

Abstract
Identification and quantification of the characteristics of stem cell preparations is critical for understanding stem cell biology and for the development and manufacturing of stem cell based therapies. We have developed image analysis and visualization software that allows effective use of time-lapse microscopy to provide spatial and dynamic information from large numbers of human embryonic stem cell colonies. To achieve statistically relevant sampling, we examined >680 colonies from 3 different preparations of cells over 5 days each, generating a total experimental dataset of 0.9 terabyte (TB). The 0.5 Giga-pixel images at each time point were represented by multi-resolution pyramids and visualized using the Deep Zoom Javascript library extended to support viewing Giga-pixel images over time and extracting data on individual colonies. We present a methodology that enables quantification of variations in nominally-identical preparations and between colonies, correlation of colony characteristics with Oct4 expression, and identification of rare events.
Keywords: Fluorescence microscopy, Stem cells, Live cell imaging, Cell therapy, Pluripotency
1. Introduction
Better understanding of stem cell biology will be aided by measurement methods that allow validation of assumptions about gene expression and morphological characteristics as criteria for assessing cell state and biological activity. Such methods will also support the development of cell therapy products, which requires methods for quantitatively assessing the quality and consistency of cells and colonies (Fink, 2009). The defining characteristics of desirable cells is often unclear (Baker, 2012), and this lack of knowledge, and the lack of robust measurement methods, complicates decision-making about starting materials, processes, and product quality. Having quantitative and relevant cell and colony characterization criteria is necessary for determining the consistency of preparations, assuring that culture processes are robust, and achieving a reliable, safe and effective product. While flow cytometry or genomics measurements provide useful data about some population characteristics at a point in time, they cannot provide spatial and dynamic information from individual cells and colonies. Tracking the relationship between cellular characteristics at a specific time and the fate of those cells in the future provides a means of determining what characteristics are meaningful for evaluating preparations and are predictive of the future response of cells (Filipczyk et al., 2015). To facilitate these goals, we have developed image analysis and visualization software that allows effective use of time-lapse microscopy to quantify spatial and dynamic differences in gene activity in a large number of human embryonic stem cell (hESC) colonies over several days under conditions designed to maintain pluripotency. We used the H9 hESC line which was modified by homologous recombination to include the gene for enhanced green fluorescent protein (EGFP) downstream of the endogenous Octamer binding transcription factor 4 (Oct4) gene according to the method of Zwaka and Thomson (2003).
We have examined Oct4 as a marker for this study because it is well known as a critical factor for maintaining pluripotency, and its expression is lost in differentiated cells (van den Berg et al., 2010; Niwa et al., 2000). The relationship between Oct4 expression and pluripotency is not a simple one and is not fully understood. While loss of pluripotency is often accompanied by Oct4 down-regulation (Pan and Thomson, 2007; Nichols et al., 1998), other factors are required (Niwa, 2007; Boyer et al., 2005). Thomson et al. (2011) demonstrated the dynamic response of Oct4 and Sox2 to differentiation factors in mouse ESCs. They showed with time-lapse imaging that Oct4 and Sox2 levels increased or decreased according to the lineage to which those cells were committing. Nanog, Sox2 and Oct4 have a complex relationship (Boyer et al., 2006; Mitsui et al., 2003) in which an assembly of Oct4 and Sox2 affect their own promoter activities and the activity of the Nanog promoter. Together, Oct4 and Sox2 promote self-renewal of ESCs by preventing differentiation (Ambrosetti et al., 2000; Chew et al., 2005; Rodda et al., 2005) at least in part by control of Nanog. High levels of Nanog help maintain ESC self-renewal (Mitsui et al., 2003; Chambers et al., 2003). At the same time, it has been observed that overexpression of Oct4 can repress its own promoter as well as that of Nanog (Pan et al., 2006) and induce differentiation (Karwacki-Neisius et al., 2013). Engineering cells for low Oct4 production stabilizes their pluripotent state and reduces their efficiency of differentiation (Karwacki-Neisius et al., 2013; Radzisheuskaya et al., 2013). These observations point to a complex interplay of pluripotency factors, and dynamic regulation of them.
Determining how to accurately interpret the presence, absence and levels of Oct4 and other factors requires the ability to quantify their dynamic responses over long times in individual cells (Filipczyk et al., 2015; Sokolik et al., 2015; Ochiai et al., 2014). Direct observation of the history of a cell or colony by tracking forward and backward in time will further our understanding of markers of cell state and our ability to correctly predict future states (Singer et al., 2014; Bajcsy et al., 2016). For the data to be meaningful, it is necessary to sample a relevant number of colonies at an appropriate level of spatial resolution with sufficient temporal interrogation without compromising the health of the cells. Handling and analyzing the very large resulting dataset requires the development and application of advanced data manipulation and visualization methods.
Here, we present imaging and analysis methods that make it possible to quantify the dynamic and spatial behavior of a reporter of stem cell pluripotency in large numbers of hESC colonies over extended periods of time. To demonstrate these methods, we observe and quantify Oct4 expression under culture conditions that are expected to retain pluripotency. The image analysis, visualization software, and analytical pipeline developed for this study allow spatial and dynamic characterization of the growth and expression of Oct4 in a large number of hESC colonies. These methods for acquisition, image analysis, and visualization allow us to quantify differences in 3 preparations of pluripotent colonies, and to identify rare behaviors in colonies. The 3 preparations were all nominally identical in the sense that there were no controlled or systematic differences between them.
