

Abstract
Endoplasmic reticulum (ER) and mitochondrial function have both been shown to be critical events in neurodegenerative diseases. The ER mediates protein folding, maturation, sorting as well acts as calcium storage. The unfolded protein response (UPR) is a stress response of the ER that is activated by the accumulation of misfolded proteins within the ER lumen. Although the molecular mechanisms underlying ER stress-induced apoptosis are not completely understood, increasing evidence suggests that ER and mitochondria cooperate to signal cell death. Similarly, calcium-mediated mitochondrial function and dynamics not only contribute to ATP generation and calcium buffering but are also a linchpin in mediating cell fate. Mitochondria and ER form structural and functional networks (mitochondria-associated ER membranes [MAMs]) essential to maintaining cellular homeostasis and determining cell fate under various pathophysiological conditions. Regulated Ca2+ transfer from the ER to the mitochondria is important in maintaining control of pro-survival/pro-death pathways. In this review, we summarize the latest therapeutic strategies that target these essential organelles in the context of neurodegenerative diseases.
Keywords: Mitochondria, ER, Calcium, Neurodegeneration, Mitochondrial Dynamics, Mitochondrial Transport, UPR
A. The Unfolded protein response
The unfolded protein response (UPR) plays a vital role in maintaining cell homeostasis as a consequence of endoplasmic reticulum (ER) stress (1, 2). Failure in assembly of protein complexes, protein degradation, formation of disulfide bonds, and protein glycosylation in the ER are common triggers of the UPR. Under moderate misfolded protein accumulation, the UPR transduces information from the ER to the nucleus and cytosol and thereby inhibits protein translation, expands the ER membrane, recruits ER chaperones to aid in the correct folding of misfolded proteins, and promotes protein degradation in order to reduce the load of unfolded or misfolded proteins (3, 4).
Three ER stress sensors mediate the UPR; PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1), and activating transcription factor 6 (ATF6) (4) (Figure 1). These sensors transmit signals from the ER to the cytoplasm or nucleus, inducing the suppression of protein translation, expression of ER molecular chaperones or activate of ER-associated degradation (ERAD) reducing the accumulation of unfolded proteins in the ER. In the first pathway, PERK directly phosphorylates and inhibits the ubiquitous eukaryotic translation initiation factor 2α (eIF2α) leading to attenuation global protein synthesis to decrease protein influx into the ER lumen (5). Phosphorylation of eIF2α also favors the selective translation of the mRNA encoding the transcription factor ATF4. Upon translocation to the nucleus, ATF4 induces the expression of ER chaperones thereby increasing the refolding of misfolded proteins. The second pathway of UPR involves the membrane protein ATF6. During ER stress, ATF6 is transported to the Golgi apparatus, where it is cleaved sequentially by the Golgi resident serine proteases S1P and S2P (site 1 and site 2 proteases, respectively) to release its cytosolic domain (6). The resulting cytosolic ATF6 fragment (ATF6f) translocates to the nucleus to induce gene expression of ER chaperones, ERAD components and XBP-1 (7). The third pathway of UPR is initiated by IRE1 that is activated upon its dimerization and auto-phosphorylation (8). IRE1 contains an N-terminal ER stress-sensing domain facing ER lumen, an ER transmembrane domain, and a serine/threonine kinase domain and a C-terminal endoribonuclease domain that faces cytosol. IRE1 binds the RING finger protein, TRAF2, and activates ASK1 (apoptosis signal–regulating kinase 1), leading to activation of JNK and p38. IRE1 degrades a subset of mRNAs, which encode for ER-localized proteins by means of regulated IRE1-dependent decay (RIDD), thereby reducing protein synthesis in the ER. Further, IRE1 activates caspase 12, which activates a cascade of caspases leading to cell death. Also, through decay of anti-caspase 2 pre-miRNAs, IRE1α activates apoptosis through up regulation of caspase 2 (1, 2, 7).
Figure 1. The Unfolded Protein Response.
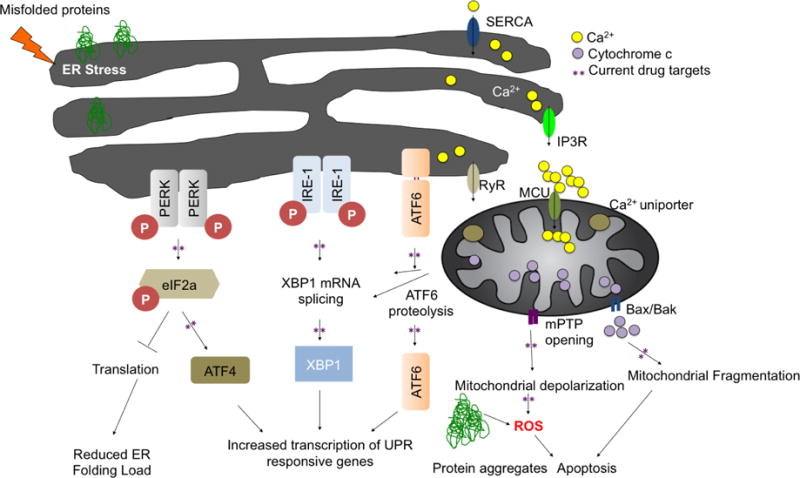
The unfolded protein response consists of three independent signaling pathways that work in parallel and are activated upon accumulation of misfolded proteins inside the ER. ER-stress sensors: RNA-activated protein kinase R (PKR)-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) are membrane resident proteins. The intracellular systems involved in regulating Ca2+ include IP3R: inositol-1,4,5-trisphosphate receptor, RyR: ryanodine receptor, SERCA: ER Ca2+-ATPase at the ER and mPTP: mitochondrial permeability transition pore Bax/Bak and mitochondrial uniporter at the mitochondria. ER stress increases Ca2+ release thus overloading mitochondria, leading to mPTP opening and cytochrome c release. Activation of the UPR leads to an overall translational block and specific activation of ER stress responsive genes, which will increase the protein folding capacity and decrease the protein-folding load in the ER.
B. ER stress and disease
Protein misfolding and abnormal aggregation is a common theme observed in most of the neurodegenerative diseases; including Huntington’s disease, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis as well as other neurodegenerative diseases involving protein defects (9–11). The abnormal protein aggregation observed in these diseases involves the accumulation of small oligomers, fibrils and large amyloid aggregates.
1. Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease causing progressive motor abnormalities and cognitive defects (12). HD is characterized by a widespread neuronal dysfunction and selective neurodegeneration, particularly in the striatum. It arises from an expanded CAG repeat, coding for a polyglutamine (polyQ) tract in the huntingtin (Htt) protein. The average polyQ length is ~18 glutamines on Htt gene, which does not confer neurotoxic activity, but expansions exceeding 35 glutamines result in disease development (13). Expression of mHtt (mutant huntingtin) in yeast and mammalian cells induces UPR (14). Furthermore, neurons of animal expressing mHtt up-regulate Rrs1, a regulator of ribosome synthesis, through ER stress, also suggests a link between ER stress and HD (15). UPR markers such as Chop, BiP, XBp1 and Herp are up regulated in postmortem samples of HD patients (16). mHtt protein associates with the ER and may play a direct role in induction of ER stress. Expanded polyQ on mHtt may compromise the proteasomal degradation of misfolded proteins in the ER, leading to ER stress (17). Furthermore, XBP1-deficient mice are more resistant to developing disease symptoms, showed improved neuronal survival and motor performance, as well as a drastic decrease in mHtt levels (18, 19). By contrast, ATF4 deficiency showed no effect, highlighting the involvement of XBP-1 in HD pathogenesis (18, 19). Treatment with ER stressors, tunicamycin or dithiothreitol (DTT), rapidly decreased BiP-GFP mobility in mHtt containing striatal cells and accelerated UPR activation compared with wild-type cells (20). Further, overexpressing GRP78 protected N2a cells, reduced formation of mHtt aggregates, inhibited caspase-12 activation and blocked cell death (21). SCAMP5 was recently identified as a novel regulator of the accumulation of mHtt in the striatum of HD patients and in cultured striatal neurons (22). In the study, the increase of SCAMP5 impaired endocytosis, which in turn enhanced mHtt aggregation (22). Thus, modulating ER stress could reduce mtHtt-induced striatal neuronal death and improve ER Ca2+ loss and reduce UPR activation thus providing novel therapeutic opportunities.
2. Alzheimer’s disease
Alzheimer’s disease (AD) manifests a gradual decline in many cognitive processes ultimately leading to dementia (23). These traits are accompanied by a selective neuronal and synaptic loss in cortical and subcortical regions, deposition of extracellular senile plaques, mainly composed of amyloid β (Aβ) peptide, presence of intracellular neurofibrillary tangles (NFT) containing hyperphosphorylated tau protein, and cerebral amyloid angiopathy in the brains of AD patients (24, 25). Accumulation of unfolded or aggregated proteins, increased oxidative stress, and metabolic disturbances are characteristic features of AD. Exogenous Aβ induces ER stress in primary neuronal cultures and activates mitochondria- and ER-mediated cell death pathways (23). Brains of AD patients have been found to have increased expression of ER chaperone, BiP, and the activated, phosphorylated form of UPR sensor PERK, as well as IRE1 and eIF2α in the temporal cortex and hippocampus (26). In AD brains, the ER stress marker GRP78/BiP positively correlated with Braak clinical staging (26). Other markers of ER stress, including cleaved caspase-12, cleaved caspase-3, cleaved caspase-4, and CHOP, were elevated in the temporal cortex of AD patients (27, 28). PERK knockdown in neuronal cells enhanced Aβ toxicity through reduced activation of eIF2α (28). Furthermore, pharmacological activation of the eIF2α pathway significantly reduced caspase-4-dependent apoptosis in Aβ treated neurons (28). These results suggest that the PERK-eIF2α pathway may play a role in cell survival, rather than apoptosis during ER stress. Interestingly, pathological events described in AD, such as neurofibrillary tangles, neuroinflammation, altered calcium signaling, and excitotoxicity, were also recently linked to the occurrence of pathological ER stress. Similarly, iPSC-derived neurons and astrocytes from APP-linked familial and sporadic AD patients accumulated Aβ oligomers, leading to ER and oxidative stress (29). ER stress may interfere with the normal trafficking of APP through the normal secretory pathway, leading to the production of Aβ, and subsequent toxicity (30). Taken together, there is mounting evidence to support that ER stress is a trigger for Aβ formation that leads to prolonged UPR activation and ER Ca2+ release. Hence, it is an attractive target for therapeutic intervention to halt or slow down the disease progression.
3. Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra in the midbrain. It is characterized by motor symptoms including bradykinesia, rigidity, resting tremor, and postural instability (31). While the etiology of PD is unknown in most cases, degenerating neuronal populations in PD exhibit α-synuclein abnormalities and mutations in that gene cause familial PD, indicating that the α-synuclein abnormalities are mechanistically linked to pathogenesis of PD (31). The genes involved in autosomal recessive PD include Parkin (PARK2), PTEN-induced putative kinase 1 (PINK1; PARK6), DJ-1 (PARK7), and ATP13A2 (PARK9), whereas the genes involved in autosomal dominant PD include α-synuclein (PARK1/4), leucine-rich repeat kinase 2 (LRRK2; PARK8), and ubiquitin carboxy-terminal hydrolase L1 (UCHL-1; PARK5) (32, 33). Many reports suggest the involvement of ER stress in the pathology of Parkinson’s disease (34, 35). In yeast, the major physical target of α-synuclein is Rab1, an essential component of the ER-to-Golgi trafficking machinery. Overexpression of Rab1 and some of its homologs rescues dopaminergic neuron loss induced by α-synuclein overexpression (36). UPR activation is noted in PD post-mortem tissues (31) and ER stress markers are up-regulated in the brain of α-synuclein transgenic mice (37, 38). The two hallmarks of ER stress – PDI and Bip levels were substantially increased also in cortical neurons derived from iPSCs of PD patients (31). Mutations in the serine/threonine kinase and GTPase LRRK2 are the most frequent genetic defect identified in PD patients, and LRRK2 partially localizes to the ER in dopaminergic neurons of PD patients (39). C. elegans, lacking the LRRK2 homolog develop spontaneous neurodegeneration and hyper-susceptibility to experimental ER stress and LRRK2-mediated neuroprotection involves the up regulation of Bip through p38 signaling (40). The diverse functional roles of LRRK2 have been under recent and extensive investigation. LRRK participates in the regulation of mitochondrial function, autophagy, and cytoskeletal dynamics (41). Most relevant to the topic of this review, LRRK2 has been shown to regulate calcium release from lysosomes through NAADP-receptors (42). It is therefore not surprising that mutations in LRRK2 result in mitochondrial depolarization and disruption in calcium homeostasis in a cultured cortical neuron model (43). Taken together, perturbation of ER homeostasis is emerging as a common pathological event triggered by genes linked to PD. The mechanisms of action for individual mutant PD genes may involve diverse pathways, but they may culminate in a final related outcome that includes pathogenic ER stress. Thus, targeting ER stress might be a potential therapeutic target for PD.
4. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is the most frequent adult-onset motor neuron disease and is characterized by selective loss of motor neurons. Approximately 90% of ALS patients have sporadic ALS (sALS) and about 10% have the inherited or familial form of ALS (fALS). Changes in ER morphology have been observed in ALS patients and the G93A-SOD1 ALS model in mice (44–46). Enhanced phosphorylation of eIF2α and increased levels of the ER foldase PDIA1 along with elevated levels of oxidized proteins and PERK, IRE1, and ATF6 were observed in spinal cords of ALS patients (44, 47, 48). Upregulation of the ER foldase ERp57, XBP-1 and ATF4 have been reported in sALS and fALS (49). Neuronal cells expressing mutants of FUS, TDP-43, C9ORF72 and in TDP-43 animal models exhibit ER stress (46, 50, 51). Impairment of protein transport between the ER and Golgi has been thought to be a major factor in driving the ER stress in ALS. Overexpression of Rab1 prevented ER stress in cells expressing mutant SOD1, TDP-43 and FUS (52). Furthermore, the presence of Rab1-positive inclusions in motor neurons of human spinal cord tissues from sALS patients implies that Rab1 is misfolded and loses its normal vesicular distribution in sALS. SOD1G85R/Perk+/− transgenic mice have an earlier activation of the UPR and apoptosis pathways than G85R transgenic mice; an enhanced and accelerated accumulation of misfolded SOD1 in SOD1G85R/Perk+/− transgenic mice was also observed (47, 53). In another study, increased expression of activated caspase-12 was observed in both the pre-symptomatic and the late symptomatic stages (54). Thus, the evidence linking ER stress and Ca2+ deregulation with misfolding and aggregation of several ALS-associated proteins suggests that prevention of dysfunctional ER would limit motor loss and improve the outcome in ALS patients.
C. Recent drug development targeting ER stress pathways
Due to the mounting evidence for UPR activation and ER stress in neurodegenerative diseases, various drug strategies are being pursued. Salubrinal was first identified as a selective inhibitor of cellular complexes that dephosphorylate eIF2α (5); Salubrinal treatment showed positive outcomes through the reduction of protein misfolding overload and thus overall ER stress in multiple disease models (55, 56). Salubrinal also inhibits the expression of proteoglycans and favors neurite outgrowth from cortical neurons and significantly reduced kainic acid-induced ER stress and neuronal death in vivo and in vitro (57, 58). It significantly increased the Grp78/Bip ER chaperone resulting in attenuation of caspase-4 dependent apoptosis in Aβ treated neurons (28). In SOD1G93A mice, Salubrinal decreased muscle strength loss and extended survival (59). GSK2606414, a inhibitor of PERK, is neuroprotective in mice against damage caused by prions, and prevents the development of cognitive deficits and other clinical manifestations of prion disease (60, 61). Even when dosed in the symptomatic phase of the prion disease, GSK2606414 significantly reduced neuronal cell death (61). Recently, an optimized version of GSK2606414, GSK2656157, was developed, has favorable pharmacokinetics and crossed the blood brain barrier when dosed orally in mice (62, 63). Using a cell-based screen for inhibitors of PERK signaling, a small molecule, ISRIB (trans-N,N′-(cyclohexane-1,4-diyl)bis(2-(4-chlorophenoxy) acetamide) that potently (IC50 = 5 nM) reverses the effects of eIF2α phosphorylation was identified (64). ISRIB was shown to reduce the viability of cells subjected to PERK-activation by chronic endoplasmic reticulum stress as well as significant enhancement in spatial and fear-associated learning (64). ISRIB reverses the effect of eIF2α phosphorylation on translation and stress granule assembly (65). So far there have been no studies of ISRIB in neurodegenerative disease models. AMG PERK44 was recently developed as a potent and selective PERK inhibitor (IC50 = 6 nM) exhibiting >160-fold selectivity for PERK over B-Raf and GCN2, as well as a panel of 387 other kinases (66). Guanabenz, an α2-adrenergic receptor agonist has been used in the treatment of hypertension and was observed to have anti-prion activity (67, 68). Further, it was shown to be beneficial in a SOD1 as well as a TDP-43 transgenic mouse model (69, 70). PPP1R15A is a regulator subunit of protein phosphatase 1 and regulates stress-induced eIF2α. Recently, Sephin1 was identified as a selective inhibitor of stress-induced PPP1R15A and targeted disease associated with accumulation of misfolded protein (71). Oral treatment of 5 mg/kg of Sephin1 per day was shown to reduce motor deficits, motor neuron loss and the molecular defects in SOD1G93A mice (71).
IRE1, the ER transmembrane sensor that activates UPR to maintain ER and cellular function promotes cell survival, but can also initiate apoptosis via decay of anti-apoptotic microRNAs (8). Recently, multiple small molecule inhibitors that selectively block IRE1–XBP1 activation have been identified. 4μ8C, an IRE1 inhibitor, that blocks substrate access to the active site of IRE1, selectively inactivates both Xbp1 splicing and IRE1-mediated mRNA degradation (72). STF-083010, a small molecule inhibitor of IRE-1, inhibited Ire1 endonuclease activity, without affecting its kinase activity, after endoplasmic reticulum stress both in vitro and in vivo in tumor models (73). KIRA6, another IRE-1 inhibitor, preserved photoreceptor numbers in a rodent model of retinal degeneration (74). Apigenin (4′,5,7-trihydroxyflavone, Baicalein and Kaempferol, pharmacologically active agents that target the expression of ATF-6, exerted neuroprotective effects AD and PD models (75–77). However, some of these molecules need to be tested in the context of AD, PD, HD and ALS.
4-Phenylbutyric acid (4-PBA) is a chemical chaperone that eliminates the accumulation of unfolded proteins in the ER. Recently, multiple analogues of 4-PBA have been developed and are in various phases on animal studies. These analogues strongly inhibit the induction of the IRE1 and ATF6 pathways and downstream pathogenic events including activation of NF-κB and phosphorylation of Akt in ER stress-exposed cells (78, 79). Tauroursodeoxycholic acid (TUDCA), a taurine conjugate of ursodeoxycholic acid (UDCA), reduces endoplasmic reticulum stress, showed neuroprotective effects in AD, PD, HD and ALS models (80). Further, in a recently published pilot study, TUDCA was well tolerated and may be effective in ALS (81).
The Sigma-1 receptor, is a chaperone protein at the ER that modulates calcium signaling through the IP3R, which has been reported to be do down regulated in the brains of PD and AD patients as well as in lumbar spinal cord of ALS patients. The Sigma-1 receptor agonists PRE-084 and SA4503 prevented neuronal loss extended survival of SOD1G93A mice and spatial learning deficits in other ND models (82). Further studies using them would help developing new potential therapeutics.
Encouraging natural protein folding machinery through the activation of heat shock transcription factor 1 (Hsf1), which is a critical regulator of chaperone protein expression, is yet another approach to target ER stress. Treatment with Arimoclomol, a co-inducer of the heat shock response, delayed disease progression and extended the lifespan of SODG93A mice, and is currently being investigated in phase II/III clinical trial for ALS patients with SOD1 mutations (83). Celastrol and Tanespimycin, which target the heat shock protein response, are other potential drugs that have shown promise in transgenic mouse model of Alzheimer’s disease as well as ALS (84–86) (Table 1). Thus, multiple studies reviewed above are promising and suggest that targeting the cellular protein quality control system of the ER is an attractive strategy for the treatment of neurodegenerative conditions.
Table 1.
Drugs targeting the Unfolded Protein Response
| Therapeutic | Mechanism | Indication | Ref |
|---|---|---|---|
| PERK arm | |||
| Salubrinal | eIF2α -phosphorylation inhibitor | AD, ALS, HD, PD | (28, 56) |
| Guanabenz | Inhibitor of eIF2α phosphatase | AD, ALS, HD | (69, 70) |
| GSK265157/GSK2606414 | PERK kinase inhibitor | AD, ALS, HD | (60, 62) |
| ISRIB | Inhibitor of eIF2α phosphatase | Prion disease | (64, 65) |
| Sephin1 | Inhibitor of eIF2α phosphatase | ALS | (71) |
| AMG PERK 44 | PERK kinase inhibitor | Not tested | (66) |
| IRE1α arm | |||
| STF-083010 | IRE1α RNase active site inhibitor | AD, ALS, HD models | (73) |
| MKC-3946 | IRE1α RNase active site inhibitor | AD, ALS, HD models | (72) |
| 4u8C | IRE1α RNase active site inhibitor | AD, ALS, HD models | (72) |
| KIRA6 | IRE1α type II kinase inhibitor | Other models | (74) |
| OICR464 | Binds to the RNase domain of murine IRE1 | Not tested | (74) |
| OICR573 | Binds to the RNase domain of murine IRE1 | Not tested | (74) |
| ATF6 arm | |||
| Apigenin | Up regulation of ATF 6 expression | PD, AD | (75, 77) |
| Baicalein | Up regulation of ATF 6 expression | PD | (76) |
| Kaempferol | Down regulation of ATF 6 expression | AD | (221) |
| Chemical chaperones | |||
| 4-phenylbutyrate (4-PBA) | Stabilizes misfolded/unfolded proteins | AD, ALS, HD | (78, 79) |
| Tauroursodeoxycholic acid (TUDCA) | Stabilizes misfolded/unfolded proteins | AD, ALS, HD | (80, 81) |
| Miscellaneous | |||
| Arimoclomol | Co-inducer of heat shock response | ALS | (83) |
| Celastrol | Co-inducer of heat shock response | AD, ALS | (84, 85) |
| Tanespimycin | Co-inducer of heat shock response | AD, ALS | (86) |
| PRE-084 | Sigma-1 receptor agonist | AD, HD, ALS | (82) |
| SA4503 | Sigma-1 receptor agonist | ALS | (82) |
D. Dysfunctional Endoplasmic Reticulum Ca2+ in neurodegeneration
The ER is an essential organelle in the cell and is involved in functions such as folding, assembly, quality control of secretory and membrane proteins, lipid biosynthesis as well as Ca2+ storage and signaling (87–89). Ca2+ homeostasis is fundamental for cell metabolism, proliferation, differentiation, and cell death. A range of Ca2+ channels are involved in Ca2+ signaling that have been discussed elsewhere (88). In the plasma membrane, ionotropic receptor-operated (ligand-gated) channels (ROCs), and voltage-gated Ca2+ channels (VGCCs) control the entry of Ca2+ into the cell. These are critical in maintaining in Ca2+ homeostasis that are reported to be altered in neurodegenerative diseases and have been reviewed elsewhere (90, 91). VGCCs comprise two groups of high voltage-activated channels, dihydropyridine-sensitive Cav1 (L-type) channels and Cav2 (P/Q-, N- and R-type) channels, as well as low voltage-activated Cav3 (T-type) channels (92). Patients with amyotrophic lateral sclerosis possess antibodies (ALS IgGs) that bind to L-type skeletal muscle VGCCs and inhibit L-type calcium current (93). Subcutaneous delivery of isradipine, inhibitior of CaV1.3 L-type Ca2+ channels, significantly protected SNc neurons in animal models of PD. Inositol-1,4,5- trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) are the two ubiquitously expressed major intracellular Ca2+ channels (ICCs) that release ER Ca2+ stores. IP3Rs constitute a family of three paralogs (IP3R1, IP3R2, IP3R3) and are critical for Ca2+ mediated processes such as gene expression, neurotransmission, memory formation, cell growth and development. Mice either lacking IP3R1 or carrying a spontaneous mutation in IP3R1 gene display severe ataxic behavior. Similar to IP3Rs, RyRs are also found in three paralogs (RyR1, RyR2, and RyR3) and are extremely important especially in neurons. Aberrant intracellular Ca2+ signaling has been observed in a variety of neurodegenerative diseases including AD, ALS, HD as well as spinocerebellar ataxia. Using single channel recordings of IP3Rs it was observed that expression of mutant PS1 and PS2 is associated with an apparent sensitization of the IP3R channel to IP3, resulting in enhanced channel gating (94). IP3R levels are reduced in the brains of patients with AD and correlates with the increased occurrence of amyloid plaques and neurofibrillary tangles (95, 96). Further, IP3R activity is altered in neurons cultured from AD mouse models (97–99). Recent studies have also shown altered RyR mediated Ca2+ release in AD models (100, 101). In patients, alterations in the ryanodine receptor calcium release channel correlated with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies (102). In HD, Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by IP3R1 (103). Disruption of IP3R binding to Htt reduced glutamate induced apoptosis by stabilizing intracellular Ca2+ homeostasis in a YAC128 HD model (104). Inhibition of RyR and IP3R through Dantrolene and xestospongin C respectively, prevented neuronal death in multiple models (89, 105–107). ER Ca2+ depletion is coupled to an increase of Ca2+ entry into the cell. STIM1 and STIM2 are plasma-membrane proteins with a luminal Ca2+ sensor and interact with Orai1 proteins (108). These tetrameric Ca2+ channels in the plasma membrane are then responsible for an increased Ca2+ entry (108). Ca2+ signaling is often abnormal in neurodegenerative diseases. Studies in AD postmortem brain tissue samples and AD animal models support a role for disruption of synaptic Ca2+ regulation in the neurotoxic action of Aβ (109). Further, similar observations have been made in HD patient’s brains and animal models as well as in PD and ALS and have been reviewed (109). The depletion of ER Ca2+ stores results in activation of store-operated channels (SOCs) at the plasma membrane, mediating store-operated calcium entry (SOCE) from the extracellular space, followed by the removal of cytosolic Ca2+ and replenishment of luminal Ca2+ through sarcoplasmic/ER Ca2+-adenosine triphosphatases Presenilins (PS1 and PS2) also govern store-operated calcium entry (SOCE), a refilling mechanism for depleted intracellular calcium stores (110). Modulation of functional PS1, by either knocking out PS1 or expressing inactive PS1, markedly potentiated SOCE, while familial Alzheimer’s disease (FAD)-linked mutant PS1 or PS2 significantly attenuated SOCE and store depletion-activated currents (111). Further, purinergic stimulation induced excess calcium release from the ER stores in SOD1G93A astrocytes, resulting from the abnormal ER calcium accumulation and independent of clearance mechanisms. Pharmacological studies suggested that store-operated calcium entry was enhanced in the SOD1G93A astrocytes (112). In transgenic Drosophila HD flies and in YAC128 mice, activity of SOCE was enhanced in neuronal cells expressing mutant Htt (113, 114). Thus, dysfunctional intracellular calcium homoeostasis is a central cause of neurodegeneration.
