

Abstract
In order to achieve enhanced and synergistic delivery of paclitaxel (PTX), a hydrophobic anticancer agent, two novel prodrug copolymers, POEG15-b-PFTS6 and POEG15-b-PFTS16 composed of hydrophilic poly(oligo(ethylene glycol) methacrylate) (POEG) and hydrophobic farnesylthiosalicylate (FTS, a nontoxic Ras antagonist) blocks, were synthesized. Both POEG-b-PFTS polymers were able to form micelles with intrinsic antitumor activity in vitro and in vivo. Employing these micelles as a carrier to load PTX, their drug loading capacity, stability, in vivo biodistribution and tumor inhibition effect were evaluated. PTX/POEG15-b-PFTS16 mixed micelles exhibited an excellent stability of 9 days at 4°C with a PTX loading capacity of 8.2%, which was more effective than PTX/POEG15-b-PFTS6 mixed micelles. In vivo biodistribution data showed that DiR-loaded POEG-b-PFTS micelles were more effectively localized in the tumor than in other organs. Moreover, both PTX/POEG-b-PFTS micelles showed significantly higher antitumor activity than Taxol in a 4T1.2 murine breast tumor model, and the tumor inhibition and animal survival followed the order of PTX/POEG15-b-PFTS16 > PTX/POEG15-b-PFTS6 > POEG15-b-PFTS16 > Taxol ≈ POEG15-b-PFTS6. Our data suggest that POEG-b-PFTS micelles are a promising anticancer drug carrier that warrants more studies in the future.
Keywords: farnesyl thiosalicylic acid (FTS), prodrug micelles, paclitaxel, doxorubicin, co-delivery
Graphical Abstract
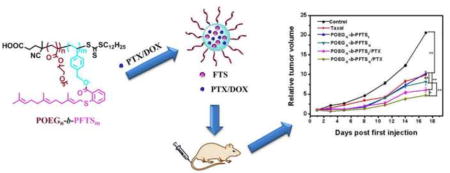
1. Introduction
Taxol is a clinical formulation of paclitaxel (PTX), which has been used to treat a range of malignant tumors. However, lack of tissue-specificity, hypersensitivity reactions and other side effects such as neuropathy limited its application [1–3]. To overcome these problems, various drug delivery systems such as liposomes, micelles, polymersomes and hydrogels have been developed [4–6]. Among them, polymeric micelles whose core-shell structures can encapsulate the hydrophobic drugs and prolong their circulation time in the blood have gained considerable attention. The small size of polymeric micelles will allow encapsulated drug to be effectively and passively delivered to tumor tissues via an enhanced permeability and retention (EPR) effect [7]. However, most polymeric micellar carriers are constructed by a large amount of pharmacologically inert materials which might increase the cost and lead to potential toxicity in vivo.
Micelles that are based on properly designed PEG-hydrophobic drug conjugates (prodrugs) possess natural amphiphilic properties and can serve as dual-functional carriers for co-delivery of other hydrophobic drugs [8–11]. Due to the antitumor activity of carrier materials, they can facilitate potential synergistic effects with the co-delivered drugs [12–14]. In addition, this system might provide a programmable loading and release of various drug components via both chemical conjugation and physical encapsulation.
It has been known that the development and growth of many tumors are related to the chronic activation of Ras signaling. S-trans,trans-farnesylthiosalicylic acid (FTS) is a nontoxic Ras antagonist, which could disassociate active Ras proteins from cell membrane selectively, leading to Ras dependent tumor regression [15–20]. Besides, FTS exhibits anti-inflammatory activity [21, 22]. Clinically, FTS (Salirasib) has been approved for phase I ~ II trials, and a phase I study reported that FTS was well tolerated and effective in relapsed hematological malignancies [23,24]. Inspired by the function of FTS, our group developed a series of prodrug micellar carriers which were based on PEG conjugates with two molecules of FTS [25–27]. It was shown that the PEG5K-FTS2 carrier well retained the biological activity of FTS upon examination in cultured tumor cells. Furthermore, PTX/PEG5K-FTS2 formulation showed a significantly higher anti-cancer activity than that of Taxol [25]. Nevertheless, the PTX loading capacity and micellar stability need to be improved.
Recent studies have shown that formulation stability, drug loading capacity and drug release rate of polymeric carriers can be regulated by modulating the size of hydrophobic cores [28–31]. Our previous work also demonstrated that a PEG conjugate with four FTS molecules (PEG5K-FTS4) could form more stable PTX-loaded micelles with higher PTX loading capacity than PEG conjugates with two FTS molecules (PEG5K-FTS2) [32]. Furthermore, in vivo study showed that PTX/PEG5K-FTS4 mixed micelles exhibited higher anti-tumor activity compared to PTX/PEG5K-FTS2 micelles. Nonetheless, due to the relatively low FTS contents in either PEG5K-FTS2- or PEG5K-FTS4-based carriers, the amounts of FTS that can be delivered to tumor tissues are limited, which limits the contribution of carriers-associated biological activity to the overall antitumor activity of drug-loaded micelles. This has promoted us to explore new prodrug micellar systems with further increased sizes of FTS-based hydrophobic core. Generally, most reports employed postmodification method by which a drug was chemically attached to a polymer backbone to construct prodrug micelles [33, 34]. However, this method often involves many synthesis steps which is a little complicated. In addition, because of steric hindrance, some reactive groups might remain in the polymers following conjugation, which might cause destruction of micelle structure or side effects in vivo through reacting with bioactive molecules [35]. Recently, polymerization of drug-based monomer has been developed as a facile strategy to obtain well-defined prodrug amphiphilic polymers [36, 37]. But most reports largely focus on the synthesis methods and the biophysical properties (e.g. micelle formation, enzymatic degradability, drug release, etc.) of prodrugs themselves [38, 39]. There is limited information about their anti-tumor activity and delivery function as prodrug carriers in vitro and in vivo. Therefore, there is still a need to develop more anti-tumor prodrug amphiphiles through development and subsequent polymerization of new drug-based monomers, and investigate their potential as a dual-functional drug delivery system.