2. Results
2.1. Imaging and visualization
Colonies were imaged with a 10× objective at 45 min intervals per field of view (FOV) over an area of approximately 4 cm2 comprising hundreds of FOVs. Three preparations were observed for approximately 120 h each, producing a total experimental data set with a storage size of 0.9 TB. Because of the size and movement of colonies, tracking and quantifying large numbers of them requires stitching together adjacent FOV into a single composite image in which colonies can span across FOV. The size of just a single composite image within these datasets (one time point ~1 gigabyte (GB) of data) precluded viewing on a typical desktop computer, in part, because of the lag time associated with unloading and loading sequential images in a time-lapse series. The data were hence visualized by a multi-resolution pyramid representation using the Deep Zoom Javascript library. Fig. 1A shows a schematic of the computational steps in the image analysis pipeline and representative images. The Deep Zoom software and data analysis pipeline provided the means of examining the entire data set through time at the level of the mosaic images down to full resolution images of each individual colony or groups of colonies in phase contrast and/or fluorescence mode over time (Fig. 1B), extracting data from the images for visualization of information, and performing quantitative evaluation (Fig. 1C).
Fig. 1.
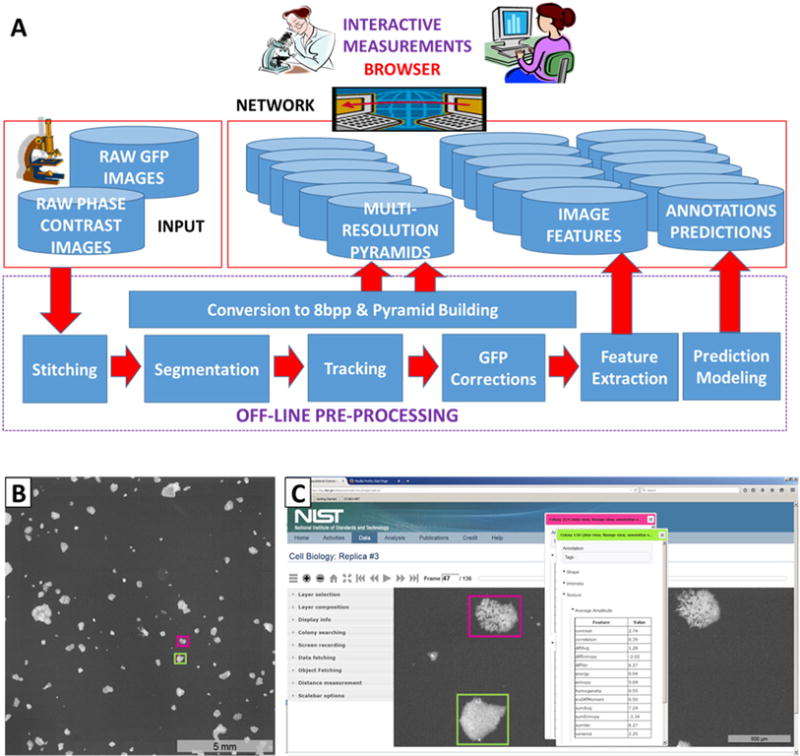
(A) Schematic of all computational steps applied to the experimental replicas. ‘GFP Corrections’ operations are to correct for uneven illumination and detection, dark current, and background fluorescence, and are performed on the stitched composite image of 360 or 396 - FOV. A multi-resolution pyramid representation of each corrected and stitched 2D image was created. Then a set of temporal gigapixel images (which is a terabyte-sized data cube) is stored as a set of pyramids on a server to allow for efficient transmission and viewing of images. (B) Composite image of Oct4-GFP fluorescence of preparation 3 at one time point created by stitching 396 - FOV together into a single composite image. This 16-bit image for this one time point in the fluorescence channel is approximately 1 GB in size. Two colonies are enclosed in boxes and visualized in greater detail in (C). (C) A view of the colonies indicated in the composite image in greater detail using the Deep Zoom tool. Any colony from any image can be viewed in detail via a mouse-click, which produces the colony identification number and data for that colony including coordinates, and features such as average GFP intensity, area, circumference, etc. The full composite image or any subset of it can be panned, visualized in time-lapse mode, and recorded for saving as one or a series of TIFF or PNG files. A video demonstration of the operation of the Deep Zoom visualization tool is provided in Supplemental Movie 1.
Given the challenges associated with maintaining stem cells in a healthy state over 5 days of imaging on a microscope incubation chamber, we first examined if cells on the microscope exhibited growth characteristics that are consistent with cells maintained in a jacketed incubator. Colonies were segmented in phase contrast, and the mask used to measure the overall culture area and growth rate. Colony growth rates on the microscope stage-top incubator were similar to that of cells maintained in a water-jacketed incubator (Supplemental Fig. 4). While this exposure regime was designed to minimize damage to cells, it resulted in a relatively low signal to noise ratio in the GFP fluorescence channel.