E. Mitochondria-associated ER membranes in neurodegenerative diseases
ER and mitochondria establish a tight interplay, which is structurally and functionally modulated through a proteinaceous tether formed at specific subdomains of the ER membrane, designated mitochondria-associated membranes or MAMs. Dysfunctional ER affects mitochondrial dynamics due to the functional and morphological connectivity between the two highly dynamic organelles. The main functions that have been characterized for ER/mitochondria contacts include control of lipid biosynthesis, mitochondrial division, Ca2+ signaling and coordinated dynamics of the two organelles (115). Many proteins are enriched at the MAMs (116, 117). VDAC1, a mitochondrial protein, is physically linked to the ER Ca2+ release channel IP3R through the molecular chaperone glucose-regulated protein 75 (grp75), optimizing calcium transfer from IP3Rs to mitochondria (Figure 2) (118). Sigma-1 receptor chaperones at the ER–mitochondrion interface regulating Ca2+ signaling and cell survival (119). Mfn2, the mitochondrial fusion protein, controls ER–mitochondria juxtaposition and calcium signaling on the MAM as well as modulates the UPR and mitochondrial function via repression of PERK (120). Bap31 is an ER protein sorting factor proposed to escort integral membrane client proteins from their biogenesis site at the Sec61 translocon to other ER complexes that determine their fate in the ER. Further, outer membrane fission protein of Fis1, makes physical contact with ER-localized Bap31. Prolonged ER stress leads to release of Ca2+ from the ER lumen at the MAM and an increased Ca2+ uptake into the mitochondrial matrix (115). Sustained Ca2+ accumulation in the mitochondrial matrix induced by ER stress triggers proapoptotic mitochondrial alteration, including permeability transition, dissipation of the electrochemical potential, matrix swelling, relocalization of Bax to mitochondria and the release of cytochrome c and apoptosis-inducing factor from mitochondria (121, 122). The spacing between the ER and mitochondria changes with different cell physiological and pathological conditions and artificial modification of this contact can lead to ER stress (123). MAM function and ER/mitochondrial communication was significantly increased in presenilin-mutant cells and in fibroblasts from patients with both the familial and sporadic forms of AD (124). Similar observations were made in human AD brain and related AD mouse and neuronal cell models (125). Furthermore, MAM is a site of production of Aβ (126). Overexpression of both wild-type and familial ALS/FTD mutant TDP-43 reduced ER–mitochondria associations (127). DJ-1, a PD associated gene, counteracts mitochondrial impairment induced by the tumor suppressor protein p53 by enhancing ER/mitochondria tethering (128). Together, the above findings highlight damage to ER/mitochondria associations as a new pathogenic mechanism in AD, PD, and ALS/FTD. Thus, correcting damaged ER/mitochondria associations may correct damage to other neurodegenerative disease-linked features.
Figure 2. ER-mitochondria contacts.
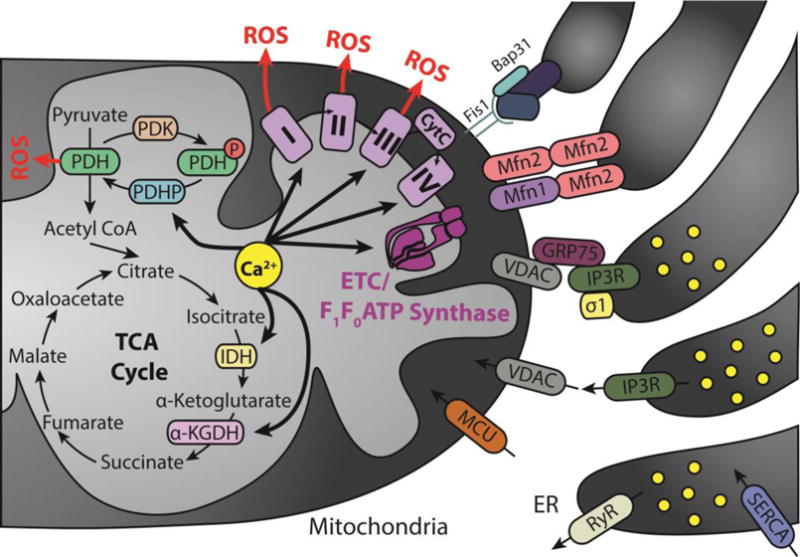
VDAC is an important regulator of Ca2+ transport in and out of the mitochondria. Accumulation of Ca2+ into the mitochondrial matrix occurs via the mitochondrial Ca2+ uniporter (MCU), which rapidly accumulates Ca2+ across the steep electrochemical gradient. Mitofusin 2 (MFN2) along with its role in mitochondrial fusion regulates mitochondrial and endoplasmic reticulum morphology and tethering. Sigma 1 receptor stabilizes IP3Rs when ER Ca2+ stores are depleted ensuring proper Ca2+ fluxes from the ER to the mitochondria. GRP75 affects the activity of ion channels by mediating the interaction of VDAC1 with IP3Rs, and thus facilitates mitochondrial Ca2+ uptake. Also, Fis1, a mitochondrial fission protein and Bap31 at ER, bridge the mitochondria–ER interface to establish a platform for apoptosis induction. Calcium directly activates pyruvate dehydrogenase phosphatase (PDHP), isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (α-KGDH), complexes I, III, and IV, as well as ATP synthase. Pyruvate dehydrogenase (PDH) complexes I, II, and III are also sites of ROS production. Abbreviations used in the illustration not mentioned above: pyruvate dehydrogenase kinase (PDK), phosphate group (P), and cytochrome c (CytC).
F. Calcium is a physiological activator of mitochondrial functions
After reaching what was considered saturation over two decades ago, advanced microscopy techniques, better biological probes, and more powerful pharmaceutical perturbations resulted in renewed interest in mitochondria biology. These tools demonstrated that mitochondria are not static organelles, but form networks that continuously undergo morphological change. In addition, mitochondria spatiotemporally modulate cell calcium levels and produce reactive oxygen species (ROS) that play essential roles in both pathological (e.g. apoptosis) and physiological (e.g. bioenergetics) signaling (129, 130). Here we focus on the role of mitochondrial calcium in neurodegeneration. Whereas calcium is already well recognized as an activator of mitochondria’s canonical metabolic function, calcium regulates mitochondria’s other functions, such as mitochondrial dynamics and ROS production that are intimately associated with neuronal function.
G. Calcium is an activator of mitochondrial metabolic function
Calcium is positive effector of mitochondrial energetics. This feature is particularly relevant in neurons as the brain utilizes approximately 20% of the body’s oxygen consumption while only comprising 2% of body mass (131, 132). Neurons specifically rely on mitochondria’s oxidative phosphorylation (OXPHOS) function due to their lower glycolytic capacity (133). This is because neurons do not generate fructose-2,6-bisphosphate, an activator of 6-phosphofructo-1-kinase, which regulates the rate-limiting step of glycolysis (134). Calcium is a potent activator of OXPPHOS, resulting in approximately a 3-fold reversible increase in respiration (135, 136). Treatment with either a competitive (La2+) or a non-competitive (ruthenium red) inhibitor of calcium transport results in a decrease of oxygen consumption by isolated mitochondria (137). The mechanisms by which calcium stimulates mitochondrial respiration through the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC) in neurons have been previously reviewed (132). In the TCA cycle, calcium directly binds and activates isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and pyruvate dehydrogenase phosphatase, an activator of pyruvate dehydrogenase (as shown in Figure 2). In parallel, calcium also activates each complex in the ETC in isolated mitochondria by approximately two-fold (138). It is unlikely that calcium directly binds complexes of the ETC (139, 140), but it may regulate post-translational modifiers. Importantly, physiological levels of calcium are necessary to respond to the energetic demands of neurons by stimulating components of both the TCA and ETC (132).
H. Calcium, ROS, and Neurodegeneration
1. Calcium is a mediator of mitochondrial ROS production
Mitochondria are a major source of cellular ROS (129); approximately 0.1% to 0.2% of oxygen consumed is associated with leakage of electrons and results in the production of ROS (141). At least 10 sites of mitochondrial ROS production have been identified including the ETC and pyruvate dehydrogenase (Figure 2). Because calcium stimulates mitochondrial metabolism through both the TCA and ETC, calcium can induce excessive ROS through stimulating mitochondrial metabolism. Not surprisingly, a higher metabolic rate increases mitochondrial ROS production (142). Other mechanisms of calcium-dependent mitochondrial ROS generation have been previously reviewed (130, 143). Calcium does not always increase mitochondrial ROS production. For example, calcium stimulation of mitochondria from brains did not result ROS production in the presence of antimycin A, a complex III inhibitor, but did in the presence of rotenone, a complex I inhibitor (144). Therefore, whether calcium induces an increase or decrease in mitochondrial ROS production depends on the source of the mitochondria, their metabolic state (brain mitochondria produce more ROS in state 4 rather than state 3), and the presence of electron transport chain inhibitors (143).
2. Excessive ROS as a therapeutic target for neurodegeneration
The Harman free radical theory from 1956 suggested that ROS accumulate spontaneously and in response to the environment (145). Neurodegenerative pathogenesis including AD, PD, ALS, and HD are associated with increasing accumulated ROS (146–148). A transcriptional profiling study of postmortem frontal cortex samples showed a decrease in expression of genes involving mitochondrial function and an increase in genes responsible for oxidative stress response after the age of 40 (149). In addition, manganese superoxide dismutase (SOD2) knockout mice exhibit neurodegeneration in the basal ganglia and the brainstem and motor deficiencies (150). SOD2 serves as the primary defense enzyme against increasing levels of mitochondrial ROS. It is therefore not surprising that lowering levels of ROS posed as an attractive therapeutic target for neurodegeneration.