In this work, we designed a novel FTS-based vinyl monomer that was easy to be synthesized and could be readily polymerized by reversible addition-fragmentation transfer (RAFT) polymerization to give well-defined diblock copolymers with a hydrophilic POEG block and a hydrophobic FTS block. The new polymers could be self-assembled into prodrug micelles and their sizes and structures were characterized. Employing these polymers as a PTX delivery carrier, the drug loading and release were further evaluated. Finally, the antitumor activity of carriers alone and PTX-loaded micelles was investigated in vitro and in vivo.
2. Materials and methods
2.1. Materials
FTS was synthesized and purified following a published literature [18]. Vinylbenzyl chloride, 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid, oligo(ethylene glycol) methacrylate OEGMA (average Mn=500), 2, 2-Azobis(isobutyronitrile) (AIBN), trypsin-EDTA solution, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and Dulbecco’s Modified Eagle’s Medium (DMEM) were all bought from Sigma-Aldrich (MO, U. S. A.). AIBN was purified by recrystallization in anhydrous ethanol. Paclitaxel was purchased from AK Scientific Inc. (CA, U. S. A.). DOX·HCl was purchased from LC Laboratories (MA, USA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were purchased from Invitrogen (NY, U. S. A.). Cell culture and animals were similarly handled as described before [32].
2.2. Synthesis of FTS-monomer
FTS (377.3 mg, 1.1 mM), vinylbenzyl chloride (167.2 mg, 1.1 mM) and K2CO3 (0.55 g, 5.5 mM) were dissolved in 5.5 mL DMF and immersed into an oil bath at 50°C. After 4 h, 5 mL water was added to the cooled mixture, and then 100 mL CH2Cl2 was added to extract the product. The organic phase was collected and washed by water and brine. After evaporation, silica gel column chromatography was used to purify the crude product with petroleum ether/diethyl ether (v/v, 8/1) as the elution. Colorless oil FTS-monomer was obtained with a 53% yield.
2.3. Synthesis of POEG macroCTA
OEGMA (3.05 g, 6.1 mmol), 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (120 mg, 0.305 mmol), AIBN (10 mg, 0.062 mmol) and 5 mL dried 1,4-dioxane were added in a Schlenk tube, and deoxygenated by free-pump-thawing for three times. Then under N2 protection, the mixture was put into an oil bath thermostated at 85°C. After 3 h, the tube was put into liquid nitrogen to quench the reaction and the mixture was then precipitated in diethyl ether for 3 times. After vacuum drying, the yellow oil product POEG was obtained. The conversion was 75% as determined by 1H NMR spectroscopy.
2.4. Synthesis of POEG-b-PFTS polymers
POEG macroCTA (140 mg, 0.0186 mmol), FTS-monomer (186 mg, 0.400 mmol), AIBN (1 mg, 0.0062 mmol) and 2 mL dried 1, 4-Dioxane were added in a Schlenk tube, and deoxygenated by free-pump-thawing for three times. The mixture was then protected under N2 and immersed into an oil bath at 90°C. After 24 h, the tube was put into liquid nitrogen to quench the reaction and the mixture was precipitated in hexane for 3 times, and dried in vacuum.
2.5. Characterization of the synthesized monomer and polymers
1H NMR spectrum was conducted on a Varian 400 FT-NMR spectrometer at 400.0 MHz with CDCl3 as the solvent. Molecular weight (Mn and Mw) and polydispersity index (Mw/Mn) of the synthesized polymers were measured by gel permeation chromatography (GPC) equipped with a Waters 2414 refractive index detector, a Waters 515 HPLC pump and a Waters 717 Plus Autosampler. THF was used as the eluent with a flowing rate of 1.0 mL/min at 35°C. A series of commercial polystyrene standards with narrow molecular weight distribution were applied to calibrate the GPC elution traces.
2.6. Preparation and characterization of PTX- and DOX-loaded micelles
PTX (5 mg/mL in dichloromethane) and POEG-b-PFTS polymers (50 mg/mL in dichloromethane) were mixed under different carrier/drug ratios. After removing the dichloromethane, a thin film was formed. PTX-loaded micelles were formed by adding DPBS solution to hydrate the thin film followed by gentle votexing. To load DOX into the POEG-b-PFTS micelles, DOX·HCl was first dissolved in a mixture of chloroform (CHCl3)/methanol (MeOH) (1:1, v/v) containing triethylamine (3 equiv) to remove HCl. DOX-loaded POEG-b-PFTS micelles were then similarly prepared. PTX loading efficiency was measured by high performance liquid chromatography (HPLC) and the DOX loading efficiency was examined by Waters Alliance 2695 Separations Module combined with Waters 2475 Fluorescence Detector (excitation, 490 nm; emission, 590 nm; gain, 3; sensitivity (FUFS), 10 000) as described before [25, 26]. Drug loading capacity (DLC) and drug loading efficiency (DLE) were calculated from the following equations:
The average diameter, size distribution and morphology of POEG-b-PFTS blank micelles and drug-loaded micelles were assessed by dynamic light scattering (DLS) and transmission electron microscopy (TEM).
2.7. Critical micelle concentration (CMC) and serum stability of POEG-b-PFTS micelles
The CMC values of POEG-b-PFTS micelles were measured with nile red as a fluorescence probe [40, 41]. In brief, 30 μL of a 0.05 mg/mL nile red solution in dichloromethane was added to each tube and then it was placed at room temperature overnight to remove solvent completely. Then 2 mL of POEG-b-PFTS micellar solution with different concentrations was added to each vial respectively. After 6 h, the emission spectra (570~720 nm) of the solutions were measured at an excitation wavelength of 550 nm and the peak intensities at 650 nm of the emission spectra were plotted versus polymer concentrations.
The serum stability of the polymers was evaluated by examining the FTS release in serum. The polymers were incubated with 50% serum (from rat) at 37 °C for 48 h. Then, methanol was added to extract the cleaved FTS for 3 times. After centrifugation at 12,000 rpm for 10 min, the supernatants were tested by MS analysis on a UPLC-QTOF MS system. FTS analysis was conducted on an Acquity UPLC BEH C18 column in negative mode with electrospray ionization.