We then performed analysis of each of 3 separate preparations of cells at the level of individual colonies, tracking colonies through time. At each time point, individual colonies were segmented, labeled with an identifying number, and evaluated for area, average GFP intensity, and other features (a total of 68 features, which are available to the user for manual browsing by clicking on the image of the colony in the Deep Zoom tool, as described in the Methods). Colonies were assigned a unique id when they merged with other colonies as they expanded. The change in area with time for hundreds of individual colonies is plotted for each of the 3 stem cell preparations (Fig. 2A–C). The integrated intensity from the GFP signal in each colony was computed at each time point and represented using an indicating color for each trajectory. During this period, colony areas increased approximately over 4 orders of magnitude, as colonies merged and the cultures became increasingly confluent (Fig. 2A–C). Further examination of panels A–C suggested that smaller, dimmer colonies that are slow growing are present in all three preparations (blue colored traces). These and additional differences and similarities were quantified through the data analysis pipeline.
Fig. 2.
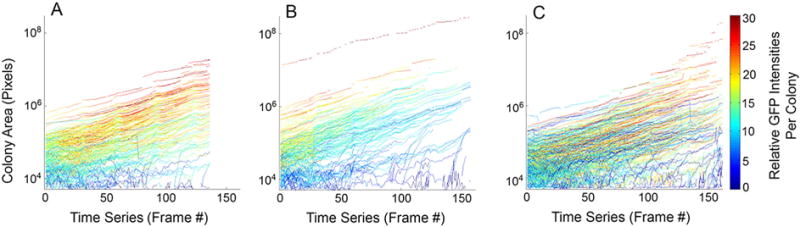
Analysis of colony area and intensity over time. (A–C) Each trajectory represents the change in area of an individual colony over time for colonies from preparations 1–3, respectively. The colors of the lines indicate average intensity of GFP fluorescence as indicated by the color bar scale. Trajectories reflect the change in the area of a colony and the average intensity of those colonies during sequential frames between colony merger events. Each pixel area = 0.394 μm2 and each frame in the time series was acquired at 45 min intervals.
Accurate analysis of colony intensities is complicated in these live cell imaging experiments because of relatively significant and uneven background fluorescence generated from the components in the media including riboflavins and residual phenol red and by uneven illumination and photobleaching. Image data were subjected to an optimized background correction procedure that involved a number of functions applied at the mosaic and sub-mosaic levels, and which was evaluated by a minimum RMS error after correction (Chalfoun et al., 2015). The background levels were not identical in the three preparations. To be sure that it was valid to quantitatively compare the 3 preparations to one another, we determined for each preparation a threshold for reliable detection of a dim fluorescent colony, and the sensitivity to detecting changes in the fluorescence intensity of a colony in the form of a signal to noise ratio (SNRcolony) (refer to Supplemental Information for a complete description). The SNRcolony and threshold for reliable detection were determined from the stitched fluorescence fields of view in the mosaic image after flatfield correction and background subtraction, thus accounting for any bias or uncertainty that may have been introduced during these processing steps. The detection threshold is the value above which GFP intensity can be reliably measured from a colony because it is unlikely to be due to random fluctuations in the background signal. The detection threshold took into account both spatial and temporal variations in background intensities and was set as 3× the standard deviation of the background intensity. The standard deviation of the background intensity was calculated from 70 × 70-pixel (1930.6 μm2) areas of background regions positioned some distance away from a colony. This area was chosen to approximate the size of a small colony, and because averaging over a smaller number of pixels is more likely to give a larger standard deviation than averaging over a larger number of pixels, this provided a sensitive estimate of background standard deviation. For each preparation the intensities of background areas were collected over 20 time frames, for a total of 100 areas used in the calculation of the mean and standard deviation in background pixel intensities. The colony GFP fluorescence signal was computed for each preparation using the mean GFP intensity of representative colonies from that preparation that appeared to be uniformly expressing GFP. The GFP signal divided by standard deviation of the background intensities (computed above) gives rise to a SNRcolony of 38 ± 6.6, 43 ± 8.5 and 26 ± 3.6 for preparations 1, 2 and 3, respectively (see Supplemental Table 1 for uncertainty calculations). The SNRcolony is different between each preparation, but the SNR is sufficiently large that we can confidently detect relative changes in colony GFP intensities such as those shown in Fig. 2.
Visual inspection of colony images revealed some colonies with complex spatial relationships of bright and dark (GFP non-expressing) cells within them, especially in preparation 3. Some colonies appear to contain only bright cells, while others contain a mixture of bright and non-expressing cells, and a few colonies contain only non-expressing cells. Representative images of these three types of colonies are shown in Fig. 3A–C. In addition, a time series of images of representative colonies of each type are shown in Supplemental Movies 2–4 (homogeneously Oct4 expressing colonies), Supplemental Movies 5–7 (heterogeneously Oct4 expressing colonies), and Supplemental Movie 8 (non-expressers). Below, we describe how we classified these colonies as ‘homogeneous’ in Oct4 expression, ‘heterogeneous’ in Oct4 expression, and ‘non-expressing’ (though the ‘non-expressing’ colonies may be weakly expressing GFP below the detection threshold of the imaging system). Time-lapse imaging and quantitative analysis of Oct4 in large numbers of colonies and over long times allowed us to address: 1) if nominally identical preparations can differ in Oct4 heterogeneity, which might indicate sensitivity to unintended differences in culture conditions; 2) if colonies change in Oct4 expression levels or heterogeneity over time, which could be due to cellular fluctuations in Oct4 expression; and 3) whether colonies that express Oct4 heterogeneously expand at a slower rate than colonies that are homogeneous in Oct4, which might indicate loss of self-renewal character.
