

Summary
Neurodegenerative diseases are distinguished by characteristic protein aggregates initiated by disease‐specific ‘seed’ proteins; however, roles of other co‐aggregated proteins remain largely unexplored. Compact hippocampal aggregates were purified from Alzheimer's and control‐subject pools using magnetic‐bead immunoaffinity pulldowns. Their components were fractionated by electrophoretic mobility and analyzed by high‐resolution proteomics. Although total detergent‐insoluble aggregates from Alzheimer's and controls had similar protein content, within the fractions isolated by tau or Aβ1–42 pulldown, the protein constituents of Alzheimer‐derived aggregates were more abundant, diverse, and post‐translationally modified than those from controls. Tau‐ and Aβ‐containing aggregates were distinguished by multiple components, and yet shared >90% of their protein constituents, implying similar accretion mechanisms. Alzheimer‐specific protein enrichment in tau‐containing aggregates was corroborated for individuals by three analyses. Five proteins inferred to co‐aggregate with tau were confirmed by precise in situ methods, including proximity ligation amplification that requires co‐localization within 40 nm. Nematode orthologs of 21 proteins, which showed Alzheimer‐specific enrichment in tau‐containing aggregates, were assessed for aggregation‐promoting roles in C. elegans by RNA‐interference ‘knockdown’. Fifteen knockdowns (71%) rescued paralysis of worms expressing muscle Aβ, and 12 (57%) rescued chemotaxis disrupted by neuronal Aβ expression. Proteins identified in compact human aggregates, bound by antibody to total tau, were thus shown to play causal roles in aggregation based on nematode models triggered by Aβ1–42. These observations imply shared mechanisms driving both types of aggregation, and/or aggregate‐mediated cross‐talk between tau and Aβ. Knowledge of protein components that promote protein accrual in diverse aggregate types implicates common mechanisms and identifies novel targets for drug intervention.
Keywords: Abeta(1‐42), acetylation (protein), aggregation (protein), Alzheimer (Disease), beta amyloid, C. elegans, microtubule‐associated protein tau, neurodegeneration, neurotoxicity, oxidation (protein), phosphorylation (protein), proteomics
Introduction
Protein aggregation has long been recognized as a common feature of most or all age‐dependent neurodegenerative diseases, and yet very little is known about which features of aggregating proteins contribute to their accrual or their neurotoxicity. Intriguing clues have come from genetic mutations that cause such diseases to recur sporadically in pedigrees, even though familial cases typically comprise only a small fraction of the total (Lio et al., 2003; Cardenas et al., 2012; Abdel‐Salam, 2014; Barmada, 2015; Heneka et al., 2015). Those mutations may render a single ‘seed’ protein susceptible to aggregation in whichever brain regions it is chiefly expressed. Examples include genetic expansion of polyglutamine (polyQ) arrays in the huntingtin protein (Huntington's disease, HD) as well as in ataxins and similar Q‐rich proteins (spinocerebellar ataxias); mutations to SOD1 that favor its aggregation (amyotrophic lateral sclerosis, ALS); mutations in α‐synuclein that cause Parkinson's disease (PD); mutations in A4/APP or presenilins that favor production of amyloid β peptide, Aβ1‐42 (Alzheimer's disease, AD); and mutated forms of tau that are predisposed to form neurofibrillary tangles (certain familial AD pedigrees, other tauopathies) (Jellinger, 2012). Some neuropathic mutations, however, may interfere more broadly with the normal clearance of diverse misfolded and aggregated proteins. Proteins harboring those mutations include ubiquilin‐1, leading to presenilin aggregation (mutated in some familial AD) (Viswanathan et al., 2011); ubiquilin‐2 which also targets ubiquitinylated proteins to proteasomes (mutated in ALS) (Zhang et al., 2014); and parkin, an E3 ubiquitin ligase required for mono‐ubiquitin addition to specific protein targets (mutated in PD) (Roy et al., 2015). Other predisposing factors can markedly elevate the risk of specific neurological diseases, for example, brain trauma, epilepsy, hypertension, obesity, type 2 diabetes, and exposure to toxic chemicals (in AD, PD) (Vosler et al., 2008; Zigman, 2013). Age is a major risk factor for almost all neuropathies, as well as an important determinant of disease progression (Brehme et al., 2014; Fjell et al., 2014).