2.8. In vitro PTX release
The kinetics of PTX release from PTX/POEG-b-PFTS mixed micelles was examined according to a previous report [42]. Briefly, 1 mL of PTX-loaded POEG-b-PFTS micelles or Taxol (1 mg PTX/mL) solution was transferred into a dialysis bag (MWCO = 3.5 kDa), and incubated in a tank with 50 mL PBS (PH = 7.4 or 5.5) containing 0.5% (w/v) Tween 80 as release medium, with gentle shaking at 37 °C. At specific time intervals, the PTX concentration in the dialysis bag was tested by HPLC at 227 nm wavelength.
2.9. In vitro cytotoxicity assay
In vitro cytotoxicity studies of POEG-b-PFTS polymers were carried out on both 4T1.2 mouse breast cancer cells and DU145 human prostate cancer cells. In brief, tumor cells were seeded into a 96-well plate at a density of 2000 cells/well and cultured overnight. Cells were challenged with various concentrations of blank POEG-b-PFTS micelles or free FTS at equivalent FTS concentrations for 72 h. MTT assay was performed as described before [25] and cell viability was calculated with untreated cells as a control. A similar study was performed with PTX- or DOX-loaded POEG-b-PFTS micelles and compared with Taxol or free DOX at same concentrations of chemotherapy drugs.
2.10. Near-infrared fluorescence optical imaging
Two hundreds μL of DiR-loaded POEG-b-PFTS micelles with a DiR concentration of 0.4 mg/mL were injected into SCID mice bearing bilateral s.c. PC-3 xenografts. At indicated times, the mice were imaged by Multispectral FX PRO system (Carestream Molecular Imaging) at a 60 s exposure time with excitation at 730 nm and emission at 835 nm. The mice were anesthetized by isoflurane inhalation before imaging. After 96 h, the mice were euthanized by CO2 overdose. The tumor and various organs were excised for ex vivo imaging.
2.11. In vivo therapeutic study
The in vivo antitumor efficacy of the POEG-b-PFTS-based drug delivery system was tested with a syngeneic 4T1.2 mouse breast cancer model [43]. 2×105 4T1.2 cells in 100 μL PBS were inoculated subcutaneously at the lower right flank of female BALB/c mice. When the tumor volume reached around 50 mm3, mice were divided into six groups (n=8) and PBS, POEG-b-PFTS blank micelles (POEG15-b-PFTS6 or POEG15-b-PFTS16), Taxol and PTX/POEG-b-PFTS micelles were i.v. injected into each group, respectively, at a PTX dose of 10 mg/kg. The treatments were performed every two days for a total of 5 times. Tumor sizes were measured every three days following the initiation of the treatment and mouse body weights were monitored as an indication of toxicity. The tumor sizes (V) were calculated by the equation: V= (length of tumor)× (width of tumor)2/2.
In addition to following changes in tumor sizes, the survival was also examined. The end point of survival will be defined as animal death or when the implanted tumor reached a volume of ~1000 mm3. The survival rate was plotted as KaplaneMeier curves and the median survival of mice was calculated.
2.12. Statistical analysis
Data are presented as mean ± standard deviation (SD). Two-tailed Student’s T test or one-way analysis of variance (ANOVA) was used to compare two groups or multiple groups, respectively, and Newman-Keuls test was performed if the overall p-value is <0.05. Survival data were generated by the KaplaneMeier method and statistical significance was evaluated by ManneWhitney U-tests. In all statistical analyses, p < 0.05 is considered statistically significant.
3. Results and Discussion
3.1. Synthesis and characterization of the POEG-b-PFTS polymers
We have previously developed a series of PEG5K-FTS2 polymers as prodrug carriers for PTX delivery [25–27]. However, due to the low percentage of hydrophobic FTS moieties in the polymers, these carriers showed limited PTX loading capacity and formulation stability. In addition, although PTX-loaded micelles exhibited high anti-tumor activity, PEG-FTS2 prodrug carriers alone showed minimal antitumor activity due to the low FTS loading capacity and thus limited amounts of FTS that can be delivered to the tumor tissues. This may limit the eventual synergistic action between PEG-FTS-based carriers and the co-delivered PTX. In order to overcome these problems, in this work, we designed and synthesized POEG-b-PFTS polymers with higher numbers of FTS moieties and investigated the impact of different amounts of FTS moieties on the delivery functions of carriers.
First, we designed and synthesized a new kind of FTS-based monomer where FTS was conjugated with vinylbenzyl chloride to form a hydrolyzable ester linkage as shown in Scheme 1. Then, the macro-chain transfer agent POEG was synthesized by RAFT polymerization of hydrophilic OEGMA monomer, which was further used to initiate the polymerization of hydrophobic FTS monomer, yielding the amphiphilic POEG-b-PFTS block copolymers. By varying the feeding ratio of FTS-monomer/macro-RAFT agent, POEG-b-PFTS copolymers with different PFTS block lengths (POEG15-b-PFTS6, POEG15-b-PFTS16, and POEG15-b-PFTS29) were prepared. Preliminary study showed that POEG15-b-PFTS29 formed precipitates in the course of micelle preparation (data not shown). This is likely due to the overly hydrophobic nature of POEG15-b-PFTS29. Therefore, subsequent studies were focused on POEG15-b-PFTS6 and POEG15-b-PFTS16 polymers. The structures of these polymers were confirmed by 1H NMR (Fig. 1), and the average degree of polymerization (DP) of the FTS monomers were calculated by comparing the intensities of Ib and Ia. The characteristics of these POEG and POEG-b-PFTS polymers as determined by 1H NMR and GPC were summarized in Table 1, which indicated the successful synthesis of the POEG-b-PFTS block copolymers with defined and controllable structures.
Scheme 1.

Synthesis of the FTS-monomer and POEG-b-PFTS polymers via RAFT polymerization.
Fig. 1.

1H NMR spectra of the POEG15, POEG15-b-PFTS6 and POEG15-b-PFTS16 polymers in CDCl3.
Table 1.