Fig. 3.
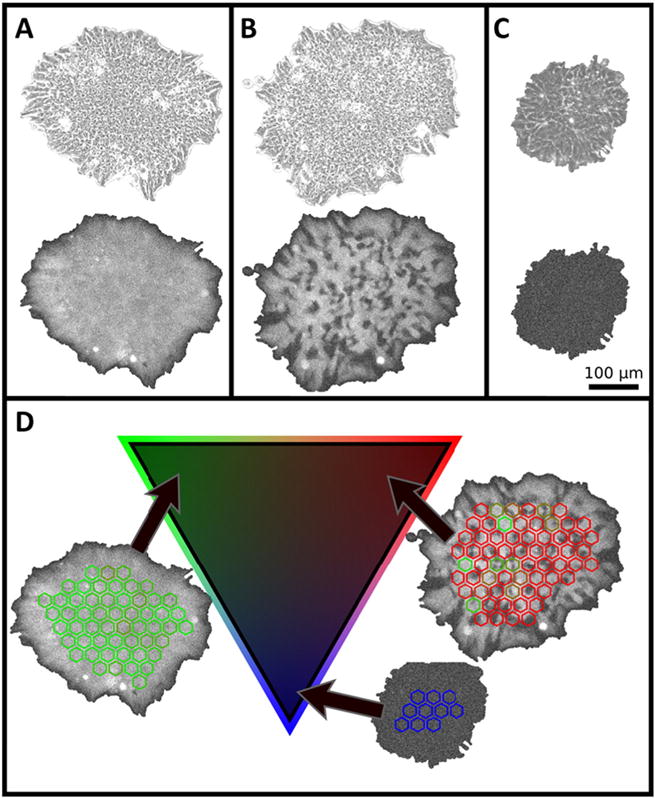
Quantifying apparent heterogeneity in Oct4 expression. (A–C) Representative colonies from preparation 3 showing phase contrast (top images) and fluorescence images of (A) a colony that is apparently homogeneous in Oct4 expression, (B) a colony that is heterogeneous in Oct4 expression, and (C) a colony that is not expressing Oct4. (D) Quantitation of heterogeneity was performed by texture analysis of 900 μm2 hexagons within colonies as depicted in the representative colony images; using a simplex algorithm, each colony in the preparation is assigned to a position on the triangle (2-simplex) according to its heterogeneity score: representative colonies that are either homogeneously bright (green), heterogeneous in GFP expression (red) or expressing little to no GFP (blue) are shown with their respective locations on the triangle.
Analysis of heterogeneity of Oct4 expression in colonies was initiated with manual examination of fluorescence images from colonies from the 3 preparations to provide a training set for texture analysis. Selected colonies were manually classified by consensus of 3 experts as containing both bright and dim cells or as homogeneously expressing or non-expressing. This approach for texture analysis is depicted in Fig. 3 and the details are provided in Supplemental Information. Briefly, colonies were divided into hexagonal areas of approximately 900 μm2 (which is approximately the size of 2 to 5 cells) and each hexagon was classified as homogeneously bright, heterogeneous in intensity, or dark (indicating non-expression). Each colony was then evaluated according to their percentage of hexagons in each class.
The 3 preparations vary in relative number of non-expressing colonies, colonies homogeneously expressing Oct4, and colonies that show heterogeneity in Oct4 expression. Fig. 4A–C indicates the number of colonies classified accordingly in each preparation. A statistical analysis of the data in Fig. 4 indicates significant differences between preparation 3 and the other 2 preparations, but no significant difference between preparation 1 and 2 (Supplemental Text and Supplemental Fig. 2). This analysis was limited to colonies of sufficient size to allow statistically relevant analysis; colonies were only considered if they encompassed an area of at least 5000 μm,2 which we estimate to be the area of about 10–20 cells. As a result, this analysis does not include colonies that remain very small and are presumably not pluripotent. The analysis indicates that for preparations 1 and 2, there are few non-fluorescent colonies of an area larger than this, and a higher number were seen in preparation 3. The large number of colonies sampled in the study allowed for an analysis and verification of the statistical robustness of these differences (Supplemental Fig. 3). After applying the automated classification of colony textures as homogeneous, heterogeneous or non-expressing in character, an analysis of the growth rates of the different colony types was performed. In preparations 1 and 2, the homogeneous and heterogeneous regions of colonies expanded at similar rates, which were faster than the expansion rates for the non-expressing regions. Preparation 3 is different in that the non-expressing regions appear to increase in area at a rate more similar to the growth rate associated with the homogeneous and heterogeneous regions (Supplemental Fig. 5).
Fig. 4.