Unfortunately, targeting ROS using generalized antioxidants has been ineffective in multiple neurodegeneration models including AD, ALS, and PD (Table 2). For example, a comprehensive randomized clinical trial for the treatment of AD using either a combination of vitamin E, vitamin C, and α-lipoic acid or coenzyme Q showed that neither treatment led to changes in biomarkers obtained from cerebrospinal fluid samples including Aβ42, tau, and p-tau. Treatment with the combination of antioxidants led to a decrease in F2-isoprostane, a marker of oxidative stress. However, this was accompanied by an accelerated decline in Mini-Mental State Examination scores (151). Furthermore, a high dose of coenzyme Q in another large randomized clinical trial showed no effect in patients with PD (152). Finally, edaravone, a potent ROS scavenger, showed no efficacy in a phase III clinical trials for ALS (153). Sadly, the lack of efficacy of antioxidants as therapeutics is not limited to neurodegenerative disorders and has been also observed in cardiovascular disorders as well (129).
Table 2.
Drug targeting mitochondrial dysfunction
| Therapeutic | Mechanism | Indication | Ref |
|---|---|---|---|
| General antioxidants | |||
| Vitamin E | General antioxidant | Alzheimer’s Disease (potentially harmful, delay of progression) Mild Cognitive Impairment (no effect) |
(151, 222, 223) |
| Vitamin C | General antioxidant | Alzheimer’s Disease Clinical Trial (potentially harmful) | (151) |
| α-lipoic acid | General antioxidant | Alzheimer’s Disease Clinical Trial (potentially harmful) | (151) |
| Coenzyme Q | General antioxidant | Alzheimer’s Disease Clinical Trial (no effect) Parkinson’s Disease Clinical Trial (no effect) ALS Clinical Trial (no effect) |
(151, 152, 224–226) |
| Curcumin | General antioxidant | Alzheimer’s Disease Clinical Trial (increased plasma Aβ) | (227) |
| Melatonin | General antioxidant | Alzheimer’s Disease Clinical Trial (no effect) | (228) |
| Pramipexole | General antioxidant | Parkinson’s Disease Clinical Trial (no effect) | (229) |
| N-Acetylcysteine | General antioxidant | Parkinson’s Disease Clinical Trial (increase brain and blood glutathione) | (230) |
| Edaravone | General antioxidant | ALS Clinical Trial (no effect) | (153) |
| Mitochondria-targeted therapies | |||
| TPP+-conjugated ROS Scavengers (MitoQ) |
Mitochondria-targeted antioxidant | Parkinson’s Disease Clinical Trial (no effect) | (164) |
| Dimebon (latrepiridine) | L-type calcium channel blocker, MPTP inhibitor | Huntington’s Disease Clinical Trial (no effect) Alzheimer’s Disease Clinical Trial (beneficial phase II, no effect phase III) |
(156–158) |
| Dexpramipexole | Mitochondria-targeted antioxidant | ALS Clinical Trial (no effect) | (231) |
| Aromatic-cationic peptides (SS31 or Bendavia, SS20) | Mitochondria-targeted antioxidant | ALS SOD1 mouse model (delayed onset) MPTP neurotoxicity in mice (neuroprotection) |
(161, 162) |
| Mdivi-1 | General inhibitor of Drp1 | Cell culture model of propofol toxicity (decreased cell death) Cell culture model of Parkinson’s Disease (rescued mitochondrial structural and functional defects) |
(197, 198) |
| P110 | Inhibition of mitochondrial Drp1 recruitment through Fis1 | Cell culture model of Parkinson’s Disease (increased mitochondrial function) Cell culture model and mouse model of Huntington’s Disease (increased mitochondrial function, mouse survival, and mouse behavior). |
(204) (205) |
| CyclosporinA | Calcineurin inhibitor, cyclophilin D inhibitor | No indication (off-target effects leading to immunosuppression). | (218) |
| Antmanide | cyclophilin D inhibitor | (220) | |
General antioxidants have multiple theoretical limitations that hinder them from use in the clinic. First, antioxidants that act as ROS scavengers, such as vitamin E or C, must be provided in stoichiometric ratios relative to excessive ROS. This translates to larger doses and for prolonged periods of time for chronic disorders. Second, ROS scavengers such as N-acetylcysteine are unstable due to auto-oxidation even prior to treatment. Auto-oxidized ROS scavengers become oxidants and therefore increase oxidative stress (154). Finally, ROS have important physiological functions including signaling of stress response, differentiation, and immunity as reviewed elsewhere (129, 155). Therefore, eliminating ROS below physiological levels may be detrimental to physiological processes and may exampling potential harmful effects of some antioxidants (151). Calcium, like ROS, has physiological roles, as discussed above. Therefore, it is not surprising that drugs such as dimebon (latrepiridine), an L-type calcium channel blocker were inefficient in clinical trials for both AD and HD (156–158).
An alternative therapeutic strategy to generalized antioxidants is mitochondria-targeted antioxidants. By targeting the sites of ROS production directly, these antioxidants may eliminate some of the limitations of generalized antioxidants. For example, the aromatic-cationic peptides SS31, or Bendavia, and SS20. These synthetic peptides are localized to the inner mitochondrial membrane rather than the mitochondrial matrix. While the mechanism of action of these peptides is not full understood, the SS31 peptide scavenges ROS through a dimethyltyrosine group, but also reduces mitochondrial ROS production and prevents opening of the mitochondrial permeability transition pore (mPTP) (159). Furthermore, SS31 inhibits cytochrome c interactions with cardiolipin, enabling cytochrome c to act as an effective electron carrier (160). In a SOD1 mutant ALS mouse model, daily injections of SS31 led to improvement in survival and motor performance associated with a decrease in neuronal cell death (161). In a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity PD mouse model, SS31 increased viability of dopaminergic neurons and a dose-dependent rescue of dopamine loss (162). Therefore, SS31 may provide effective treatment for neurodegenerative diseases. In addition to the aromatic-cationic peptides, attachment of the lipophilic triphenylphosphonium cation (TPP+) to a general antioxidant enabled its delivery to the mitochondrial matrix. One limitation of this mechanism is that compound delivery requires an active mitochondrial membrane potential (159). Mitochondria produce excessive ROS when their membrane potential is compromised and ATP production is inefficient. Therefore, antioxidants must to target the mitochondria regardless of their membrane potential to effectively eliminate the production of excessive ROS. Nevertheless TPP+-conjugated ROS scavengers have been developed including MitoVitE (mitocopherol), MitoTEMPOL (TPP+ conjugated to 4-hydroxy-2,2,6,6,-tetramethyl-piperidine-1-oxyl), SkQ1 (TPP+ conjugated to plastoquinone), MitoPBN (TPP+ conjugated to phenoxy-butyl-nitrone), and MitoQ (TPP+ conjugated to coenzyme Q) (163). The most studied, MitoQ, showed cardioprotection in several animal models and was shown to be well tolerated in a Phase I clinical study (164). However, MitoQ failed to show efficacy in PD patients (164). While potentially more efficacious than generalized antioxidants, the relevance of mitochondria-targeted antioxidants in humans is yet to be shown.
I. Calcium regulates mitochondrial morphology and intracellular transport
Rather than pharmacologically targeting ROS as an output of dysfunctional mitochondria, another therapeutic strategy involves directly targeting dysfunctional mitochondria and improving overall mitochondrial health. Neurons’ complex shape and energetic demands pose a challenge for mitochondria. Neurons have three main structural domains: the cell body (soma), a long axon, and branch-like dendrites. Areas such as postsynaptic and presynaptic terminals are remote from the cell body yet require a higher ATP supply and a more responsive calcium buffering system (165). Release of neurotransmitters at presynaptic termini is mediated by calcium flow through voltage-gated channels and an increase of cytoplasmic calcium (Ca2+cyto). To restore presynaptic termini, mitochondria must sequester Ca2+cyto to prepare for an additional neuronal firing (166). Mitochondrial biogenesis and degradation primarily occurs in the cell body. Therefore, mitochondria must have dynamic mechanisms to regulate mitochondrial number, shape, and subcellular localization, to meet the energetic and calcium buffering demands of specialized cells like neurons (165). The previous view of mitochondria as static, uniformly shaped organelles fails to explain this biology.
1. Calcium regulates mitochondrial morphology
One of the observations that reignited mitochondrial biology research is that within a cell, thousands of mitochondria form a dynamic network (167). The functionality of this network is dependent both on the shape and localization of mitochondria. The number and morphology of mitochondria are both mediated by two opposing processes that are at the core of mitochondrial dynamics: fission and fusion (Figure 3A). Fission, the process by which a single mitochondrion divides into two or more mitochondria increases mitochondrial number and regulated mitochondrial quality control. Damage segregates asymmetrically and fission separates damaged from healthy mitochondria (168). Mitochondrial fission is primarily mediated by dynamin-related protein 1 (Drp1), a large GTPase that constricts the mitochondrial membrane to lead to scission (169, 170). Conversely, mitochondrial fusion is the stitching of two or more mitochondria to form a single mitochondrion. Fusion facilitates complementation of mitochondrial DNA and proteins (168). Three large GTPases drive mitochondrial fusion: mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) that stitch the outer mitochondrial membrane, and optic atrophy 1 (Opa1), responsible for joining the inner mitochondrial membrane (171, 172). Maintaining a balance between these two processes is essential for maintaining mitochondrial network homeostasis and cell viability (168).
Figure 3. Calcium regulates mitochondrial dynamics and transport.
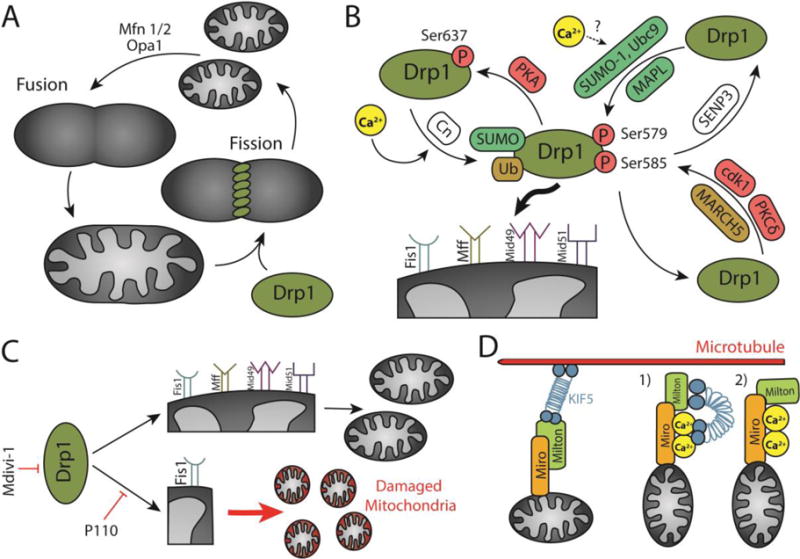
A) Mitochondrial fission/fusion cycle. Mitochondrial fusion is mediated by the outer mitochondrial membrane GTPases Mfn1 and Mfn2, and the inner mitochondrial membrane GTPase Opa1. Mitochondrial fission is regulated by the cytosolic GTPase Drp1 that anchors on the outer mitochondrial membrane. B) Post-translational modifications of Drp1 determine its localization in the cytosol or the mitochondrial membrane. PKA, cdk1, and PKCδ phosphorylate Drp1. Calcineurin (Cn) dephosphorylates Drp1. SUMO-1, Ubc9, and MAPL SUMOylate Drp1. SENP3 deSUMOylates Drp1. MARCH 5 ubiquitinates Drp1. Calcium is an activator of calcineurin and potentially Ubc9, thus promoting Drp1 translocation to the mitochondria. Drp1 is anchored to the mitochondrial membrane through four adaptors: Fis1, Mff, Mid49, and Mid51. C) Drp1 drives both physiological fission and Fis1-dependent pathological fission. While mdivi-1 is a general inhibitor of Drp1, P110 selectively inhibits pathological fission. D) Mitochondria traverse microtubules through motor adaptor complexes including Miro, Milton, and KIF5. Calcium detaches mitochondria from microtubules through two potential mechanisms: 1) KIF1 detachment from microtubules and attachment to calcium-bound Miro, and 2) Milton detachment from KIF5 and attachment to calcium-bound Miro.