Characterizations of the POEG15 Macro-RAFT agent, POEG15-b-PFTS6 and POEG15-b-PFTS16 block copolymers.
| Block compolymer | Monomer conversiona | Hydrophilic/Hydroph obic ratiob | Mna (NMR) | Mnc (GPC) | Mwc (GPC) | Mw/Mnc (GPC) |
|---|---|---|---|---|---|---|
| POEG15 | 75 | - | 7500 | 7130 | 8140 | 1.14 |
| POEG15-b-PFTS6 | 75 | 7500/2844 | 10700 | 9160 | 10260 | 1.12 |
| POEG15-b-PFTS16 | 70 | 7500/7584 | 15500 | 13700 | 17600 | 1.30 |
Notes:
Calculated by NMR results.
The block molecular weight ratio of OEG/FTS in polymers.
Measured by GPC with THF as the eluent, and the molecular weights and their distribution were calculated with polystyrene standards.
3.2. Physicochemical characterization of blank and PTX-loaded POEG-b-PFTS micelles
A simple film hydration method was used for preparing blank POEG-b-PFTS and PTX-loaded POEG-b-PFTS micelles. Fig. 2 & Table 2 showed the CMCs of POEG15-b-PFTS6 and POEG15-b-PFTS16 micelles. It was interesting to note that incorporation of sixteen FTS units led to a 3-fold decrease in CMC compared to POEG15-b-PFTS6. The low CMC of POEG15-b-PFTS16 (~2.7 mg/L) shall provide a good stability for the micelles upon dilution in blood stream after administration. DLS showed that POEG15-b-PFTS6 and POEG15-b-PFTS16 formed micelles with a diameter of 30 nm and 130 nm in aqueous solution, respectively (Table 2). TEM (Fig. 3A and 3C) and cryo-EM (Fig. S1) images further confirmed the spherical shape of POEG-b-PFTS micelles. Besides, the FTS loading is calculated to be 20.1% for POEG15-b-PFTS6 and 36.7% for POEG15-b-PFTS16 prodrug micelles (Table 2), which is significantly higher than that of previously reported PEG-FTS conjugates (11.3~20.4%) [25, 26, 32].
Fig. 2.

Plots of fluorescence intensity at 650 nm versus concentrations of POEG15-b-PFTS6 (A) and POEG15-b-PFTS16 copolymers.
Table 2.
Size, CMC and FTS loading capacity of POEG15-b-PFTS6 and POEG15-b-PFTS16 micelles
| Micelles | Size (nm)a | PDIb | DLC(%)c | CMCd (mg/L) |
|---|---|---|---|---|
| POEG15-b-PFTS6 | 39.2 ± 0.4 | 0.26 ± 0.01 | 20.1 | 8.6 |
| POEG15-b-PFTS16 | 138.7 ± 3.3 | 0.33 ± 0.04 | 36.7 | 2.7 |
Measured by dynamic light scattering particle sizer.
PDI = polydispersity index.
FTS loading capacity.
CMC = critical micelle concentration.
Fig. 3.

TEM images of PTX-free POEG15-b-PFTS6 micelles (A) and POEG15-b-PFTS16 micelles (C), and PTX-loaded POEG15-b-PFTS6 micelles (B) and POEG15-b-PFTS16 micelles (D) using negative staining. Scale bar is 100 nm.
Since FTS is linked to the polymer by an ester bond, the serum stability of the polymers was evaluated by examining the FTS cleavage percentage in 50% serum. As shown in Fig. S2, only 0.4% and 0.2% FTS were cleaved from the POEG15-b-PFTS6 and POEG15-b-PFTS16 polymers in the serum, respectively, which indicated the good stability of POEG-b-PFTS polymers in serum. This is likely due to the fact that FTS is tightly packed in the hydrophobic core, limiting the digestion by the serum esterases.
We then examined and compared the PTX loading capacity and colloidal stability of the PTX-loaded POEG-b-PFTS micelles. As shown in Table 3 & Fig. 3B&D, PTX could be loaded into both POEG-b-PFTS micelles at a carrier/drug ratio as low as 10/1 (mg/mg). At this ratio, the POEG15-b-PFTS16 can achieve a PTX loading capacity of 8.2% and the PTX-loaded micelles were stable with no obvious changes in sizes or precipitation for 9 days at 4°C. In addition, upon increasing the carrier amount, the drug encapsulation efficiency and colloidal stability were further improved. It is also apparent that PTX-loaded POEG15-b-PFTS16 micelles showed better colloidal stability than POEG15-b-PFTS6 formulation at all carrier/drug ratios examined (Table 3). This improvement may be due to enhanced carrier/drug interactions provided by increased number of FTS hydrophobic motifs. In addition to PTX, several other commonly used chemotherapeutic agents including docetaxel, doxorubicin (DOX), gefitinib, and imatinib could be effectively loaded into POEG-b-PFTS prodrug carriers (Table 1 and data not shown). Table S1 and Fig. S3 showed the physicochemical characterization and TEM imaging of DOX-loaded POEG-b-PFTS micelles. Thus, the POEG-b-PFTS prodrugs may represent a versatile carrier for delivery of different kinds of anticancer drugs.
Table 3.
Physicochemical characterizations of PTX-loaded PEG-b-FTS micelles.
| Micelles | Mass ratio (mg: mg)a | Size (nm)b | PDIc | DLC(%)d | DLE(%)e | Stabilityf |
|---|---|---|---|---|---|---|
| POEG15-b-PFTS6 : PTX | 10: 1 | 32.8 ± 0.3 | 0.26 ± 0.01 | 4.6 | 51.1 | 5 d |
| POEG15-b-PFTS6 : PTX | 15: 1 | 32.8 ± 0.2 | 0.24 ± 0.02 | 4.3 | 68.7 | 8 d |
| POEG15-b-PFTS6 : PTX | 30: 1 | 37.8 ± 0.2 | 0.34 ± 0.03 | 2.8 | 87.4 | 12 d |
| POEG15-b-PFTS16 : PTX | 10: 1 | 115.6 ± 1.9 | 0.32 ± 0.04 | 8.2 | 90.5 | 9 d |
| POEG15-b-PFTS16 : PTX | 15: 1 | 113.9 ± 2.1 | 0.32 ± 0.04 | 5.4 | 85.9 | 13 d |
| POEG15-b-PFTS16 : PTX | 30: 1 | 130.6 ± 1.7 | 0.39 ± 0.03 | 3.1 | 96.7 | 23 d |
PTX concentration in micelles was kept at 1 mg/mL.