Factors that confer risk for progressive neurodegenerative diseases, although diverse, are all consistent with a simple unifying hypothesis: Protein aggregation leads to neural damage and is exacerbated by aging and other pro‐inflammatory triggers (David et al., 2010; Dillin & Cohen, 2011). Aggregates are neurotoxic (or more generally cytotoxic) in at least some forms (Bucciantini et al., 2002); it remains unresolved whether the greater threat is posed by relatively soluble and diffuse aggregates (typically oligomeric) or insoluble, compact conglomerates (Tai et al., 2013). Protein aggregates have been shown to form even during normal aging of nematodes (David et al., 2010; Ayyadevara et al., 2014). Postsynthetic modifications such as hyperphosphorylation (Rudrabhatla et al., 2011) and oxidation (Boyd‐Kimball et al., 2006) strongly promote aggregation and may also accrue with age.
We recently characterized protein aggregates formed in a C. elegans model of HD, expressing Q40::YFP (a 40‐glutamine tract fused in‐frame to yellow fluorescent protein) in body‐wall muscle. We purified co‐aggregating proteins by YFP‐antibody affinity and analyzed them by high‐resolution proteomics (Ayyadevara et al., 2014). We demonstrated in this model that several proteins identified in compact Q40‐containing foci promote aggregation by disrupting proteasomal function, and knockdown of their expression markedly reduced the extent of aggregation in the same HD model and in two C. elegans lines expressing a human Aβ1–42 transgene (Ayyadevara et al., 2014).
We now extend this experimental approach to affected hippocampal tissue from subjects with dementia and histopathology typical of AD, and we compare them to the same regions from normal controls spanning the same age range. The hippocampus, a cortical structure implicated in memory formation, is especially vulnerable in AD. We find higher levels in AD of hippocampal aggregates that bind antibody against Aβ1–42 or tau; these immuno‐purified conglomerates contain many proteins that differ markedly and reproducibly in quantity and post‐translational modifications between AD and normal controls. Intriguingly, we find roughly twofold increases in oxidation, phosphorylation, and acetylation of specific proteins enriched in AD aggregates. Selected results were confirmed in additional proteomic analyses, targeted knockdowns, and imaging of proximity ligation products.
Results
Proteomic analysis of pure aggregates from Alzheimer and control hippocampus
We first isolated and compared compact‐aggregate fractions from pools of caudal hippocampus obtained from AD vs. normal controls (NC). After differential centrifugation to remove debris and organelles, tissue homogenates were gently mixed with magnetic beads (Dynabeads™) coated with antibody to either Aβ1–42 or tau. Complexes containing those proteins were bound, rinsed thoroughly, and recovered by elution from the beads. The two immuno‐adsorbed subfractions as well as total large aggregates (prior to affinity isolation) were each partitioned by solubility in 1% sarcosyl to separate ‘compact’ (sarcosyl‐insoluble) aggregates from those that dissolved in this strong ionic detergent, in the presence of 0.3‐m β‐mercaptoethanol as reducing agent (Ayyadevara et al., 2014). A typical 1‐D acrylamide gel is shown in Fig. 1A for sarcosyl‐insoluble fractions. Most, but not all, Aβ‐ and tau‐associated proteins are more abundant in compact aggregates derived from AD samples (lanes 2 and 4) than in those derived from NC samples (lanes 1 and 3), whereas total signal for large aggregates did not differ in overall intensity between groups (compare lanes 5 and 6). Note, however, that the band at ~30 kDa did not differ appreciably in intensity between AD and NC, providing a sort of fortuitous, unidentified negative control for bands that did alter.
Figure 1.