Calcium mediates mitochondrial network dynamics by regulating mitochondrial morphology through modulating the expression and localization of several factors, including Drp1, the canonical driver of mitochondrial fission. Drp1 is post-translationally regulated by multiple modifiers including cyclin-dependent kinase I (cdk1)/cyclin B phosphorylation at Ser585 and Ser616, cyclic AMP-dependent protein kinase (PKA) phosphorylation at Ser637, calcineurin dephosphorylation at Ser637, delta protein kinase C (PKCδ) phosphorylation at Ser579, SUMOylation by SUMO-conjugating enzyme Ubc9, SUMO-1, and mitochondrial anchored RING-finger containing protein (MAPL), deSUMOylation by SUMO1 specific protease 3 (SENP3), and mitochondrial E3 ubiquitin ligase MARCH5 (173–177). These post-translational modifications are summarized in Figure 3B.
The role of calcium in modulating localization of Drp1 is best shown in the context of the phosphorylation state of Drp1 at Ser637 (178–180). Drp1 is primarily localized in the cytoplasm when Ser637 is phosphorylated. Upon activation by calcium, calcineurin dephosphorylates Drp1 at Ser637, which in turn signals Drp1 localization to the mitochondrial membrane, where it induces mitochondrial fission. In an in vivo mouse heart model, activation of PKA with forskolin induces phosphorylation of Drp1 at Ser637. Addition of BayK8644, an L-type Ca2+ channel activator, together with forskolin reduces phosphorylated Drp1 at Ser637 (178). In a HeLa cell culture model, depolarizing mitochondria with arachidonic acid or carbonylcyanide-p-trifluoromethoxy phenylhydrazone increases cytosolic calcium and results in fragmented mitochondria. However, addition of cyclosporine A, an inhibitor of calcineurin, both reduces calcineurin catalytic activity and decreases mitochondrial fission (180). It is important to note that in these two examples, calcium-induced mitochondrial fission is induced by stress. However, mitochondrial fission is an essential physiological process. Drp1 homozygote knockout mice are embryonic lethal with developmental abnormalities in the forebrain (181, 182) and neural-specific Drp1 homozygote knockout mice have brain hypoplasia and die shortly following birth. Primary neuronal culture from the forebrain of these mice have defective synapse formation due to inability of mitochondria to distribute properly (181). These findings suggest that calcium, through the activation of calcineurin, may have a role in the activation of Drp1 and mitochondrial fission under physiological conditions as well. It is possible that levels of dephosphorylated Drp1 at Ser637 and therefore frequency of mitochondrial fission may dependent on localized calcium concentrations.
Calcium regulation of other post-translational modifiers of Drp1 remains an unexplored area of research. For example, calcium may regulate SUMOylation of Drp1 (183, 184). However, in these reports, calcium did not directly regulate SUMOylation, but instead, mediated a switch from SUMOylation to acetylation through calcineurin-dependent dephosphorylation (184). A calcium-dependent PKC signaling pathway was recently found to regulate trapping of Ubc9, a SUMOylating enzyme, to dendritic spines in cultured hippocampal neurons (185). Ubc9-dependent SUMOylation of Drp1 is associated in greater accumulation of Drp1 at the mitochondrial membrane (186), yet increased mitochondrial fission at dendritic spines due to calcium-dependent Ubc9 localization has not been reported. Therefore, calcium regulation of mitochondrial fission through phosphorylation-independent post-translational modification of Drp1 still remains to be explored.
More recent reports have attributed calcium-dependent changes in mitochondrial morphological shape to processes may be independent of Drp1-mediated mitochondrial fission (187, 188). In cortical astrocytes, treatment with a calcium ionophore, 4Br-A23187, increases intracellular calcium levels and leads to a decrease in mitochondrial length (187). Yet, calcineurin inhibitors, CsA and FK506, did not inhibit 4Br-32187-mediated reduction in mitochondrial length (187), indicating a calcineurin-independent mechanism. However, calcineurin inhibitors attenuate the 4Br-A23187-induced increase in mitochondrial number, suggesting that calcium-mediated changes in mitochondrial morphology can occur independently of a complete fission event. The fission-independent remodeling process is mediated through reactive oxygen species (ROS) as treatment with the antioxidant N-acetylcysteine (NAC) does not increase mitochondrial in combination with 4Br-A23187 treatment but attenuates mitochondrial length shortening (188). Therefore, it is not clear from these findings whether mitochondrial remodeling occurs independently of the canonical fission protein Drp1. For example, it is possible that calcium- and ROS-mediated remodeling occurs through another post-translational modification of Drp1.
2. Mitochondrial dynamics as a therapeutic target
As mentioned earlier, mitochondrial dynamics, or the continuous cycle of mitochondrial division and amalgamation, is essential for maintaining a balanced mitochondrial network. Uncontrolled fission, through which mitochondria become fragmented and therefore too small to function properly, leads to cell death (168), and is a hallmark of numerous neurodegenerative disorders including HD, AD, PD, dementia, ataxia, and hypertension-induced encephalopathy (189–195). Excessive mitochondrial fission is therefore an attractive therapeutic target for neurodegenerative disorders. Efforts using small molecules to inhibit fission by targeting its main mediator Drp1 have led to the development of a specific inhibitor: mdivi-1 (Figure 3C) (196). Treatment with mdivi-1 in a cell culture model rescued propofol-induced toxicity in stem cell-derived neurons as well as functional and structural mitochondrial defects in a PINK1 mutant Parkinson’s cell culture model (197, 198). Unfortunately, general toxicity and developmental defects are associated with mdivi-1 in animal models (199–203). The inherit limitation of these general inhibitors is their attenuation of the less-understood physiological fission. While excessive fission is pathological, tightly-regulated physiological fission is necessary for cell survival (168). However, the mechanism distinguishing between physiological and pathological fission are only now being uncovered (129). A peptide inhibitor of Drp1 activation specifically under stress conditions has been recently developed: P110 (Figure 3C) (204). The cytosolic Drp1 anchors onto to the mitochondrial outer membrane by four separate adaptors: mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial dynamics proteins of 49 and 51 kDa (Mid49 and Mid51) (129). P110 specifically inhibits the interaction between Drp1 and Fis1 and thus rescues mitochondrial function and morphology in a 1-methyl-4-phenylpryridium neuronal cell culture model of Parkinson’s disease (204). In addition, P110 showed efficacy in both cell culture and animal models of HD (205). In patient-derived iPSCs and neurons, P110 inhibited excessive fission and restored mitochondrial function. In an R6/2 mouse model of HD, P110 improved mouse behavior and increased survival (205). Importantly, P110 had no effect on mitochondria under basal conditions (205). Small peptide inhibitors are increasingly used as pharmacological tools both in basic science research and in the clinic due to their increased conformational flexibility, selectivity, cell permeability, bioavailability, and their ability to cross the blood-brain barrier opening a new therapeutic avenue for pathological protein-protein interactions, such as the one between Fis1 and Drp1 (206–208).
3. Calcium as a regulator of mitochondrial motility
To meet the energetic and buffering challenge posed by the complexity of neuronal morphology, mitochondria must also migrate to various areas of the cell in addition to changing shape. Mitochondria rely on microtubules for both anterograde (away from the cell body) and retrograde (towards the cell body). Anterograde transport of mitochondria is primarily mediated through kinesin-1 (KIF5). KIF5 forms a complex with mitochondrial Rho GTPase (Miro 1 and Miro 2) and Milton (TRAK1 and TRAK2) (Figure 4D) (209). A recent report showed that mitochondrial bidirectional mitochondrial migration to either dendrites or axons is mediated through interactions with either TRAK1 or TRAK2 (210). TRAK1 interactions with both KIF5 and dynein/dynactin mediate mitochondrial transport to axons whereas TRAK2 interactions with dynein/dynactin promote mitochondrial migration to dendrites (210). In the context of microtubule-driven mitochondrial transport, calcium acts as a “stop sign” (211–213). In both H9C2 cardiac cells and primary neurons, overexpressing Miro increases mitochondrial movement at basal cytoplasmic calcium levels. In contrast, at high cytoplasmic calcium levels, increasing Miro overexpression results in more frequent arrest of mitochondrial motility. This arrest is mediated by two EF-hands that act as calcium binding domains (213). Two models were suggested for calcium-mediated attenuation of mitochondrial movement along microtubules: 1) upon calcium binding to Miro through the EF-hands, KIF5 detaches from microtubules and binds Miro (212), and 2) upon calcium binding to Miro, TRAK2 detaches from KIF5 (Figure 4D) (211). Therefore, calcium plays an important role in regulating mitochondrial localization and proteins associated with mitochondrial transport pose as attractive therapeutic targets.
J. Calcium as a regulator of mitochondrial permeability transition pore opening
Calcium is a potent activator of the mitochondrial permeability transition pore (mPTP) (214). At the onset of cellular pathology and associated calcium overload, mPTP opening leads to matrix expansion and membrane rupturing. Following these events, mitochondria discharge a multiple proapoptotic factors, excessive ROS, and stored calcium, abrogating the mitochondria’s calcium buffering capability (214). While the structure of the mPTP has been under great controversy, a recently suggested and more consistent model includes a dimer of F0F1 ATP synthase under the regulation of cyclophilin D and calcium (215). Pathological opening of the mPTP is associated with a variety of neurodegenerative disorders, including AD, PD, and HD (216). Cyclophilin D’s most well known inhibitor, Cyclosporin A (CsA), inhibits mPTP opening (217). Unfortunately as discussed above, CsA is also an inhibitor of calcineurin. CsA’s inhibitory interactions with calcineurin elicits immunosuppressive effects, making it a less ideal drug candidate (218). Novel candidates for CsA have been proposed, including alisporivir, SCY-635, and NIM811 as reviewed elsewhere (219). A highly specific cyclophilin D inhibitor, Antmanide, had no effect in the absence of cyclophilin D on cells in culture and isolated mitochondria. Combined with its functional efficacy, Antmanide serves as a promising drug candidate for the treatment of mPTP-mediated neurodegenerative pathologies (220). Since calcium-mediated pathological mPTP opening is associated with a range of neurodegenerative disorders, compounds that target the regulation of mPTP activity pose as strong therapeutic candidates.
Concluding Remarks
Endoplasmic reticulum (ER) stress and mitochondrial dysfunction play an important role in a range of neurodegenerative disorders, including AD, PD, HD and ALS. These diseases are characterized by accumulation of misfolded proteins and aggregates as well as by mitochondrial dysfunction. Although the contribution of these key organelles is in these disorders is now well recognized, the mechanisms by which they contribute to neurodegeneration remains unclear. However, increasing evidence obtained using disease models supports that alterations in ER homeostasis cause accumulation of disease-associated misfolded proteins, which in turn may initiate the ER stress response and contribute to synaptic and neuronal dysfunction in neurodegenerative diseases. Furthermore, calcium directly modulates mitochondrial function, dynamics and mobility. Therefore, signaling components of the UPR, ER Ca2+ signaling as well as direct regulators of mitochondrial functions are emerging as potential targets for intervention, and so the treatment of a range of neurodegenerative disorders might become possible.
Highlights.
The unfolded protein response plays a vital role in maintaining cell homeostasis as a consequence of endoplasmic reticulum stress.
ER and mitochondrial dysfunction are common themes in neurodegenerative diseases.