Measured by dynamic light scattering particle sizer.
PDI = polydispersity index.
DLC = drug loading capacity.
DLE = drug loading efficiency.
Data mean that there was no noticeable PTX precipitation during the follow-up period at 4 °C.
3.3. In vitro drug release
The release profile of PTX-formulated POEG-b-PFTS micelles was investigated with a dialysis method in DPBS (pH 7.4) with Taxol formulation as a control. As shown in Fig. 4, 45% of PTX was rapidly released from Taxol formulation in the first hour and around 60% of the total drug was released within 12 h. In comparison, less than 20% of PTX incorporated in the POEG-b-PFTS micelles was released in the first hour and a slow kinetics of release persisted for an extended period of 60 hours. The performance of PTX cumulative release follows the order of PTX/POEG15-b-PFTS16 > PTX/POEG15-b-PFTS6 > Taxol. Similar results were obtained when this experiment was conducted with an acidic (pH=5.5) release medium (Fig. S4). This sustained drug release is likely due to the strong carrier-drug interaction between PTX and POEG-b-PFTS carriers. POEG15-b-PFTS16 with more FTS units provided enhanced hydrophobic interaction, resulting in the most favorable release kinetics of PTX.
Fig. 4.

Cumulative PTX release profile from two PTX-loaded POEG-b-PFTS micelles with Taxol as the control. PBS containing 0.5% (w/v) Tween 80 was used as the release medium. PTX concentration was fixed at 1 mg PTX/mL. Values reported are the means ± SD for triplicate samples. *P < 0.05 (POEG15-b-PFTS16 vs Taxol).
3.4. In vitro cytotoxicity of prodrug micelles and PTX/DOX-loaded micelles
The cytotoxicity of POEG15-b-PFTS6 and POEG15-b-PFTS16 was examined in 4T1.2 mouse breast cancer cells and DU-145 human prostate cancer cells with free FTS as a control. Cell viability was determined by MTT assay after 3 days of treatment with untreated cells as a control. As shown in Fig. 5, free FTS exhibited a concentration-dependent tumor inhibition effect. The two POEG-b-PFTS prodrug micelles were less active than free FTS at equivalent amount of FTS in both 4T1.2 and DU145 tumor cells (Fig. 5A, B). The cytotoxicity of POEG-b-PFTS was unlikely due to the surfactant effect since polymer micelles exhibited little hemolytic activity at even much higher concentrations (data not shown). The cytotoxicity shall likely come from the FTS cleaved from the polymer following intracellular uptake. The less effectiveness of POEG-b-PFTS in cytotoxicity was likely due to the limited FTS release in a relative short period of treatment. We can expect an enhanced cell killing effect with an extended treatment since the POEG-b-PFTS carriers alone showed significant tumor inhibition effect in a later in vivo study (Fig. 8).
Fig. 5.

MTT cytotoxicity of two POEG-b-PFTS prodrug micelles in 4T1.2 mouse breast cancer cell line (A) and DU-145 human prostate cancer cell line (B) with free FTS as the control. Cells were treated with different micelles for 72 h and values reported are the means ± SD for triplicate samples. *P < 0.05, **P < 0.01 (POEG15-b-PFTS6 vs FTS); #P < 0.05, ##P < 0.01 (POEG15-b-PFTS6 vs POEG15-b-PFTS16).
Fig. 8.

(A) Antitumor activity of Taxol, POEG-b-PFTS prodrug micelles and PTX-loaded POEG-b-PFTS micelles in a syngeneic murine breast cancer model (4T1.2). Five injections were given on days 1, 2, 5, 8 and 11 and each point represents the mean of tumor size (n = 5). *P < 0.05; **P < 0.01. (B) Changes of body weight in mice receiving different treatments.
Figure 6A&B show the cytotoxicity of Taxol and PTX-loaded POEG-b-PFTS micelles in 4T1.2 and DU145 cell lines. PTX formulated in POEG-b-PFTS micelles showed a cell killing effect that was comparable to that of Taxol control. The cytotoxicity of DOX-loaded micelles were also evaluated with free DOX·HCl as the control. As presented in Fig. 6C&D, DOX-loaded POEG-b-PFTS micelles exhibited similar cell killing effect compared to free DOX·HCl.
Fig. 6.

MTT cytotoxicity assay of PTX-loaded POEG-b-PFTS micelles in 4T1.2 (A) and DU-145 cell line (B), and DOX-loaded POEG-b-PFTS micelles in 4T1.2 (C) and DU-145 cell line (D) after 72 h treatment. Data are presented as the means ± SD for triplicate samples. *P < 0.05, **P < 0.01 (POEG-b-PFTS/PTX vs Taxol, POEG-b-PFTS/DOX vs DOX); #P < 0.05, ##P < 0.01 (POEG15-b-PFTS6/DOX vs DOX).
3.5. Near-infrared fluorescence optical imaging
In vivo biodistribution of POEG-b-PFTS micelles was investigated by near-infrared fluorescent optical imaging in a PC-3 human prostate cancer xenograft model. A highly penetrating hydrophobic fluorescence dye DiR was loaded into the POEG-b-PFTS micelles for tissue imaging. As shown in Fig. 7A, DiR-loaded POEG-b-PFTS micelles were largely concentrated at tumor sites. The signals could be detected at as early as 1 hour after injection and peaked at about 24 h. However, substantial amounts of micelles were still retained at tumor sites even 96 h later. No obvious tumor accumulation was detected in the mice treated with free DiR dye [44].
Fig. 7.

(A) In vivo and (B) ex vivo NIRF imaging of DiR-loaded POEG15-b-PFTS6 and POEG15-b-PFTS16 micelles (denoted as PF6 and PF16) in prostate cancer PC-3 xenograft-bearing mice. (C) Quantitative fluorescence intensities of tumors and major organs from ex vivo images.