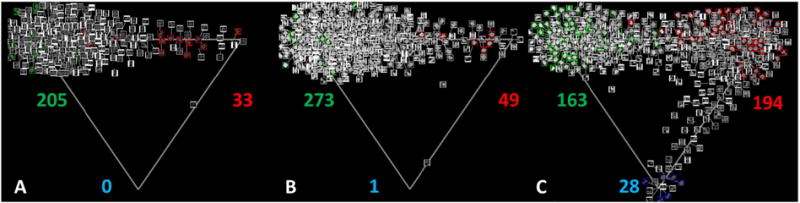
Relative abundance of the different colony types for the 3 preparations. Representation of colonies in (A) preparation 1, (B) preparation 2 and (C) preparation 3, respectively, projected on a triangle (2-simplex) with each vertex corresponding to one of the three pure colony types. In each triangle, the upper left vertex corresponds to ‘homogeneous’ colonies, the upper right vertex corresponds to ‘heterogeneous’ colonies, and the lower vertex corresponds to ‘non-expressing’ colonies. Colonies from time frames 18, 52, and 85 were used in this analysis. The location of each colony within the triangle was determined by the automated classifier. The number of each colony type in each triangle is indicated with the color coded number: # of ‘homogeneous’ colonies in green, # of ‘heterogeneous’ colonies in red, and # of ‘non-expressing’ colonies in blue. The red and green framed colonies are examples of manually annotated colonies that were used to train the automated classifier. Access to the full size colony images and an interactive visualization of the automated classification of hexagonal sub-regions is available at https://isg.nist.gov/deepzoomweb/analysis.
The use of time-lapse measurements allows exploration of how or whether heterogeneity in Oct4 expression can change within the preparations and within individual colonies over time. Inspection of the lineage data shown in Supplemental Fig. 6 suggests that most colonies remained quite stable in their expression of Oct4 even when colonies merge with other colonies of different textural character. A quantitative analysis of the temporal changes in colony character was performed based on the simplex analysis described in Figs. 3 and 4. The position of a colony on the triangle can change over time if its Oct4 expression character changes, i.e. if it becomes more heterogeneous, homogeneous or dark over time. The positional change of individual colonies over time in the classification triangle were measured, and colonies were scored according to how much change in GFP expression character they experienced. The mathematical details of the scoring method are provided in Supplemental Information and is graphically depicted in Supplemental Fig. 6. The result of this analysis is a temporal fluctuation score for each colony. The low values of the score correspond to colonies that are stable in their GFP expression character. The application of this analysis for each preparation is shown in Fig. 5. Most colonies exhibit stable fluctuation scores (0.05–0.1), which indicate a strong tendency for colonies to maintain their degree of ‘homogeneous’, ‘heterogeneous’ and ‘non-expressing’ character. But some colonies, especially in preparation 3, showed larger fluctuation scores, suggesting significant changes in the spatial heterogeneity of Oct4 expression over time.
Fig. 5.
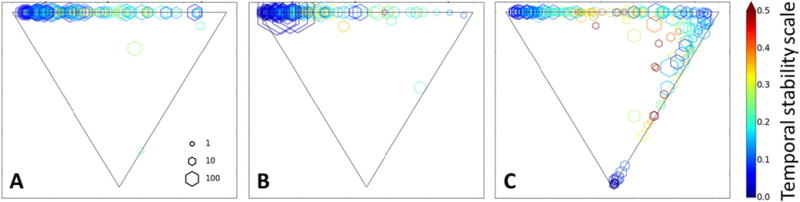
(A–C) Simplex triangles for preparation 1, 2 and 3 respectively. The mean ‘homogeneous’, ‘heterogeneous’, and ‘non-expressing’ intensity and textural character of a colony over time is indicated by its location in the triangle. The upper left vertex corresponds to ‘homogeneous’; colonies, the upper right vertex corresponds to ‘heterogeneous’ colonies, and the lower vertex corresponds to ‘non-expressing’ colonies. The color of the marker relates to the scale bar on the right and indicates the magnitude of the temporal fluctuations of Oct4 character exhibited by the colony. The size of the marker indicates the colony area (i.e., the average number of hexagons associated with the colony over its lifetime) as shown in the scale in the lower right hand corner of (A). The minimum temporal fluctuation scores for homogeneous and heterogeneous colonies that appear stable are 0.05 and 0.1, respectively (as determined from Supplemental Movies 2–4 and 5–7).
These colonies were examined closely with the Deep Zoom visualization tool to identify if these results reflected artifacts or interesting biological behavior. Three instances of unusual behavior were observed through this analysis. In one case, a few non-expressing cells separated from an otherwise homogeneously Oct4 expressing colony and formed a non-Oct4 expressing colony (Supplemental Movie 9). In another, a colony segregated into 3 distinct regions, 2 of which are homogeneously bright and 1 of which is non-expressing (Supplemental Movie 10). In a third case, a colony exhibits a transition from homogeneously bright to dark, apparently suppressing the expression of Oct4 over several days (Supplemental Movie 11). In total, we examined over 680 colonies and identified 6 examples of transitions of colonies from Oct4 expressing to Oct4 non-expressing. We observed no transitions of non-expressing colonies to Oct4 expressing colonies. Given that each colony was observed for approximately 20 h, this suggests that under these culturing conditions, the inter-conversion rate from Oct4-expressing to Oct4 non-expressing is 0.01 d−1; i.e., 1 out of 100 colonies suppressed Oct4 expression per day under these conditions.