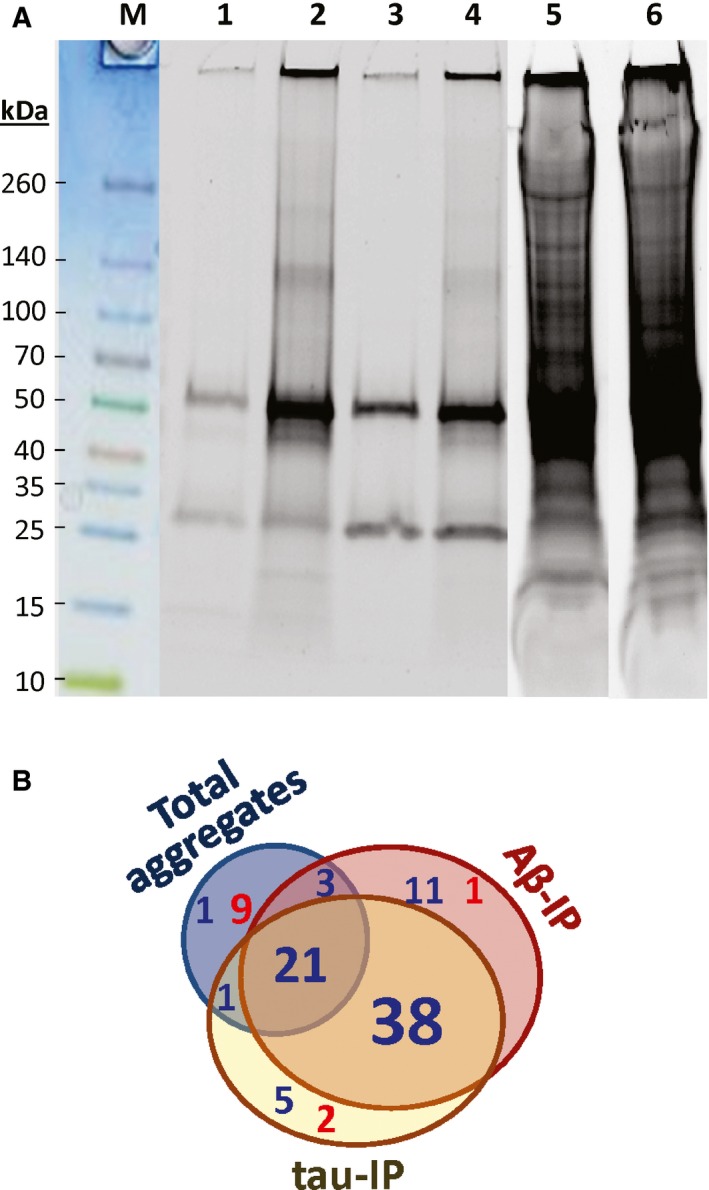
Proteins in detergent‐insoluble AD‐derived aggregates are more abundant and diverse than those from controls. (A) Aggregates insoluble in 1% sarcosyl, isolated from hippocampi of normal controls (3 subjects pooled, lanes 1, 3, and 5) or AD (pool of 3, lanes 2, 4, 6). Lanes 1 and 2: pulldown with antibody to Aβ1–42; lanes 3 and 4: pulldown with antibody to tau; lanes 5 and 6: total large insoluble aggregates. (B) Venn diagram of proteins found to be significantly enriched (blue font) or depleted (red) in aggregates from AD relative to controls. More than half of the proteins listed in Table 1 (59/100) were significantly more abundant in both Aβ and tau pulldowns from AD than from controls.
Proteomic analyses of insoluble fractions were conducted as detailed in Experimental procedures. For each fraction, corresponding to lanes 1–6 of Fig. 1A, proteins were separated by preparative gel electrophoresis. Gels were sliced, and the slices (including insoluble material at the top) were robotically excised for trypsin digestion prior to peptide analysis by mass spectroscopy (LC‐MS/MS). No insoluble material was detected after digestion.
Positively identified proteins and their modifications are listed in Table 1. Data columns 1–6, corresponding to lanes 1–6 of Fig. 1, show actual spectral counts, which are proportional to relative protein abundances after adjustment for protein size (Byrum et al., 2013). For Aβ immuno‐pulldown (IP), 134 proteins differed significantly (at chi‐squared P < 0.05) between AD and NC, far exceeding the 15 expected by chance (=5% of ~300 proteins with sufficient counts to reach significance; see Experimental procedures for details, and Table 1, for examples). In tau‐IP, 121 proteins differed between AD and NC (at P < 0.05) vs. 11 expected by chance. For total sarcosyl‐insoluble aggregates, which are much more abundant overall, 115 proteins altered significantly whereas 44 would be expected at random.
Table 1.