Tumors and major organs were harvested for ex vivo imaging and fluorescence quantification at 96 h post-injection. Significantly larger amounts of signals were observed in the tumor tissues compared to other major organs including lung, heart, liver, kidney and spleen (Fig. 7B). The enhanced micelle accumulation at tumor site other than other major organs/tissues was likely attributed to a strong enhanced permeability and retention (EPR) effect due to the small size (less or around 100 nm) of POEG-b-PFTS micelles and their excellent stability in blood stream. Fig. 7C showed that higher DiR signals were observed in tumors with DiR/POEG15-b-PFTS16 treatment compared to POEG15-b-PFTS6 formulation, suggesting POEG15-b-PFTS16 might serve as a better drug delivery system because of its enhanced stability and more effective tumor targeting in vivo.
3.6. In vivo therapeutic efficacy
The anti-tumor activity of PTX/POEG-b-PFTS mixed micelles was investigated by using an aggressive 4T1.2 mouse tumor model. It was demonstrated in Fig. 8A that Taxol formulation at a dose of 10 mg PTX/kg had a moderate tumor inhibition effect. Interestingly, POEG15-b-PFTS6 and POEG15-b-PFTS16 prodrug micelles alone showed comparable or further enhanced tumor inhibition effect compared to Taxol at day 17. It is likely that the antitumor activity of POEG-b-PFTS comes from the released FTS following the enzyme-mediated cleavage of the ester bond. This is quite different from our previous studies with PEG-FTS conjugates, in which the carriers alone showed minimal antitumor activity [25]. This is attributed to the fact that our new carriers allow delivery of sufficient amounts of FTS to the tumors due to the increased FTS moieties in POEG-b-PFTS polymers. It is also apparent that incorporation of PTX into POEG-b-PFTS micelles led to further improved anti-tumor activity. The superior anti-tumor activity of PTX/POEG-b-PFTS is likely attributed to the small size of the micelles, which allows them to efficiently penetrate and accumulate in the poorly vascularized tumors. Moreover, the potential combination effect of cleaved FTS and co-delivered PTX may also play a role in tumor inhibition. Overall, POEG15-b-PFTS16 micelles were more active than POEG15-b-PFTS6 micelles either by themselves or as mixed micelles with PTX (P<0.05). More FTS units in POEG15-b-PFTS16 provided stronger carrier-drug interaction to achieve an improved stability (Table 3) and better targeting (Fig. 7) in vivo. In addition, more FTS will be delivered to tumors with POEG15-b-PFTS16 system than with POEG15-b-PFTS6 system. All of the micellar formulations were well tolerated and no significant changes were found in mouse body weights (Fig. 8B).
In addition to following changes in tumor sizes, the survivals of the treated mice were examined. As shown in Fig. 9, control mice with PBS treatment had a short survival time of around 18 days. Substantial survival advantage was observed in mice treated with PTX/POEG-b-PFTS mixed micelles. The median survival of mice in PTX/POEG-b-PFTS group (35 days) is significantly longer than that of Taxol (24 days, P<0.05) and blank micelles (25 days, P < 0.05). These results suggested that PTX/POEG-b-PFTS micelles could effectively inhibit the tumor growth and increase the survival of tumor-bearing mice.
Fig. 9.
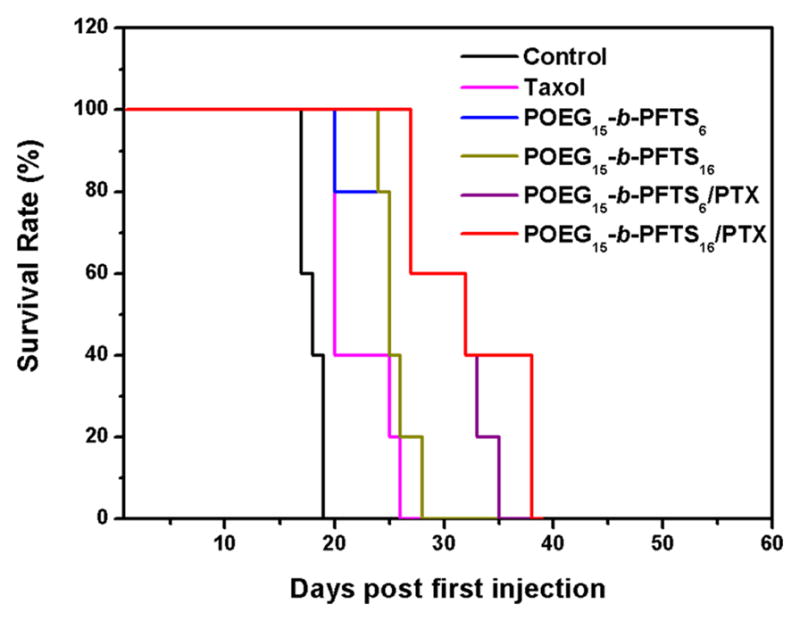
Kaplan-Meier survival curves for 4T1.2 tumor-bearing mice receiving different treatments. Mice were treated with Taxol, POEG-b-PFTS prodrug micelles, PTX-loaded POEG-b-PFTS micelles, and saline on days 1, 3, 5, 8, 11 respectively.
4. Conclusions
In this work, we reported two well characterized POEG-b-PFTS prodrug micelle carriers (POEG15-b-PFTS6 and POEG15-b-PFTS16) that consist of multiple FTS-based hydrophobic domains and a POEG hydrophilic segment for efficient entrapment of water-insoluble chemotherapy drugs. Our POEG-b-PFTS prodrug micelles well retained the biological activity of FTS in vitro and showed significant tumor inhibition effect in a 4T1.2 mouse tumor model. Besides, these POEG-b-PFTS nanocarriers could efficiently penetrate into and accumulate at the tumors. Both carriers were effective in formulating PTX and demonstrated a slow kinetics of drug release. Finally, in vivo delivery of PTX via our prodrug carriers, particularly POEG15-b-PFTS16, led to significant inhibition of 4T1.2 tumors, much more effectively than carriers alone and Taxol. Future studies include more comprehensive evaluation of the therapeutic efficacy in various tumor models. The potential of POEG-b-PFTS nanocarriers in delivery of other anticancer agents will also be examined. Finally, the toxicity profiles of POEG-b-PFTS nanocarriers, particularly their biodegradability, will be studied.