3. Discussion
Live cell imaging allows examination of spatial and temporal information about gene expression in colonies and between colonies (Singer et al., 2014; Bajcsy et al., 2016; Halter et al., 2011; Barbaric et al., 2014; Chan et al., 2009). Meaningful quantification requires sampling a large population of cells and colonies, and because the area of individual colonies can be larger than the FOV with a 10× objective lens, tracking a colony over time as it grows and moves requires that many adjacent fields be examined and stitched together. For each time point in this study, hundreds of FOV were corrected and stitched together to form a single mosaic image of approximately 1 GB in size. The handling and visualization of that amount of image data were enabled by the Deep Zoom tools demonstrated in Supplemental Movie 1.
The processing, visualization and analysis pipeline presented here will make it possible to address additional questions about Oct4, and to query other fluorescent indicators of pluripotency and differentiation over time. This approach will allow systematic quantitative examination of the effects of experimental parameters such as culture conditions, and in combination with other analyses, can help to insure correct interpretation of markers of pluripotency and differentiation. These methods allow quantification of the reproducibility of cell preparations under nominally identical culture conditions. Variations in culture conditions will exist from laboratory to laboratory, and during the development of manufacturing processes. It is critical to have robust quantitative criteria that allow the comparison of media and other conditions and parameters that may affect a preparation. Although the three preparations of cells used here were handled under nominally identical conditions, we observed differences in the numbers of colonies that did not express GFP and colonies that contained both expressing and non-expressing cells. Nevertheless, we observed consistent rates of population doubling for the 3 preparations, including preparation 2 where the size of seeded colonies was unintentionally much larger than in the other preparations. We found that the doubling rates for all preparations were equivalent to doubling rates for the cells in a jacketed incubator (Supplemental Fig. 1), suggesting that their continuous location on the incubator-microscope stage and their intermittent exposure to light did not affect cell viability.
The differences in the expression of Oct4 that were observed are puzzling. Variation in Oct4 expression in cells within the same colony has been reported previously (Hough et al., 2009; Šustáčková et al., 2011; Silva and Smith, 2008; Gerrard et al., 2005). It has been suggested that non-Oct4 expressing cells that exist within what appear to be otherwise pluripotent colonies are also pluripotent (Šustáčková et al., 2011), and that heterogeneity in Oct4 expression may be the result of culture conditions (Hough et al., 2014), or stochastic fluctuations (Hough et al., 2009). We observed that most colonies have a similar GFP intensity throughout the colonies and assigned these a ‘homogeneous’ label, but in preparation 3, many colonies show marked heterogeneity in Oct4 expression within the colony. An analysis of change in area of GFP homogeneous, heterogeneous and non-expressing regions of colonies suggests that homogeneous and heterogeneous areas proliferate at a similar rate, which is greater than the rate of growth of non-expressing cells (Supplemental Fig. 5). These results suggest that colonies that are heterogeneous in Oct4 expression have similar proliferation rates to colonies uniformly expressing Oct4 and may retain their pluripotency, but further analysis might indicate an altered potential for differentiation.
The development of technology to provide and quantify the results of time-lapse imaging of live colonies using fluorescent protein reporters can shed light onto the temporal progression of colonies and expression of critical genes, and increase our understanding of the control mechanisms of pluripotency and differentiation. This study would not have been possible without the computational methods described here that will enable the acquisition, handling and analysis of very large image datasets.
4. Materials and methods
4.1. Generation of Oct4-GFP reporter cells
H9 human embryonic stem cell line (WiCell, Madison, WI) was genetically engineered to express EGFP under the influence of the Oct4 promoter by homologous recombination using a plasmid developed in the laboratory of James Thomson and obtained from Addgene (Cambridge, MA) (Zwaka and Thomson, 2003). Briefly, H9 hESCs were grown as a monolayer on Matrigel™ in mTeSR medium (Stem Cell Technologies, Vancouver, BC). The targeting vector, pOCTF3-AK (Addgene #21165) was linearized and introduced to the cells using Nucleofector (Lonza, Walkersville, MD), allowing the insertion of an IRES-EGFP, IRES-neo, and SV40 polyadenylation sequence into the 3′ untranslated region of the human POU5F1 gene. After G418 selection for 2 weeks, surviving clones were picked and re-cloned to generate populations of GFP-positive cells. Among the clones, the ones which expressed GFP in an undifferentiated state and shut down GFP expression upon differentiation were selected and expanded for further study.
4.2. Cell handling
Oct4-GFP H9 hESCs were maintained on Matrigel™ (BD, Franklin Lakes, NJ) in 6-well tissue culture plates (BD) in the presence of TesR-E8™ (E8) medium (Stem Cell Technologies). Cells were passaged every 4 to 5 days according to a published protocol (Chen et al., 2011). Briefly, cells in 6-well plates were washed twice with 1 ml of 0.5 mM EDTA in PBS, incubated in 1 ml of fresh EDTA solution for 3 min, and suspended as aggregates of cells in 8 ml of E8 medium. Cell aggregates were then seeded on Matrigel™-coated plates. Three separate dynamic imaging experiments were performed with different preparations of cells. Preparation 1 cells were seeded for imaging on the 3rd passage after thawing. Preparation 2 came from the same thaw, and were passaged for 4 times before imaging. Preparation 3 were from a different thaw and were passage 5 times before imaging. Cells were seeded in 6-well plates in phenol red-free E8 medium (preparations 1 and 2) or in E8 medium containing approximately 1.3 mg/ml of phenol red (preparation 3). E8 medium was prepared according to the published recipe (DMEM/F12, sodium selenium (14 μg/l), insulin (19.4 mg/l), transferrin (10.7 mg/l), NaHCO3 (543 mg/l) (all from Life Technologies, Carlsbad, CA), FGF2 (100 μg/l), TGF-β1(2 μg/l) (both from R&D Systems, Minneapolis, MN), and L-ascorbic acid-2-phosphate magnesium (64 mg/l) (Sigma Aldrich, St. Louis, MO) (Chen et al., 2011).