Relative protein abundances in immuno‐purified (IP) vs. total sarcosyl‐insoluble aggregates from AD vs. NC hippocampus. Spectral counts are shown for insoluble fractions: totals in black, oxidized peptides red, phosphorylations green, and acetylations blue

Ten proteins (yellow highlighting in Table 1) clearly distinguish between Aβ1–42‐ and tau‐affinity IP in the control samples, some by >30‐fold (tau, apolipoprotein D, and clusterin). The two IPs from AD tissue likewise differ markedly from one another in the abundance of 5 proteins (α‐enolase, sequestosome‐1, tau, and talins 1 and 2), highlighted in blue. Such striking differences demonstrate that distinct aggregate populations were isolated by the two antibodies.
Nevertheless, these data also provide evidence of extensive overlap between proteins co‐aggregating with Aβ1–42 vs. tau. Of the 100 proteins listed in Table 1 were significantly more abundant (at χ2 P < 0.05) in aggregates from AD hippocampus than from NC, after both Aβ‐ and tau‐affinity IP (Fig. 1B). These include many proteins associated with axonal transport (tau, dynactins, dynamins, dynein 1, filamins, GFAP, cofilins, microtubule‐associated proteins MAP1A & B, and neurofilament chains NFL, NFM, & NFH), neurotransmission (band 4.1‐like proteins, β‐SNAB, synapsins, synemin, and syntaxins), other neuronal processes (amine oxidase B, contactin 1, excitatory AA transporter 2, Na+/K+ transporter ATPase α2, tenascin receptor), redox control (peroxiredoxin 1, carbonyl reductase, and Mu‐class GSTs), signal transduction (14‐3‐3 proteins, elongation factor 1α, Gα, β proteins, 6‐phosphofructokinase C), and proteostasis (heat‐shock proteins, MAP1 complex, peptidyl‐prolyl isomerase, sequestosome‐1, UBA‐1, polyubiquitin B, ubiquitin C‐terminal hydroxylase, vinculin). In contrast to those AD‐associated increases, α and β synucleins, characteristic of Parkinson inclusions (Park et al., 2002), were depleted in tau‐ and Aβ‐IP aggregates derived from AD hippocampus.
All of the above AD‐NC changes were peculiar to the tau‐ and Aβ‐pulldown fractions and were severely blunted in total aggregates, isolated by sarcosyl insolubility without IP. Of the 59 proteins with higher representation in AD‐derived tau‐ and Aβ‐IP aggregates, 21 were more modestly (but significantly) enriched with AD in total large aggregates (data columns 5 and 6), whereas 9 were depleted in AD samples. Total aggregate protein far exceeds the sum of IP fractions, as is apparent in Fig. 1A (note lighter exposure for lanes 5 and 6), Table 1, and Supplemental Spreadsheets, reflecting the age‐dependent accrual of protein aggregates even without a pathogenic ‘seed’ protein (David et al., 2010; Ayyadevara et al., 2014). However, pronounced AD/NC bias was rare in total aggregates, where it was seen only for apolipoprotein E, neurochondrin, secernin‐1, and tau, with an intermediate shift also evident for filamins A and C, β‐SNAB, and carbonyl reductase.
Peptides modified by oxidation, phosphorylation, or acetylation were identified and, with few exceptions, were substantially enriched in Aβ1–42‐ and tau‐affinity pulldowns from AD (Table 1, colored numbers, summarized in the bottom rows and in Fig. S1; see also (Boyd‐Kimball et al., 2006)). Oxidations, the most common modifications (typically 25–44% of total hits), increased nearly threefold in AD relative to normal controls (>1.7‐fold even after adjusting for 66% more aggregates observed in AD), in both Aβ1–42‐ and tau‐affinity aggregates (for each IP, P = 0.006 by 2‐tailed paired t‐tests; P < 10−6 by Fisher exact tests) but not in total aggregates from AD subjects (AD ≈ NC). Oxidation of AD‐aggregate proteins was 31% more frequent in Aβ1–42‐IP aggregates than in tau‐IP aggregates (2‐tailed paired t‐test P < 0.008), as might be expected from the predominantly extracellular sites of Aβ plaque. The extent of oxidation thus varies among aggregate proteins, reflecting variation in susceptibility and also differs between aggregate classes. For example, oxidized sites were found in 22–23% of all neurofilament‐chain peptides from Aβ‐IP aggregates but were seen in only 1–2% of the same peptides from either total or tau‐IP aggregates (chi‐squared P < 10−30 for each difference). In contrast, elongation factor 1α is far more oxidized in total aggregates (27–31%) than in Aβ‐ or tau‐IP aggregates (0–13%). Oxidized proteins are thought to be more vulnerable to aggregation and were found to be elevated in a C. elegans model of AD (Boyd‐Kimball et al., 2006) and in hippocampi of AD and aging Down's patients (Butterfield et al., 2014; Di Domenico et al., 2014). However, this report is the first (to our knowledge) to identify and quantify proteins and modifications from specific affinity‐purified aggregates, and the first to show unequal distribution of oxidized proteins among aggregate types, which must reflect differing properties of those classes (see Discussion).