Supplementary Material
Statement of significance.
Polymerization of drug-based monomer represents a facile and precise method to obtain well-defined polymeric prodrug amphiphiles. Currently, most reports largely focus on the synthesis methods and the biophysical properties. There is limited information about their anti-tumor activity and delivery function as prodrug carriers in vitro and in vivo. In this manuscript, we report the development of two novel prodrug copolymers, POEG15-b-PFTS6 and POEG15-b-PFTS16 composed of hydrophilic poly(oligo(ethylene glycol) methacrylate) (POEG) and hydrophobic farnesylthiosalicylate (FTS, a nontoxic Ras antagonist) blocks. Both POEG-b-PFTS polymers were able to self-assemble into nano-sized micelles with intrinsic antitumor activity in vitro and in vivo. More importantly, POEG-b-PFTS polymers were effective in forming stable mixed micelles with various anticancer agents including PTX, DOX, docetaxel, gefitinib, and imatinib. Delivery of PTX via our new carrier led to significantly improved antitumor activity, suggesting effective PTX/FTS combination therapy. We believe that our study shall be of broad interest to the readers in the fields of biomaterials and drug delivery.
Acknowledgments
This work was supported by NIH Grants No. RO1CA173887, No. R01GM102989, and No. R21CA173887.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weiss RB, Donehower R, Wiernik P, Ohnuma T, Gralla R, Trump D, Baker J, Van Echo D, Von Hoff D, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 2.Feng SS, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58:4087–4114. [Google Scholar]
- 3.El-Rayes BF, Ibrahim D, Shields AF, LoRusso PM, Zalupski MM, Philip PA. Phase I study of liposomal doxorubicin (Doxil) and cyclophosphamide in solid tumors. Invest New Drugs. 2005;23:57–62. doi: 10.1023/B:DRUG.0000047106.20104.a7. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Li S, Tu T, Qi X, Xiong Y, Du S, Shen Y, Tu J, Sun C. Paclitaxel-loaded cholesterol-conjugated polyoxyethylene sorbitol oleate polymeric micelles for glioblastoma therapy across the blood-brain barrier. Polym Chem. 2015;6:2740–2751. [Google Scholar]
- 5.Vashist A, Vashist A, Gupta Y, Ahmad S. Recent advances in hydrogel based drug delivery systems for the human body. J Mater Chem B. 2014;2:147–166. doi: 10.1039/c3tb21016b. [DOI] [PubMed] [Google Scholar]
- 6.Gu Y, Zhong Y, Meng F, Cheng R, Deng C, Zhong Z. Acetal-linked paclitaxel prodrug micellar nanoparticles as a versatile and potent platform for cancer therapy. Biomacromolecules. 2013;14:2772–2780. doi: 10.1021/bm400615n. [DOI] [PubMed] [Google Scholar]
- 7.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Duncan R, Vicent MJ. Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev. 2013;65:60–70. doi: 10.1016/j.addr.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Lu J, Huang Y, Zhao W, Chen Y, Li J, Gao X, Venkataramanan R, Li S. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10:2880–2890. doi: 10.1021/mp300729y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu J, Huang Y, Zhao W, Marquez RT, Meng X, Li J, Gao X, Venkataramanan R, Wang Z, Li S. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials. 2013;34:1591–1600. doi: 10.1016/j.biomaterials.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Zhang X, Lu J, Huang Y, Li J, Li S. Targeted delivery of curcumin to tumors via PEG-derivatized FTS-based micellar system. AAPS J. 2014;16:600–608. doi: 10.1208/s12248-014-9595-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi Y, Zhao J, Feng SS. Targeted co-delivery of docetaxel, cisplatin and herceptin by vitamin E TPGS-cisplatin prodrug nanoparticles for multimodality treatment of cancer. J Control Release. 2013;169:185–192. doi: 10.1016/j.jconrel.2013.01.035. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS Nano. 2013;7:9518–9525. doi: 10.1021/nn405674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noh I, Kim HO, Choi J, Choi Y, Lee DK, Huh YM, Haam S. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763–774. doi: 10.1016/j.biomaterials.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer. Methods Enzymol. 2008;439:467–489. doi: 10.1016/S0076-6879(07)00432-6. [DOI] [PubMed] [Google Scholar]
- 16.Niv H, Gutman O, Henis YI, Kloog Y. Membrane interactions of a constitutively active GFP-Ki-Ras 4B and their role in signaling evidence from lateral mobility studies. J Biol Chem. 1999;274:1606–1613. doi: 10.1074/jbc.274.3.1606. [DOI] [PubMed] [Google Scholar]
- 17.Haklai R, Weisz MG, Elad G, Paz A, Marciano D, Egozi Y, Ben-Baruch G, Kloog Y. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37:1306–1314. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 18.Marom M, Haklai R, Ben-Baruch G, Marciano D, Egozi Y, Kloog Y. Selective inhibition of Ras-dependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270:22263–22270. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 19.Kloog Y, Cox AD. RAS inhibitors: potential for cancer therapeutics. Mol Med Today. 2000;6:398–402. doi: 10.1016/s1357-4310(00)01789-5. [DOI] [PubMed] [Google Scholar]
- 20.Gana-Weisz M, Halaschek-Wiener J, Jansen B, Elad G, Haklai R, Kloog Y. The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid chemosensitizes human tumor cells without causing resistance. Clin Cancer Res. 2002;8:555–565. [PubMed] [Google Scholar]
- 21.Mor A, Aizman E, Kloog Y. Celecoxib enhances the anti-inflammatory effects of farnesylthiosalicylic acid on T Cells independent of prostaglandin E2 production. Inflammation. 2012;35:1706–1714. doi: 10.1007/s10753-012-9488-8. [DOI] [PubMed] [Google Scholar]