After initial seeding of cells, plates were held in the incubator for 24 h and then moved to the microscope stage for 24 h prior to begin image collection. Day 1 of image collection is therefore day 3 after seeding. Plates were removed from the microscope stage for feeding every 24 h and replaced on the stage within 45 min of their removal, which was the time interval between imaging time points. Fiduciary marks on the plates facilitated alignment of images collected before and after feeding, and image stitching routines compensated for misalignments as described below.
4.3. Time-lapse imaging
For imaging, live cells were maintained at 37 °C and 5% CO2 with 80% humidity, in a custom built incubation chamber (Kairos Instruments, Pittsburgh, PA) attached to a Zeiss 200 M microscope (Carl Zeiss USA, Thornwood, NY) with a Zeiss 10×, 0.3NA objective (Zeiss part number 420341-9911-000) and an automated stage (Ludl Electronic Products, Hawthorne, NY). The microscope was equipped with a CoolSNAP HQ CCD camera (Photometrics, Tucson, Arizona), an LED epi-light source providing excitation light (470 nm, 25 mW; Thorlabs, Newton, NJ). LED light was passed through a filter cube with an excitation filter (470 nm ± 20 nm), emission filter (525 nm ± 20 nm), and a dichroic mirror centered at 495 nm (HE38 GFP filter set, Zeiss, part number 489038-9901-000). A fluorescent glass, Schott 475 GG glass was purchased as a 1 inch diameter round filter (Edmund Optics, Barrington, NJ; Part # 46-075; Lot # 035480), was used daily as a benchmark to assess microscope performance. A complete description for the use of this material is published (Halter et al., 2014). Stage, filters and shutters were controlled by the Zeiss Axiovision software. Every 45 min each FOV was exposed to light from a halogen lamp for phase contrast imaging, followed by excitation light for GFP fluorescence imaging. This series of images was taken for 120 h. Because of the large size of colonies (hundreds of microns after several days), and to assure adequate sampling of colonies, we imaged a large contiguous area of the culture plate well. The stage was programmed to move from field to field with an overlap of adjacent fields of 10% for preparations 1 and 2 and 20% for preparation 3 to facilitate stitching of all fields into a single composite image. Each composite image consisted of either 320 FOV (16 × 22) or 396 FOV (18 × 22). A spatial calibration target was used to determine that each pixel is equivalent to an area of 0.394 μm2.
Imaging live cells over long periods of time presents challenges in both data collection and analysis. To achieve long-term viability of cells and prevent light-induced damage, cells were exposed to a relatively low level of excitation light, which was determined as measured with a photodiode to be approximately 1.85 mW/2.3 mm2 or 0.8 mW/mm2. The growth of cells on the microscope was compared to the growth of cells kept in a jacketed incubator, and found to be commensurate (see Supplemental Fig. 1). While this exposure regime apparently prevented damage to cells, it resulted in a relatively low signal to noise. A systematic study of the effect of binning multiple pixels on the signal to noise ratio (Chalfoun et al., 2015) indicated that 4 × 4 binning provided a signal to noise ratio for each data point within a colony to be significantly (5× on average) above non-colony background levels. This provides evidence that the smallest feature size that can be detected with a signal to noise ratio > 5 in the GFP fluorescence channel is 4 × 4 pixels (6.3 μm2).
4.4. Image data visualization
Image data were saved in TIFF format. The composite images created by stitching together individual FOV contained several hundred colonies at early time points. Each composite image consists of about 22,912 × 20,775 pixels with a depth of 16 bits per pixel which is equivalent to approximately 1 GB of data. All image analysis was performed on the stitched multi-field composite images. Data for the three preparations consisted of 135, 141 and 161 time points respectively, resulting in a total of 0.9 TB of data for the entire experiment. The large amount of data precluded routine visualization on a desktop computer.
The Deep Zoom based visualization was enabled for the composite images of (a) raw GFP and phase contrast intensities, (b) their side row-time views, (c) corrected and corrected plus binned GFP intensities, and (d) the segmentation masks. It was extended to support viewing images along the temporal axis (3D data), to provide adaptive scale measurements during on-the-fly zooming and panning, to view 3D data from orthogonal viewpoints, and to sample any user-specified subset of the 3D image cube. The user interfaces replicated the experience of Google Maps browsing for images and enabled not only various downloading options for off-line processing but also interactive measurements of cell colonies in the browser (https://isg.nist.gov/deepzoomweb/) (Bajcsy et al., 2016).
4.5. Image processing steps and validation
The image processing pipeline is shown in Fig. 1A (schematic of all computational steps applied to the experimental replicas) and includes several software-based measurements that have been evaluated for their accuracy: image stitching, image segmentation, colony tracking, background correction, extraction of image analysis derived features, and predictive modeling of Oct4 heterogeneous, homogeneous, and non-expressing colony textural character. Each of these computational procedures is described in the Supplemental Information. Flat field correction was enabled by using a highly concentrated solution of fluorescein (Model and Burkhardt, 2001).