Phosphorylations were seen in 285 proteins, 12% of those detected. Summing over all aggregate proteins, phosphorylations were 3.2‐ and 3.5‐fold more frequent within Aβ‐ and tau‐containing aggregates, respectively, from AD vs. NC samples (1.9‐ and 2.1‐fold after adjustment for aggregate quantities) but were scarcely elevated in total aggregates (P < 1E–7 by Fisher's exact tests). Although tau‐IP aggregates were purified with antibody to native tau, the increase of tau peptides in AD samples was largely due to higher abundance of tau phosphopeptides in AD; the fraction of phosphorylated tau peptides was >52% for AD, vs. 22% for NC (P < 10−11). Among other proteins recovered from aggregates, phosphorylated peptides comprised 0–8% of total peptides (e.g., 3 neurofilament chains and GFAP), averaging twofold to threefold higher levels in AD than NC samples. Acetylations were only half as frequent as phosphorylations in these aggregates, but were likewise enriched in samples derived from AD relative to NC: 3.3‐fold after Aβ‐IP, 2.8‐fold after tau‐IP (2‐ and 1.7‐fold with adjustment for aggregate quantities; each P < 0.005), but <1.3‐fold in total large aggregates (Table 1, last 4 rows). These data could imply that acetylation, phosphorylation, and oxidation all promote aggregation of at least some proteins. Other explanations, such as increased susceptibility to modification of proteins in specific aggregates, cannot be excluded by the data presented. In view of the consistent lack of AD enrichment for similar modifications in total aggregate proteins, however, a purely artefactual explanation appears unlikely.
Meta‐analysis of aggregate proteins that are differentially represented in AD vs. controls
We analyzed the 89 most differentially represented proteins for functional‐annotation term enrichment using GO/pathway analysis (DAVID ver. 6.7, https://david.ncifcrf.gov/; Nature Protocols 2009; 4: 44). The top GO and KEGG terms identified were as follows:
| Term | N | Enrichment | FDR (Benj.) |
|---|---|---|---|
| Cytoskeleton | 39 | 4.9 | 4.4E–16 |
| Acetylation | 45 | 4.1 | 6.0E–16 |
| Cytoplasm | 49 | 3.5 | 1.2E–15 |
| Phosphoprotein | 67 | 2.2 | 1.5E–14 |
| Neural projection | 20 | 10.1 | 3.8E–12 |
| 14‐3‐3 proteins | 6 | 216 | 5.1E–11 |
| Actin binding | 17 | 9.4 | 1.8E–09 |
| P‐ser interaction | 6 | 202 | 1.1E–08 |
| Intracellular transport | 19 | 5.4 | 5.1E–06 |
| GTP binding | 7 | 24.8 | 1.2E–05 |
| ATPase, G type | 5 | 69.4 | 3.9E–05 |
| Disease mutation | 22 | 3.3 | 4.1E–05 |
| Microtubule | 9 | 9.3 | 1.3E–04 |
| Neurodegeneration | 5 | 14.8 | 4.4E–03 |
These highly significant enrichments corroborate processes that were also suggested by the differential protein abundances of Table 1. Specifically, acetylation and phosphorylation of proteins (and proteins that recognize other phosphorylated proteins) are important to aggregation, as are pathologic genetic mutations. The cell structures most closely associated with aggregate components are the cytoskeleton, cytoplasm, axons, and dendrites, with particular involvement of microtubules, neurofilaments and actin‐binding proteins. Related processes include neurodegeneration and heritable diseases, as well as intracellular transport, a variety of ATPases, and GTP binding proteins.