Targeting these pathways provides an opportunity to develop new therapeutics for neurodegenerative disease.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants HL52141 and NIAAA11147 to Dr Mochly-Rosen. OSK is supported by a Smith Stanford Graduate Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
All authors have read and approved the final article. Authors reports no conflicts
References
- 1.Scheper W, Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta neuropathologica. 2015;130(3):315–31. doi: 10.1007/s00401-015-1462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutation research. 2005;569(1–2):29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 3.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews Molecular cell biology. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 4.Hampton RY. ER stress response: getting the UPR hand on misfolded proteins. Current biology: CB. 2000;10(14):R518–21. doi: 10.1016/s0960-9822(00)00583-2. [DOI] [PubMed] [Google Scholar]
- 5.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 6.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Molecular cell. 2000;6(6):1355–64. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 7.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nature cell biology. 2005;7(8):766–72. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends in cell biology. 2013;23(11):547–55. doi: 10.1016/j.tcb.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1–40 peptide in brain endothelial cells. Biochimica et biophysica acta. 2013;1832(12):2191–203. doi: 10.1016/j.bbadis.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Current opinion in cell biology. 2011;23(2):239–52. doi: 10.1016/j.ceb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell death and differentiation. 2006;13(3):385–92. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 12.Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. The Journal of clinical investigation. 2013;123(12):5371–88. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nature reviews Neuroscience. 2005;6(12):919–30. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 14.Vidal R, Caballero B, Couve A, Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Current molecular medicine. 2011;11(1):1–12. doi: 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- 15.Carnemolla A, Fossale E, Agostoni E, Michelazzi S, Calligaris R, De Maso L, Del Sal G, MacDonald ME, Persichetti F. Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. The Journal of biological chemistry. 2009;284(27):18167–73. doi: 10.1074/jbc.M109.018325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho KJ, Lee BI, Cheon SY, Kim HW, Kim HJ, Kim GW. Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience. 2009;163(4):1128–34. doi: 10.1016/j.neuroscience.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 17.Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes & development. 2008;22(23):3308–19. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuleta A, Vidal RL, Armentano D, Parsons G, Hetz C. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington’s disease. Biochemical and biophysical research communications. 2012;420(3):558–63. doi: 10.1016/j.bbrc.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 19.Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, Glimcher LH, Hetz C. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Human molecular genetics. 2012;21(10):2245–62. doi: 10.1093/hmg/dds040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lajoie P, Snapp EL. Changes in BiP availability reveal hypersensitivity to acute endoplasmic reticulum stress in cells expressing mutant huntingtin. Journal of cell science. 2011;124(Pt 19):3332–43. doi: 10.1242/jcs.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Y, Lv H, Liao M, Xu X, Huang S, Tan H, Peng T, Zhang Y, Li H. GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neuroscience letters. 2012;516(2):182–7. doi: 10.1016/j.neulet.2012.03.074. [DOI] [PubMed] [Google Scholar]
- 22.Noh JY, Lee H, Song S, Kim NS, Im W, Kim M, Seo H, Chung CW, Chang JW, Ferrante RJ, Yoo YJ, Ryu H, Jung YK. SCAMP5 links endoplasmic reticulum stress to the accumulation of expanded polyglutamine protein aggregates via endocytosis inhibition. The Journal of biological chemistry. 2009;284(17):11318–25. doi: 10.1074/jbc.M807620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. Journal of chemical neuroanatomy. 2004;28(1–2):67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. Journal of neuroinflammation. 2009;6:41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijholt DA, de Graaf TR, van Haastert ES, Oliveira AO, Berkers CR, Zwart R, Ovaa H, Baas F, Hoozemans JJ, Scheper W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell death and differentiation. 2011;18(6):1071–81. doi: 10.1038/cdd.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G. Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nature cell biology. 2000;2(12):863–70. doi: 10.1038/35046500. [DOI] [PubMed] [Google Scholar]
- 27.Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PloS one. 2011;6(10):e26420. doi: 10.1371/journal.pone.0026420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Lee do Y, Lee KS, Lee HJ, Kim do H, Noh YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC, Jeong Y, Kim DK, Lee WB, Kim SS. Activation of PERK signaling attenuates Abeta-mediated ER stress. PloS one. 2010;5(5):e10489. doi: 10.1371/journal.pone.0010489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, Nakano R, Watanabe D, Maruyama K, Hori O, Hibino S, Choshi T, Nakahata T, Hioki H, Kaneko T, Naitoh M, Yoshikawa K, Yamawaki S, Suzuki S, Hata R, Ueno S, Seki T, Kobayashi K, Toda T, Murakami K, Irie K, Klein WL, Mori H, Asada T, Takahashi R, Iwata N, Yamanaka S, Inoue H. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell stem cell. 2013;12(4):487–96. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Barbero-Camps E, Fernandez A, Baulies A, Martinez L, Fernandez-Checa JC, Colell A. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. The American journal of pathology. 2014;184(7):2066–81. doi: 10.1016/j.ajpath.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schule B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, Jaenisch R, Lindquist S. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342(6161):983–7. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics and pathogenesis. Annual review of pathology. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- 33.Burke WJ. Recent advances in the genetics and pathogenesis of Parkinson’s disease. Neurology. 2002;59(7):1118. doi: 10.1212/wnl.59.7.1118. [DOI] [PubMed] [Google Scholar]
- 34.Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y. Endoplasmic reticulum stress and Parkinson’s disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxidative medicine and cellular longevity. 2013;2013:239854. doi: 10.1155/2013/239854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mercado G, Valdes P, Hetz C. An ERcentric view of Parkinson’s disease. Trends in molecular medicine. 2013;19(3):165–75. doi: 10.1016/j.molmed.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313(5785):324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belal C, Ameli NJ, El Kommos A, Bezalel S, Al’Khafaji AM, Mughal MR, Mattson MP, Kyriazis GA, Tyrberg B, Chan SL. The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant alpha-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Human molecular genetics. 2012;21(5):963–77. doi: 10.1093/hmg/ddr502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, Battistin L, Spillantini M, Missale C, Spano P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. Journal of neurochemistry. 2011;116(4):588–605. doi: 10.1111/j.1471-4159.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- 39.Melrose HL. LRRK2 and ubiquitination: implications for kinase inhibitor therapy. The Biochemical journal. 2015;470(3):e21–4. doi: 10.1042/BJ20150785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan Y, Cao P, Smith MA, Kramp K, Huang Y, Hisamoto N, Matsumoto K, Hatzoglou M, Jin H, Feng Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PloS one. 2011;6(8):e22354. doi: 10.1371/journal.pone.0022354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Esteves AR, Cardoso SM. LRRK2 at the Crossroad Between Autophagy and Microtubule Trafficking: Insights into Parkinson’s Disease. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2016 doi: 10.1177/1073858415616558. [DOI] [PubMed] [Google Scholar]
- 42.Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, Bloor-Young D, Patel S, Woodman PG, Churchill GC, Hilfiker S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Human molecular genetics. 2012;21(3):511–25. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherra SJ, 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. The American journal of pathology. 2013;182(2):474–84. doi: 10.1016/j.ajpath.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaronen M, Goldsteins G, Koistinaho J. ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Frontiers in cellular neuroscience. 2014;8:402. doi: 10.3389/fncel.2014.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes & development. 2008;22(11):1451–64. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK, Atkin JD. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PloS one. 2013;8(11):e81170. doi: 10.1371/journal.pone.0081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Human molecular genetics. 2011;20(5):1008–15. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. The Journal of biological chemistry. 2006;281(40):30152–65. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 49.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes & development. 2009;23(19):2294–306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farg MA, Soo KY, Walker AK, Pham H, Orian J, Horne MK, Warraich ST, Williams KL, Blair IP, Atkin JD. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiology of aging. 2012;33(12):2855–68. doi: 10.1016/j.neurobiolaging.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 51.Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, Davis-Dusenbery BN, Ziller M, Oakley D, Ichida J, Di Costanzo S, Atwater N, Maeder ML, Goodwin MJ, Nemesh J, Handsaker RE, Paull D, Noggle S, McCarroll SA, Joung JK, Woolf CJ, Brown RH, Eggan K. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell stem cell. 2014;14(6):781–95. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soo KY, Halloran M, Sundaramoorthy V, Parakh S, Toth RP, Southam KA, McLean CA, Lock P, King A, Farg MA, Atkin JD. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta neuropathologica. 2015;130(5):679–97. doi: 10.1007/s00401-015-1468-2. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Popko B, Roos RP. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Human molecular genetics. 2014;23(10):2629–38. doi: 10.1093/hmg/ddt658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vijayalakshmi K, Alladi PA, Ghosh S, Prasanna VK, Sagar BC, Nalini A, Sathyaprabha TN, Raju TR. Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiology of disease. 2011;41(3):695–705. doi: 10.1016/j.nbd.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Gong T, Wang Q, Lin Z, Chen ML, Sun GZ. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochemical and biophysical research communications. 2012;427(3):461–5. doi: 10.1016/j.bbrc.2012.08.068. [DOI] [PubMed] [Google Scholar]
- 56.Romero-Ramirez L, Nieto-Sampedro M, Barreda-Manso MA. All roads go to Salubrinal: endoplasmic reticulum stress, neuroprotection and glial scar formation. Neural regeneration research. 2015;10(12):1926–7. doi: 10.4103/1673-5374.169619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barreda-Manso MA, Yanguas-Casas N, Nieto-Sampedro M, Romero-Ramirez L. Salubrinal inhibits the expression of proteoglycans and favors neurite outgrowth from cortical neurons in vitro. Experimental cell research. 2015;335(1):82–90. doi: 10.1016/j.yexcr.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 58.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(4):901–8. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nature neuroscience. 2009;12(5):627–36. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 60.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) Journal of medicinal chemistry. 2012;55(16):7193–207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 61.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Science translational medicine. 2013;5(206):206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- 62.Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E, Mencken T, Gaul N, Goetz A, Stanley T, Hassell AM, Gampe RT, Atkins C, Kumar R. Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. ACS medicinal chemistry letters. 2013;4(10):964–8. doi: 10.1021/ml400228e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishnamoorthy J, Rajesh K, Mirzajani F, Kesoglidou P, Papadakis AI, Koromilas AE. Evidence for eIF2alpha phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications. Cell cycle. 2014;13(5):801–6. doi: 10.4161/cc.27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, Walter P. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. eLife. 2015:4. doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith AL, Andrews KL, Beckmann H, Bellon SF, Beltran PJ, Booker S, Chen H, Chung YA, D’Angelo ND, Dao J, Dellamaggiore KR, Jaeckel P, Kendall R, Labitzke K, Long AM, Materna-Reichelt S, Mitchell P, Norman MH, Powers D, Rose M, Shaffer PL, Wu MM, Lipford JR. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK) Journal of medicinal chemistry. 2015;58(3):1426–41. doi: 10.1021/jm5017494. [DOI] [PubMed] [Google Scholar]
- 67.Norez C, Vandebrouck C, Antigny F, Dannhoffer L, Blondel M, Becq F. Guanabenz, an alpha2-selective adrenergic agonist, activates Ca2+-dependent chloride currents in cystic fibrosis human airway epithelial cells. European journal of pharmacology. 2008;592(1–3):33–40. doi: 10.1016/j.ejphar.2008.06.103. [DOI] [PubMed] [Google Scholar]
- 68.Tribouillard-Tanvier D, Beringue V, Desban N, Gug F, Bach S, Voisset C, Galons H, Laude H, Vilette D, Blondel M. Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions. PloS one. 2008;3(4):e1981. doi: 10.1371/journal.pone.0001981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiology of disease. 2014;71:317–24. doi: 10.1016/j.nbd.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang HQ, Ren M, Jiang HZ, Wang J, Zhang J, Yin X, Wang SY, Qi Y, Wang XD, Feng HL. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2014;277:132–8. doi: 10.1016/j.neuroscience.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 71.Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, Sigurdardottir A, Bertolotti A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348(6231):239–42. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(15):E869–78. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117(4):1311–4. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schurer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158(3):534–48. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li S, Pu XP. Neuroprotective effect of kaempferol against a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biological & pharmaceutical bulletin. 2011;34(8):1291–6. doi: 10.1248/bpb.34.1291. [DOI] [PubMed] [Google Scholar]