- 22.Mor A, Aizman E, Chapman J, Kloog Y. Immunomodulatory properties of farnesoids: the new steroids? Curr Med Chem. 2013;20:1218–1224. doi: 10.2174/0929867311320100002. [DOI] [PubMed] [Google Scholar]
- 23.Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6:1435–1437. doi: 10.1097/JTO.0b013e318223c099. [DOI] [PubMed] [Google Scholar]
- 24.Badar T, Cortes JE, Ravandi F, O’Brien S, Verstovsek S, Garcia-Manero G, Kantarjian H, Borthakur G. Phase I study of S-trans, trans-farnesylthiosalicylic acid (salirasib), a novel oral RAS inhibitor in patients with refractory hematologic malignancies. Clinical Lymphoma Myeloma and Leukemia. 2015;15:433–438. doi: 10.1016/j.clml.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Lu J, Huang Y, Zhao W, Chen Y, Li J, Gao X, Venkataramanan R, Sun M, Stolz DB, Zhang L, Li S. PEG-farnesylthiosalicylate conjugate as a nanomicellar carrier for delivery of paclitaxel. Bioconjug Chem. 2013;24:464–472. doi: 10.1021/bc300608h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Huang Y, Zhao W, Liu H, Marquez R, Lu J, Zhang P, Zhang Y, Li J, Gao X, Venkataramanan R, Xu L, Li S. Targeted delivery of anticancer agents via a dual function nanocarrier with an interfacial drug-interactive motif. Biomacromolecules. 2014;15:4326–4335. doi: 10.1021/bm501339j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Huang Y, Ghazwani M, Zhang P, Li J, Thorne SH, Li S. Tunable pH-responsive polymeric micelle for cancer treatment. ACS Macro Lett. 2015;4:620–623. doi: 10.1021/acsmacrolett.5b00165. [DOI] [PubMed] [Google Scholar]
- 28.Mi Y, Liu Y, Feng SS. Formulation of docetaxel by folic acid-conjugated d-α-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS 2k) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32:4058–4066. doi: 10.1016/j.biomaterials.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 29.Aliabadi HM, Lavasanifar A. Polymeric micelles for drug delivery. Expert Opin Drug Deliv. 2006;3:139–162. doi: 10.1517/17425247.3.1.139. [DOI] [PubMed] [Google Scholar]
- 30.Huynh VT, de Souza P, Stenzel MH. Polymeric micelles with pendant dicarboxylato chelating ligands prepared via a michael addition for cis-platinum drug delivery. Macromolecules. 2011;44:7888–7900. [Google Scholar]
- 31.Wang J, Sun J, Chen Q, Gao Y, Li L, Li H, Leng D, Wang Y, Sun Y, Jing Y. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33:6877–6888. doi: 10.1016/j.biomaterials.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, Huang Y, Zhao W, Chen Y, Zhang P, Li J, Venkataramanan R, Li S. PEG-farnesyl thiosalicylic acid telodendrimer micelles as an improved formulation for targeted delivery of paclitaxel. Mol Pharm. 2014;11:2807–2814. doi: 10.1021/mp500181x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du JZ, Du XJ, Mao CQ, Wang J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J Am Chem Soc. 2011;133:17560–17563. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Yu F, Chen Y, Oupický D. Polymeric drugs: advances in the development of pharmacologically active polymers. J Control Release. 2015;219:369–382. doi: 10.1016/j.jconrel.2015.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delplace V, Couvreur P, Nicolas J. Recent trends in the design of anticancer polymer prodrug nanocarriers. Polym Chem. 2014;5:1529–1544. [Google Scholar]
- 36.Smith D, Pentzer EB, Nguyen ST. Bioactive and therapeutic ROMP polymers. J Macromol Sci: Polym Rev. 2007;47:419–459. [Google Scholar]
- 37.Hasegawa U, Van Der Vlies AJ, Wandrey C, Hubbell JA. Preparation of well-defined ibuprofen prodrug micelles by RAFT polymerization. Biomacromolecules. 2013;14:3314–3320. doi: 10.1021/bm4009149. [DOI] [PubMed] [Google Scholar]
- 38.Zuwala K, Smith AA, Postma A, Guerrero-Sanchez C, Ruiz-Sanchis P, Melchjorsen J, Tolstrup M, Zelikin AN. Polymers fight HIV: potent prodrugs identified through parallel automated synthesis. Adv Healthcare Mater. 2015;4:46–50. doi: 10.1002/adhm.201400148. [DOI] [PubMed] [Google Scholar]
- 39.Smith AA, Wohl BM, Kryger MB, Hedemann N, Guerrero-Sanchez C, Postma A, Zelikin AN. Macromolecular prodrugs of ribavirin: concerted efforts of the carrier and the drug. Adv Healthcare Mater. 2014;3:1404–1407. doi: 10.1002/adhm.201300637. [DOI] [PubMed] [Google Scholar]
- 40.Mynar JL, Goodwin AP, Cohen JA, Ma Y, Fleming GR, Frechet JMJ. Two-photon degradable supramolecular assemblies of linear-dendritic copolymers. Chem Commun. 2007:2081–2082. doi: 10.1039/b701681f. [DOI] [PubMed] [Google Scholar]
- 41.Klaikherd A, Nagamani C, Thayumanavan S. Multi-stimuli sensitive amphiphilic block copolymer assemblies. J Am Chem Soc. 2009;131:4830–4838. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y, Lu J, Gao X, Li J, Zhao W, Sun M, Stolz DB, Venkataramanan R, Rohan LC, Li S. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjug Chem. 2012;23:1443–1451. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang P, Huang Y, Liu H, Marquez RT, Lu J, Zhao W, Zhang X, Gao X, Li J, Venkataramanan R, Li S. A PEG-Fmoc conjugate as a nanocarrier for paclitaxel. Biomaterials. 2014;35:7146–7156. doi: 10.1016/j.biomaterials.2014.04.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho H, Indig GL, Weichert J, Shin HC, Kwon GS. In vivo cancer imaging by poly (ethylene glycol)-b-poly (ε-caprolactone) micelles containing a near-infrared probe. Nanomedicine: NBM. 2012;8:228–236. doi: 10.1016/j.nano.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.