Proteomic analyses of individual cortical samples
The above proteomic studies compared pooled hippocampal tissue from 3 AD cases to a similar pool from 3 normal controls. To assess within‐ and between‐group variation in the protein composition of aggregates, we compared proteomic data for tau‐IP samples from caudal hippocampus, analyzed individually for 4 AD vs. 4 age‐matched control (AMC) subjects, 61–85 years of age (Table S1). This comparison was not intended to precisely replicate that of pooled samples, which had been fractionated by gel electrophoresis, as individual samples were not similarly prefractionated—an economy that reduced the yield of identified proteins. Nevertheless, many proteins that were most differentially abundant in tau‐IP aggregates from AD vs. control pools also differed significantly between AD vs. AMC groups when individual aggregate samples were analyzed. In this analysis, 25 proteins were significantly enriched in aggregates from AD tissue, at t‐test P < 0.05. Table 2 lists 20 AD‐enriched proteins from this comparison, with AD/AMC ratios >1.5, and their P values (in 1‐tailed heteroscedastic t‐tests, appropriate to 1‐way comparisons for samples of this size) (“tau‐IP 1” columns). Table 2 also includes data from a technical repeat comparing 4 AD to 4 NC samples using a different LC‐MS platform and protocol (“tau‐IP 2” columns). The correlation coefficient between technical replicates for AD/AMC ratios of all 25 tau‐IP proteins was R = 0.80 (P < 1E–6), or 0.83 (P < 4E–5) considering only 11 proteins with P ≤ 0.08. The last column, tau pools, shows corresponding hits for pooled samples (Table 1).
Table 2.
Proteins in tau immuno‐purified hippocampus aggregates: Differential abundance for AD vs. AMC individuals

In the primary analysis, 235 ± 50 (mean ± SD) proteins were identified per individual in tau‐IP aggregates, and a similar number (264 proteins) were present in enough individuals to have the potential to indicate a significant shift between AD and AMC. Of these, 12–13 proteins are expected to appear differential by chance alone at α ≤ 0.05, whereas 25 were enriched in AD aggregates by this criterion. Because up to half of the candidate proteins might be false positives, we limited Table 2 to 20 proteins showing pronounced, replicable shifts. Proteins were omitted due to either low AD/AMC ratios (1.2 for tubulin chains α‐4A and β‐4B) or lack of support from other comparisons (elongation factor Tu, ADP/ATP translocase 2, and 2‐oxoglutarate dehydrogenase E1). Proteins less abundant in AD than AMC aggregates (glutamate dehydrogenase, aspartate aminotransferase, stress‐70/mortalin, and keratins I‐13 and I‐14) were excluded by our predetermined focus on AD increases.
Proteins significantly and reproducibly enriched in tau‐IP aggregates from AD relative to AMC (Table 2, “tau‐IP 1”) include amyloid precursor protein (APP or A4), astrocytic phosphoprotein PEA‐15, formin‐like protein 20‐like, heterogeneous nuclear ribonucleoprotein [hnRNP] A2/B1, and taste receptor 2‐38 (all found only in AD); ATP synthase (α and γ subunits), calcium/calmodulin‐dependent protein kinase II‐β, ubiquitin‐40S ribosomal protein, 14‐3‐3 protein η (eta), microtubule‐associated protein 1B, vimentin, α‐internexin, glucose‐6‐phosphate isomerase, HSP90‐α and HSP90‐β, α‐crystallin B chain, vesicle‐fusing ATPase, dynamin‐3, and GFAP‐1.
We extended this comparison to a larger sample (8 AD and 7 NC) spanning a wider age range (46–92 years) and analyzed 73 candidate proteins by multivariate regression against each of the potentially relevant parameters (Table 2, multivariate regression columns). Effects of gender and race were not tested due to insufficient sample size for one or more groups; likewise, ApoE genotype (a strong risk factor for AD) was not included in regressions due to its close coupling to AD in our sample (see Table S1). Tau‐IP levels of 3 proteins showed nominally significant associations with postmortem interval (PMI), and levels of 4 proteins covaried with age (close to the frequencies expected by chance). In contrast, AD severity based on immunohistochemical features (Braak et al., 2006) was significantly linked to aggregate tau‐IP levels of 13 proteins (18%) after adjustment for age and PMI effects—substantially more frequently than expected by chance.