- 76.Mu X, He G, Cheng Y, Li X, Xu B, Du G. Baicalein exerts neuroprotective effects in 6-hydroxydopamine-induced experimental parkinsonism in vivo and in vitro. Pharmacology, biochemistry, and behavior. 2009;92(4):642–8. doi: 10.1016/j.pbb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Patil SP, Jain PD, Sancheti JS, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective and neurotrophic effects of Apigenin and Luteolin in MPTP induced parkinsonism in mice. Neuropharmacology. 2014;86:192–202. doi: 10.1016/j.neuropharm.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H, Nakajima S, Kato H, Gu L, Yoshitomi T, Nagai K, Shinmori H, Kokubo S, Kitamura M. Selective, potent blockade of the IRE1 and ATF6 pathways by 4-phenylbutyric acid analogues. British journal of pharmacology. 2013;170(4):822–34. doi: 10.1111/bph.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mimori S, Okuma Y, Kaneko M, Kawada K, Hosoi T, Ozawa K, Nomura Y, Hamana H. Protective effects of 4-phenylbutyrate derivatives on the neuronal cell death and endoplasmic reticulum stress. Biological & pharmaceutical bulletin. 2012;35(1):84–90. doi: 10.1248/bpb.35.84. [DOI] [PubMed] [Google Scholar]
- 80.Vang S, Longley K, Steer CJ, Low WC. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Global advances in health and medicine: improving healthcare outcomes worldwide. 2014;3(3):58–69. doi: 10.7453/gahmj.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elia AE, Lalli S, Monsurro MR, Sagnelli A, Taiello AC, Reggiori B, La Bella V, Tedeschi G, Albanese A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. European journal of neurology. 2016;23(1):45–52. doi: 10.1111/ene.12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peviani M, Salvaneschi E, Bontempi L, Petese A, Manzo A, Rossi D, Salmona M, Collina S, Bigini P, Curti D. Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiology of disease. 2014;62:218–32. doi: 10.1016/j.nbd.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 83.Kalmar B, Lu CH, Greensmith L. The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacology & therapeutics. 2014;141(1):40–54. doi: 10.1016/j.pharmthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 84.Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, March A, Ait-Ghezala G, Mullan MJ. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. Journal of neuroinflammation. 2010;7:17. doi: 10.1186/1742-2094-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neuro-degenerative diseases. 2005;2(5):246–54. doi: 10.1159/000090364. [DOI] [PubMed] [Google Scholar]
- 86.Ho SW, Tsui YT, Wong TT, Cheung SK, Goggins WB, Yi LM, Cheng KK, Baum L. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Translational neurodegeneration. 2013;2(1):24. doi: 10.1186/2047-9158-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ouyang YB, Giffard RG. ER-Mitochondria Crosstalk during Cerebral Ischemia: Molecular Chaperones and ER-Mitochondrial Calcium Transfer. International journal of cell biology. 2012;2012:493934. doi: 10.1155/2012/493934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koch GL. The endoplasmic reticulum and calcium storage. BioEssays: news and reviews in molecular, cellular and developmental biology. 1990;12(11):527–31. doi: 10.1002/bies.950121105. [DOI] [PubMed] [Google Scholar]
- 89.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harbor perspectives in biology. 2011;3(6) doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li M, Lester HA. Ion channel diseases of the central nervous system. CNS drug reviews. 2001;7(2):214–40. doi: 10.1111/j.1527-3458.2001.tb00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shirwany NA, Payette D, Xie J, Guo Q. The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatric disease and treatment. 2007;3(5):597–612. [PMC free article] [PubMed] [Google Scholar]
- 92.Cataldi M. The changing landscape of voltage-gated calcium channels in neurovascular disorders and in neurodegenerative diseases. Current neuropharmacology. 2013;11(3):276–97. doi: 10.2174/1570159X11311030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith RG, Alexianu ME, Crawford G, Nyormoi O, Stefani E, Appel SH. Cytotoxicity of immunoglobulins from amyotrophic lateral sclerosis patients on a hybrid motoneuron cell line. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(8):3393–7. doi: 10.1073/pnas.91.8.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58(6):871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young LT, Kish SJ, Li PP, Warsh JJ. Decreased brain [3H]inositol 1,4,5-trisphosphate binding in Alzheimer’s disease. Neuroscience letters. 1988;94(1–2):198–202. doi: 10.1016/0304-3940(88)90295-9. [DOI] [PubMed] [Google Scholar]
- 96.Duncan RS, Goad DL, Grillo MA, Kaja S, Payne AJ, Koulen P. Control of intracellular calcium signaling as a neuroprotective strategy. Molecules. 2010;15(3):1168–95. doi: 10.3390/molecules15031168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(2):508–13. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferreiro E, Resende R, Costa R, Oliveira CR, Pereira CM. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiology of disease. 2006;23(3):669–78. doi: 10.1016/j.nbd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 99.Kasri NN, Kocks SL, Verbert L, Hebert SS, Callewaert G, Parys JB, Missiaen L, De Smedt H. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knock-out cells. Cell calcium. 2006;40(1):41–51. doi: 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 100.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(30):9458–70. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26(19):5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O’Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92(2):499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 103.Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39(2):227–39. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington’s disease mouse model. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(5):1257–66. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen X, Wu J, Lvovskaya S, Herndon E, Supnet C, Bezprozvanny I. Dantrolene is neuroprotective in Huntington’s disease transgenic mouse model. Molecular neurodegeneration. 2011;6:81. doi: 10.1186/1750-1326-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PloS one. 2012;7(12):e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Staats KA, Van Rillaer M, Scheveneels W, Verbesselt R, Van Damme P, Robberecht W, Van Den Bosch L. Dantrolene is neuroprotective in vitro, but does not affect survival in SOD1(G(9)(3)A) mice. Neuroscience. 2012;220:26–31. doi: 10.1016/j.neuroscience.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 108.Dziadek MA, Johnstone LS. Biochemical properties and cellular localisation of STIM proteins. Cell calcium. 2007;42(2):123–32. doi: 10.1016/j.ceca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 109.Bezprozvanny IB. Calcium signaling and neurodegeneration. Acta naturae. 2010;2(1):72–82. [PMC free article] [PubMed] [Google Scholar]
- 110.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. The Journal of cell biology. 2000;149(4):793–8. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27(3):561–72. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 112.Kawamata H, Ng SK, Diaz N, Burstein S, Morel L, Osgood A, Sider B, Higashimori H, Haydon PG, Manfredi G, Yang Y. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34(6):2331–48. doi: 10.1523/JNEUROSCI.2689-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chemistry & biology. 2011;18(6):777–93. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Czeredys M, Gruszczynska-Biegala J, Schacht T, Methner A, Kuznicki J. Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington’s disease. Frontiers in molecular neuroscience. 2013;6:42. doi: 10.3389/fnmol.2013.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nature reviews Molecular cell biology. 2012;13(10):607–25. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, Verfaillie T, Matus S, Smith MA, Mott JL, Agostinis P, Hetz C, Samali A. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell death & disease. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell death and differentiation. 2012;19(11):1880–91. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poston CN, Duong E, Cao Y, Bazemore-Walker CR. Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochemical and biophysical research communications. 2011;415(2):355–60. doi: 10.1016/j.bbrc.2011.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131(3):596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 120.Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, Diaz-Ramos A, Hernandez-Alvarez MI, Sebastian D, Mauvezin C, Palacin M, Zorzano A. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. The EMBO journal. 2013;32(17):2348–61. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27(3):285–99. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 122.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407–18. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM) Biochimica et biophysica acta. 2013;1833(1):213–24. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 124.Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. The EMBO journal. 2012;31(21):4106–23. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M, Graff C, Winblad B, Galter D, Behbahani H, Pizzo P, Glaser E, Ankarcrona M. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7916–21. doi: 10.1073/pnas.1300677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schreiner B, Hedskog L, Wiehager B, Ankarcrona M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. Journal of Alzheimer’s disease: JAD. 2015;43(2):369–74. doi: 10.3233/JAD-132543. [DOI] [PubMed] [Google Scholar]
- 127.Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J, Dickson DW, Petrucelli L, Mitchell JC, Shaw CE, Miller CC. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nature communications. 2014;5:3996. doi: 10.1038/ncomms4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ottolini D, Cali T, Negro A, Brini M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Human molecular genetics. 2013;22(11):2152–68. doi: 10.1093/hmg/ddt068. [DOI] [PubMed] [Google Scholar]
- 129.Kornfeld OS, Hwang S, Disatnik M-H, Chen C-H, Qvit N, Mochly-Rosen D. Mitochondrial Reactive Oxygen Species at the Heart of the Matter: New Therapeutic Approaches for Cardiovascular Diseases. Circulation Research. 2015;116(11):1783–99. doi: 10.1161/circresaha.116.305432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 131.Mink JW, Blumenschine RJ, Adams DB. RATIO OF CENTRAL NERVOUS-SYSTEM TO BODY METABOLISM IN VERTEBRATES – ITS CONSTANCY AND FUNCTIONAL BASIS. American Journal of Physiology. 1981;241(3):R203–R12. doi: 10.1152/ajpregu.1981.241.3.R203. [DOI] [PubMed] [Google Scholar]
- 132.Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol-London. 2015;593(16):3447–62. doi: 10.1113/Jp270254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15294–9. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nature cell biology. 2009;11(6):747–U105. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 135.Potter VR, Siekevitz P, Simonson HC. Latent adenosinetriphosphatase activity in resting rat liver mitochondria. The Journal of biological chemistry. 1953;205(2):893–908. [PubMed] [Google Scholar]
- 136.Chance B. The Energy-Linked Reaction of Calcium with Mitochondria. The Journal of biological chemistry. 1965;240:2729–48. [PubMed] [Google Scholar]
- 137.Reed KC, Bygrave FL. Inhibition of Mitochondrial Calcium-Transport by Lanthanides and Ruthenium Red. Biochemical Journal. 1974;140(2):143–55. doi: 10.1042/bj1400143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of Calcium on the Oxidative Phosphorylation Cascade in Skeletal Muscle Mitochondria. Biochemistry. 2013;52(16):2793–809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Panov AV, Scaduto RC. Influence of Calcium on Nadh and Succinate Oxidation by Rat-Heart Submitochondrial Particles. Arch Biochem Biophys. 1995;316(2):815–20. doi: 10.1006/abbi.1995.1109. [DOI] [PubMed] [Google Scholar]
- 140.Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. Febs Lett. 2000;466(1):130–4. doi: 10.1016/S0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- 141.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46(9):1283–97. doi: 10.1016/j.freeradbiomed.2009.02.008. Epub 2009/02/28. doi: S0891-5849(09)00081-1 [pii] 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 142.Sohal RS, Allen RG. Relationship between metabolic rate, free radicals, differentiation and aging: a unified theory. Basic life sciences. 1985;35:75–104. doi: 10.1007/978-1-4899-2218-2_4. [DOI] [PubMed] [Google Scholar]
- 143.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Frontiers in Bioscience-Landmark. 2009;14:1197–218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sousa SC, Maciel EN, Vercesi AE, Castilho RF. Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. Febs Lett. 2003;543(1–3):179–83. doi: 10.1016/s0014-5793(03)00421-6. [DOI] [PubMed] [Google Scholar]
- 145.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 146.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 147.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3):205–14. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 148.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58(1):39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 149.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429(6994):883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 150.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9782–7. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, Cotman C, Cottrell B, Montine TJ, Thomas RG, Aisen P, Study ADC. Antioxidants for Alzheimer Disease A Randomized Clinical Trial With Cerebrospinal Fluid Biomarker Measures. Arch Neurol-Chicago. 2012;69(7):836–41. doi: 10.1001/archneurol.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parkinson Study Group QEI. Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, Haas R, Juncos JL, Nutt JG, Voss TS, Ravina B, Shults CM, Helles K, Snively V, Lew MF, Griebner B, Watts A, Gao S, Pourcher E, Bond L, Kompoliti K, Agarwal P, Sia C, Jog M, Cole L, Sultana M, Kurlan R, Richard I, Deeley C, Waters CH, Figueroa A, Arkun A, Brodsky M, Ondo WG, Hunter CB, Jimenez-Shahed J, Palao A, Miyasaki JM, So J, Tetrud J, Reys L, Smith K, Singer C, Blenke A, Russell DS, Cotto C, Friedman JH, Lannon M, Zhang L, Drasby E, Kumar R, Subramanian T, Ford DS, Grimes DA, Cote D, Conway J, Siderowf AD, Evatt ML, Sommerfeld B, Lieberman AN, Okun MS, Rodriguez RL, Merritt S, Swartz CL, Martin WR, King P, Stover N, Guthrie S, Watts RL, Ahmed A, Fernandez HH, Winters A, Mari Z, Dawson TM, Dunlop B, Feigin AS, Shannon B, Nirenberg MJ, Ogg M, Ellias SA, Thomas CA, Frei K, Bodis-Wollner I, Glazman S, Mayer T, Hauser RA, Pahwa R, Langhammer A, Ranawaya R, Derwent L, Sethi KD, Farrow B, Prakash R, Litvan I, Robinson A, Sahay A, Gartner M, Hinson VK, Markind S, Pelikan M, Perlmutter JS, Hartlein J, Molho E, Evans S, Adler CH, Duffy A, Lind M, Elmer L, Davis K, Spears J, Wilson S, Leehey MA, Hermanowicz N, Niswonger S, Shill HA, Obradov S, Rajput A, Cowper M, Lessig S, Song D, Fontaine D, Zadikoff C, Williams K, Blindauer KA, Bergholte J, Propsom CS, Stacy MA, Field J, Mihaila D, Chilton M, Uc EY, Sieren J, Simon DK, Kraics L, Silver A, Boyd JT, Hamill RW, Ingvoldstad C, Young J, Thomas K, Kostyk SK, Wojcieszek J, Pfeiffer RF, Panisset M, Beland M, Reich SG, Cines M, Zappala N, Rivest J, Zweig R, Lumina LP, Hilliard CL, Grill S, Kellermann M, Tuite P, Rolandelli S, Kang UJ, Young J, Rao J, Cook MM, Severt L, Boyar K. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 2014;71(5):543–52. doi: 10.1001/jamaneurol.2014.131. [DOI] [PubMed] [Google Scholar]