Supplementary Material
Acknowledgments
We would like to thank Michael Majurski, Jing Gao, and Mylene Simon for data processing and web application development. We would also like to thank Steven Lund and James Filliben for help with statistical analysis. MH acknowledges support from the ISAC Scholars Program.
Abbreviations
- hESC
human embryonic stem cell
- EGFP
enhanced green fluorescent protein
- FOV
field of view
- TB
terabyte
- GB
gigabyte
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.scr.2016.05.012
Disclaimer: Commercial products are identified in this document in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the products identified are necessarily the best available for the purpose.
References
- Ambrosetti DC, Schöler HR, Dailey L, Basilico C. Modulation of the activity of multiple transcriptional activation domains by the DNA binding domains mediates the synergistic action of Sox2 and Oct-3 on the fibroblast growth factor-4 enhancer. J Biol Chem. 2000;275:23387–23397. doi: 10.1074/jbc.M000932200. [DOI] [PubMed] [Google Scholar]
- Bajcsy P, et al. Interactive measurements of terabyte-sized 3D images. IEEE Computer. 2016 Jul; in press. [Google Scholar]
- Baker M. Reprogramming: faithful reporters. Nat Methods. 2012;9:231–234. [Google Scholar]
- Barbaric I, et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem cell reports. 2014;3:142–155. doi: 10.1016/j.stemcr.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Mathur D, Jaenisch R. Molecular control of pluripotency. Current Opinion in Genetics & Development. 2006;16:455–462. doi: 10.1016/j.gde.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfoun J, et al. Background intensity correction for terabyte-sized time-lapse images. J Microsc. 2015;257:226–237. doi: 10.1111/jmi.12205. [DOI] [PubMed] [Google Scholar]
- Chambers I, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Chan EM, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew JL, et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol. 2005;25:6031–6046. doi: 10.1128/MCB.25.14.6031-6046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipczyk A, et al. Network plasticity of pluripotency transcription factors in embryonic stem cells. Nat Cell Biol. 2015;17:1235–1246. doi: 10.1038/ncb3237. [DOI] [PubMed] [Google Scholar]
- Fink DW. FDA regulation of stem cell-based products. Science. 2009;324:1662. doi: 10.1126/science.1173712. [DOI] [PubMed] [Google Scholar]
- Gerrard L, Zhao D, Clark AJ, Cui W. Stably transfected human embryonic stem cell clones express OCT4-specific green fluorescent protein and maintain self-renewal and pluripotency. Stem Cells. 2005;23:124–133. doi: 10.1634/stemcells.2004-0102. [DOI] [PubMed] [Google Scholar]
- Halter M, et al. Cell cycle dependent TN-C promoter activity determined by live cell imaging. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2011;79:192–202. doi: 10.1002/cyto.a.21028. [DOI] [PubMed] [Google Scholar]
- Halter M, et al. An automated protocol for performance benchmarking a widefield fluorescence microscope. Cytometry Part A: the journal of the International Society for Analytical Cytology. 2014;85:978–985. doi: 10.1002/cyto.a.22519. [DOI] [PubMed] [Google Scholar]
- Hough SR, Laslett AL, Grimmond SB, Kolle G, Pera MF. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS One. 2009;4:e7708. doi: 10.1371/journal.pone.0007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough SR, et al. Single-cell gene expression profiles define self-renewing, pluripotent, and lineage primed states of human pluripotent stem cells. Stem Cell Reports. 2014;2:881–895. doi: 10.1016/j.stemcr.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacki-Neisius V, et al. Reduced Oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by Oct4 and Nanog. Cell Stem Cell. 2013;12:531–545. doi: 10.1016/j.stem.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- Model MA, Burkhardt JK. A standard for calibration and shading correction of a fluorescence microscope. Cytometry. 2001;44:309–316. doi: 10.1002/1097-0320(20010801)44:4<309::aid-cyto1122>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nichols J, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Niwa H, Miyazaki JI, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Sugawara T, Sakuma T, Yamamoto T. Stochastic promoter activation affects Nanog expression variability in mouse embryonic stem cells. Scientific reports. 2014;4:7125. doi: 10.1038/srep07125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- Pan G, Li J, Zhou Y, Zheng H, Pei D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006;20:1730–1732. doi: 10.1096/fj.05-5543fje. [DOI] [PubMed] [Google Scholar]
- Radzisheuskaya A, et al. A defined Oct4 level governs cell state transitions of pluripotency entry and differentiation into all embryonic lineages. Nat Cell Biol. 2013;15:579–590. doi: 10.1038/ncb2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda DJ, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- Silva J, Smith A. Capturing pluripotency. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, et al. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell. 2014;55:319–331. doi: 10.1016/j.molcel.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolik C, et al. Transcription factor competition allows embryonic stem cells to distinguish authentic signals from noise. Cell systems. 2015;1:117–129. doi: 10.1016/j.cels.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šustáčková G, et al. Differentiation-independent fluctuation of pluripotency-related transcription factors and other epigenetic markers in embryonic stem cell colonies. Stem Cells Dev. 2011;21:710–720. doi: 10.1089/scd.2011.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M, et al. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell. 2011;145:875–889. doi: 10.1016/j.cell.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg DL, et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.