Positive associations with AD severity were found for α‐internexin (P < 2 × 10−6), plectin (P < 0.0007), Ca++/calmodulin‐dependent protein kinase II‐β (P < 0.003), vimentin (P < 0.014), hnRNP A2/B1 (P < 0.02), formin‐like protein 20‐like (P < 0.03), glycogen phosphorylase B (P < 0.04), sequestosome‐1 (P < 0.04), and epiplakin (P < 0.05). Negative regressions were nominally significant for stress‐70/mortalin (P < 0.014), syntaxin‐1B (P < 0.013), α2‐macroglobulin (P < 0.03), immunoglobulin superfamily member 8 (P < 0.03), and ketamine reductase mu‐crystallin 1 (P < 0.05).
In situ confirmation of protein–protein associations and their enrichment in AD aggregates
We used antibodies specific to several of the proteins identified in tau‐IP aggregates to assess whether colocalization by fluorescence microscopy would support or refute the associations implied by our proteomic data. Representative immunostained images are shown in Fig. 2 (upper panels) wherein light green and yellow (e.g., arrows) imply coincidence between phospho‐tau (P‐tau) imaged as green, and dynactin shown as red. P‐tau [Ser202, Thr205] was detected in most AD but none of the AMC neurons, consistent with our proteomic data (threefold more P‐tau peptides in AD, summed over all phosphorylation sites; Table 1). Dynactins were equally abundant in AD and AMC neuron bodies, again in agreement with proteomic results (Table 1, ‘total’ aggregate columns). These observations imply that a substantial fraction of dynactin colocalizes with some portion of P‐tau (presumably intracellular, as in control neurons of panel D). In the absence of any P‐tau signal for AMC tissue, it is not possible to draw any conclusions regarding P‐tau interaction with dynactin in normal cells.
Figure 2.
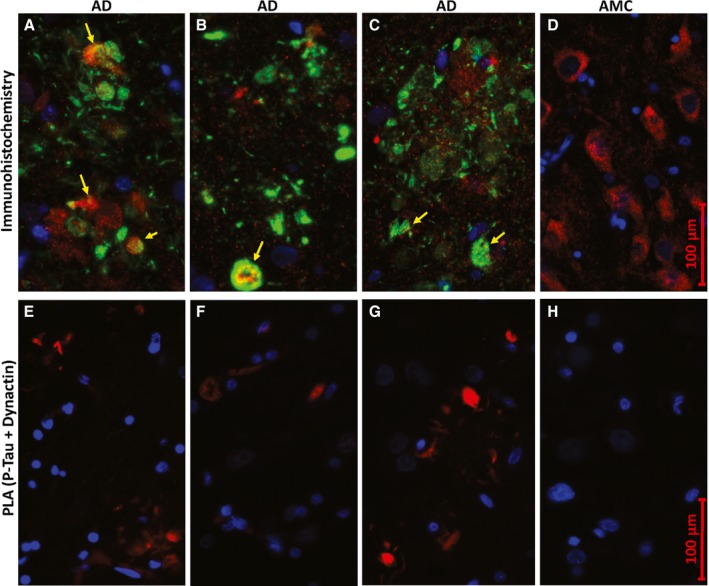
P‐tau coincides with dynactin in situ. Images show hippocampus sections from Alzheimer disease (A–C, E–G) or age‐matched controls (D, H). (A–D) Conventional immunohistochemistry, visualizing proteins with fluor‐tagged secondary antibodies, after incubation with antibodies to P‐tau (Ser202, Thr205; green) and dynactin/p50 (red); nuclei (blue) were stained with DAPI. Light green and yellow (arrows) indicate superposition of red and green fluors. (E–H) Proximity ligation amplification (PLA); red product indicates close proximity (<40 nm separation) of P‐tau (Ser202, Thr205) to dynactin/p50.
Proximity ligation amplification (DuoLink PLA, Fig. 2 lower panels) provides a much more stringent test of co‐localization, as rolling‐circle amplification primers coupled to two distinct secondary antibodies can greatly amplify their product (detected as red fluorescence) only if they are situated within 40 nm of each other. PLA results place tighter limits on the coincidence of P‐tau and dynactin signals in AD, noted above, and corroborate the data of Table 1 indicating enrichment of dynactins in tau‐IP aggregates prepared from AD but not AMC hippocampus.