- 153.Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M, Hamada C, Kondo K, Yoneoka T, Akimoto M, Yoshino H, Edaravone ALSSG Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):610–7. doi: 10.3109/21678421.2014.959024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kim S, Joe Y, Jeong SO, Zheng M, Back SH, Park SW, Ryter SW, Chung HT. Endoplasmic reticulum stress is sufficient for the induction of IL-1 beta production via activation of the NF-kappa B and inflammasome pathways. Innate Immunity. 2014;20(8):799–815. doi: 10.1177/1753425913508593. [DOI] [PubMed] [Google Scholar]
- 155.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Molecular cell. 2012;48(2):158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Group HIotHS, European Huntington’s Disease N. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013;70(1):25–33. doi: 10.1001/2013.jamaneurol.382. [DOI] [PubMed] [Google Scholar]
- 157.Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D, dimebon i Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372(9634):207–15. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- 158.Jones RW. Dimebon disappointment. Alzheimers Res Ther. 2010;2(5):25. doi: 10.1186/alzrt49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. The AAPS Journal. 2006;8:E521–E31. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. British journal of pharmacology. 2014;171:2017–28. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. Journal of neurochemistry. 2006;98(4):1141–8. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 162.Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11(9):2095–104. doi: 10.1089/ARS.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochimica et biophysica acta. 2014;1842(8):1282–94. doi: 10.1016/j.bbadis.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, Protect Study G A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25(11):1670–4. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 165.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nature reviews Neuroscience. 2012;13(2):77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22(14):5840–7. doi: 10.1523/JNEUROSCI.22-14-05840.2002. doi: 20026597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28(4):693–705. doi: 10.1016/0092-8674(82)90049-6. Epub 1982/04/01. [DOI] [PubMed] [Google Scholar]
- 168.Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM. The Dynamin-related GTPase, Dnm1p, Controls Mitochondrial Morphology in Yeast. The Journal of cell biology. 1998;143(2):333–49. doi: 10.1083/jcb.143.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Smirnova E, Shurland D-L, Ryazantsev SN, van der Bliek AM. A Human Dynamin-related Protein Controls the Distribution of Mitochondria. The Journal of cell biology. 1998;143(2):351–8. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–62. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 172.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Annals of the New York Academy of Sciences. 2010;1201:21–5. doi: 10.1111/j.1749-6632.2010.05615.x. Epub 2010/07/24. [DOI] [PubMed] [Google Scholar]
- 173.Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, Chang C-R, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. The FASEB Journal. 2009;23(11):3917–27. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. Embo Rep. 2009;10(7):748–54. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Molecular cell. 2015;59(6):941–55. doi: 10.1016/j.molcel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 176.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. The Journal of cell biology. 2007;178(1):71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jahani-Asl A, Slack RS. The phosphorylation state of Drp1 determines cell fate. Embo Rep. 2007;8(10):912–3. doi: 10.1038/sj.embor.7401077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. Embo Rep. 2007;8:939–44. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. Journal of Biological Chemistry. 2007;282(30):21583–7. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 180.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15803–8. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto YI, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature cell biology. 2009;11(8):958–U114. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 182.Wakabayashi J, Zhang ZY, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. Journal of Cell Biology. 2009;186(6):805–16. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Calcium regulation of mitochondria motility and morphology. 2009 doi: 10.1016/j.bbabio.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 184.Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311(5763):1012–7. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 185.Loriol C, Casse F, Khayachi A, Poupon G, Chafai M, Deval E, Gwizdek C, Martin S. mGlu5 receptors regulate synaptic sumoylation via a transient PKC-dependent diffusional trapping of Ubc9 into spines. Nature communications. 2014;5:5113. doi: 10.1038/ncomms6113. [DOI] [PubMed] [Google Scholar]
- 186.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. The Journal of cell biology. 2007;177(3):439–50. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tan AR, Cai AY, Deheshi S, Rintoul GL. Elevated intracellular calcium causes distinct mitochondrial remodelling and calcineurin-dependent fission in astrocytes. Cell calcium. 2011;49(2):108–14. doi: 10.1016/j.ceca.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 188.Deheshi S, Dabiri B, Fan S, Tsang M, Rintoul GL. Changes in mitochondrial morphology induced by calcium or rotenone in primary astrocytes occur predominantly through ros-mediated remodeling. Journal of neurochemistry. 2015;133(5):684–99. doi: 10.1111/jnc.13090. [DOI] [PubMed] [Google Scholar]
- 189.Hroudova J, Singh N, Fisar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. BioMed research international. 2014;2014:175062. doi: 10.1155/2014/175062. Epub 2014/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. The Journal of pharmacology and experimental therapeutics. 2012;342(3):619–30. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23(2):64–71. doi: 10.1016/j.tcb.2012.10.006. Epub 2012/11/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cronin-Furman EN, Borland MK, Bergquist KE, Bennett JP, Jr, Trimmer PA. Mitochondrial quality, dynamics and functional capacity in Parkinson’s disease cybrid cell lines selected for Lewy body expression. Molecular neurodegeneration. 2013;8:6. doi: 10.1186/1750-1326-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Reddy PH. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today. 2014;19(7):951–5. doi: 10.1016/j.drudis.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Human molecular genetics. 2011;20(7):1438–55. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Brustovetsky N. Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Developmental Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. The Journal of biological chemistry. 2010;285(15):11740–52. doi: 10.1074/jbc.M109.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Twaroski DM, Yan Y, Zaja I, Clark E, Bosnjak ZJ, Bai X. Altered Mitochondrial Dynamics Contributes to Propofol-induced Cell Death in Human Stem Cell-derived Neurons. Anesthesiology. 2015;123(5):1067–83. doi: 10.1097/ALN.0000000000000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Akita M, Suzuki-Karasaki M, Fujiwara K, Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y, Suzuki-Karasaki Y. Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced apoptosis. International journal of oncology. 2014;45(5):1901–12. doi: 10.3892/ijo.2014.2608. [DOI] [PubMed] [Google Scholar]
- 200.Wang J, Hansen K, Edwards R, Van Houten B, Qian W. Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochemical and biophysical research communications. 2015;456(1):7–12. doi: 10.1016/j.bbrc.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox biology. 2013;1(1):542–51. doi: 10.1016/j.redox.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kim B, Kim J-S, Yoon Y, Santiago MC, Brown MD, Park J-Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2013;305(8):R927–R38. doi: 10.1152/ajpregu.00502.2012. [DOI] [PubMed] [Google Scholar]
- 203.Ji-Yeong Y, Sung-Hun M, Hyo-Jin P, Jin-Woo K, Yong-Hee L, Soo-Yong P, Pil-Soo JEONG HP, Dong-Seok L, Sun-Uk K, CHANG Kyu-Tae, K D-B. Mdivi-1, mitochondrial fission inhibitor, impairs developmental competence and mitochondrial function of embryos and cells in pigs. The Journal of reproduction and development. 2015;61(2):81. doi: 10.1262/jrd.2014-070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Qi X, Qvit N, Su Y-C, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. Journal of cell science. 2013;126:789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Guo X, Disatnik M-h, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes. Huntington disease – associated neurodegeneration. 2013:123. doi: 10.1172/JCI70911DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122–8. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 207.Corzo G, Escoubas P. Pharmacologically active spider peptide toxins. Cell Mol Life Sci. 2003;60(11):2409–26. doi: 10.1007/s00018-003-3108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Qvit N, Kornfeld OS. Development of a Backbone Cyclic Peptide Library as Potential Antiparasitic. Therapeutics Using Microwave Irradiation. 2016;(107):e53589. doi: 10.3791/53589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cai Q, Sheng ZH. Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron. 2009;61(4):493–6. doi: 10.1016/j.neuron.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77(3):485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 211.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–55. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136(1):163–74. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(52):20728–33. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell calcium. 2014;56(6):437–45. doi: 10.1016/j.ceca.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bernardi P. The mitochondrial permeability transition pore: A mystery solved? Frontiers in Physiology. 2013;4:1–12. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell calcium. 2011;50(3):222–33. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 217.Alam MR, Baetz D, Ovize M. Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. Journal of Molecular and Cellular Cardiology. 2015;78:80–9. doi: 10.1016/j.yjmcc.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 218.Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-H. [DOI] [PubMed] [Google Scholar]
- 219.Sweeney ZK, Fu J, Wiedmann B. From chemical tools to clinical medicines: non-immunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds. Journal of medicinal chemistry. 2014;57:7145–59. doi: 10.1021/jm500223x. [DOI] [PubMed] [Google Scholar]
- 220.Azzolin L, Antolini N, Calderan A, Ruzza P, Sciacovelli M, Marin O, Mammi S, Bernardi P, Rasola A. Antamanide, a derivative of Amanita phalloides, is a novel inhibitor of the mitochondrial permeability transition pore. PloS one. 2011;6(1):e16280. doi: 10.1371/journal.pone.0016280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kim JK, Choi SJ, Cho HY, Hwang HJ, Kim YJ, Lim ST, Kim CJ, Kim HK, Peterson S, Shin DH. Protective effects of kaempferol (3,4′,5,7-tetrahydroxyflavone) against amyloid beta peptide (Abeta)-induced neurotoxicity in ICR mice. Bioscience, biotechnology, and biochemistry. 2010;74(2):397–401. doi: 10.1271/bbb.90585. [DOI] [PubMed] [Google Scholar]
- 222.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ, Alzheimer’s Disease Cooperative Study G Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352(23):2379–88. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 223.Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, Prieto S, Chen P, Loreck DJ, Trapp G, Bakshi RS, Mintzer JE, Heidebrink JL, Vidal-Cardona A, Arroyo LM, Cruz AR, Zachariah S, Kowall NW, Chopra MP, Craft S, Thielke S, Turvey CL, Woodman C, Monnell KA, Gordon K, Tomaska J, Segal Y, Peduzzi PN, Guarino PD. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311(1):33–44. doi: 10.1001/jama.2013.282834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Investigators NN-P. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68(1):20–8. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- 225.Shults CW, Beal MF, Fontaine D, Nakano K, Haas RH. Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q10 in parkinsonian patients. Neurology. 1998;50(3):793–5. doi: 10.1212/wnl.50.3.793. [DOI] [PubMed] [Google Scholar]
- 226.Kaufmann P, Thompson JL, Levy G, Buchsbaum R, Shefner J, Krivickas LS, Katz J, Rollins Y, Barohn RJ, Jackson CE, Tiryaki E, Lomen-Hoerth C, Armon C, Tandan R, Rudnicki SA, Rezania K, Sufit R, Pestronk A, Novella SP, Heiman-Patterson T, Kasarskis EJ, Pioro EP, Montes J, Arbing R, Vecchio D, Barsdorf A, Mitsumoto H, Levin B, Group QS. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann Neurol. 2009;66(2):235–44. doi: 10.1002/ana.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol. 2008;28(1):110–3. doi: 10.1097/jcp.0b013e318160862c. [DOI] [PubMed] [Google Scholar]
- 228.Singer C, Tractenberg RE, Kaye J, Schafer K, Gamst A, Grundman M, Thomas R, Thal LJ, Alzheimer’s Disease Cooperative S A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep. 2003;26(7):893–901. doi: 10.1093/sleep/26.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schapira AH, McDermott MP, Barone P, Comella CL, Albrecht S, Hsu HH, Massey DH, Mizuno Y, Poewe W, Rascol O, Marek K. Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol. 2013;12(8):747–55. doi: 10.1016/S1474-4422(13)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Holmay MJ, Terpstra M, Coles LD, Mishra U, Ahlskog M, Oz G, Cloyd JC, Tuite PJ. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin Neuropharmacol. 2013;36(4):103–6. doi: 10.1097/WNF.0b013e31829ae713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, Hardiman O, Bozik ME, Ingersoll EW, Archibald D, Meyers AL, Dong Y, Farwell WR, Kerr DA, investigators E Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12(11):1059–67. doi: 10.1016/S1474-4422(13)70221-7. [DOI] [PubMed] [Google Scholar]