubMed] [Google Scholar]
- Guo D, Zhou H, Wu Y, Zhou F, Xu G, Wen H, Zhang X. Involvement of ERK1/2/NF-kappaB signal transduction pathway in TF/FVIIa/PAR2-induced proliferation and migration of colon cancer cell SW620. Tumour Biol. 2011;32:921–930. doi: 10.1007/s13277-011-0194-1. [DOI] [PubMed] [Google Scholar]
- Hachem JP, Houben E, Crumrine D, Man MQ, Schurer N, Roelandt T, Choi EH, Uchida Y, Brown BE, Feingold KR, Elias PM. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol. 2006;126:2074–2086. doi: 10.1038/sj.jid.5700351. [DOI] [PubMed] [Google Scholar]
- Hou L, Kapas S, Cruchley AT, Macey MG, Harriott P, Chinni C, Stone SR, Howells GL. Immunolocalization of protease-activated receptor-2 in skin: receptor activation stimulates interleukin-8 secretion by keratinocytes in vitro. Immunology. 1998;94:356–362. doi: 10.1046/j.1365-2567.1998.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SK, Kim HJ, Youm JK, Ahn SK, Choi EH, Sohn MH, Kim KE, Hong JH, Shin DM, Lee SH. Mite and cockroach allergens activate protease-activated receptor 2 and delay epidermal permeability barrier recovery. J Invest Dermatol. 2008;128:1930–1939. doi: 10.1038/jid.2008.13. [DOI] [PubMed] [Google Scholar]
- Kawagoe J, Takizawa T, Matsumoto J, Tamiya M, Meek SE, Smith AJ, Hunter GD, Plevin R, Saito N, Kanke T, Fujii M, Wada Y. Effect of protease-activated receptor-2 deficiency on allergic dermatitis in the mouse ear. Jpn J Pharmacol. 2002;88:77–84. doi: 10.1254/jjp.88.77. [DOI] [PubMed] [Google Scholar]
- Kim HY, Goo JH, Joo YA, Lee HY, Lee SM, Oh CT, Ahn SM, Kim NH, Hwang JS. Impact on inflammation and recovery of skin barrier by nordihydroguaiaretic Acid as a protease-activated receptor 2 antagonist. Biomol. Ther. (Seoul) 2012;20:463–469. doi: 10.4062/biomolther.2012.20.5.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Saijoh K, Kuk C, Liu AC, Khan S, Shirasaki F, Takehara K, Diamandis EP. Human tissue kallikrein expression in the stratum corneum and serum of atopic dermatitis patients. Exp Dermatol. 2007;16:513–519. doi: 10.1111/j.1600-0625.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- Lee SE, Jeong SK, Lee SH. Protease and protease-activated receptor-2 signaling in the pathogenesis of atopic dermatitis. Yonsei Med J. 2010;51:808–822. doi: 10.3349/ymj.2010.51.6.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest. 2004;113:651–657. doi: 10.1172/JCI21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane SR, Plevin R. Intracellular signalling by the G-protein coupled proteinase-activated receptor (PAR) family. Drug Dev Res. 2003;59:367–374. doi: 10.1002/ddr.10305. [DOI] [Google Scholar]
- Madison KC. Barrier function of the skin: “la raison d’être” of the epidermis. J Invest Dermatol. 2003;121:231–241. doi: 10.1046/j.1523-1747.2003.12359.x. [DOI] [PubMed] [Google Scholar]
- Morita E, Takahashi H, Niihara H, Dekio I, Sumikawa Y, Murakami Y, Matsunaka H. Stratum corneum TARC level is a new indicator of lesional skin inflammation in atopic dermatitis. Allergy. 2010;65:1166–1172. doi: 10.1111/j.1398-9995.2010.02361.x. [DOI] [PubMed] [Google Scholar]
- Morita H, Tsuchiya T, Kishibe K, Noya S, Shiro M, Hirasawa Y. Antimitotic activity of lobaric acid and a new benzofuran, sakisacaulon A from Stereocaulon sasakii. Bioorg Med Chem Lett. 2009;19:3679–3681. doi: 10.1016/j.bmcl.2009.03.170. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112:S118–S127. doi: 10.1016/j.jaci.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Su MJ, Bikle DD, Mancianti ML, Pillai S. 1,25-Dihydroxyvitamin D3 potentiates the keratinocyte response to calcium. J Biol Chem. 1994;269:14723–14729. [PubMed] [Google Scholar]
- Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109:1207–1217. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]