A total of 5 protein–protein interactions implicated by proteomic tau‐IP results were tested and confirmed by PLA, in some cases coupled with conventional immunofluorescent counterstaining (Figs 2, 3, 4, 5). Consistent and fairly uniform co‐localization of total tau with 14‐3‐3 proteins was observed in AD (Fig. 3A–C) but not AMC hippocampus (Fig. 3D), whereas tau clusters differed widely in the extent of labeling with internexin‐tau PLA product, also seen only in AD (Fig. 3E–H).
Figure 3.
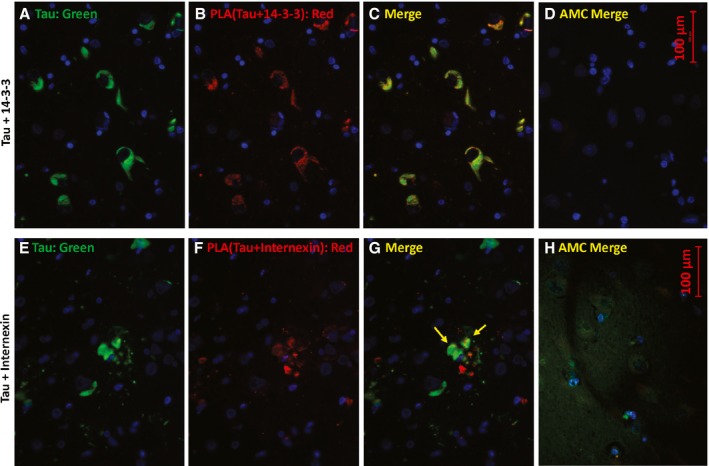
Total tau colocalizes with 14‐3‐3 and a subset of internexin in AD hippocampus. Sections were immunostained for total tau (A, E; green), or by proximity ligation amplification (PLA) for total tau in conjunction with 14‐3‐3 proteins (B; red) or with internexin (F; red). C and G are merged images of A+B and E+F, respectively. D and H are the corresponding merged images for AMC hippocampus. Note that in H, the tau (green) exposure was increased ~10‐fold to illustrate the diffuse distribution of normally phosphorylated tau in control tissue.
Figure 4.
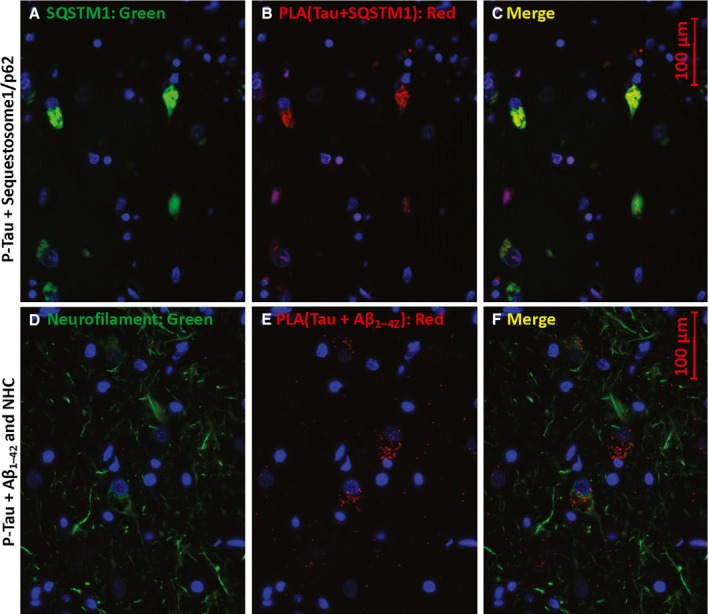
P‐tau proximity to sequestosome‐1 and Aβ1–42/APP. Sections were immunostained for sequestosome‐1 (SQSTM1, p62; A) or neurofilament heavy chain (NHC, D) and for PLA product (red) showing P‐tau (Thr205) proximal to SQSTM1 (B) or to Aβ1–42 (E). C and D are merged images of A+B and D+E, respectively.
Figure 5.
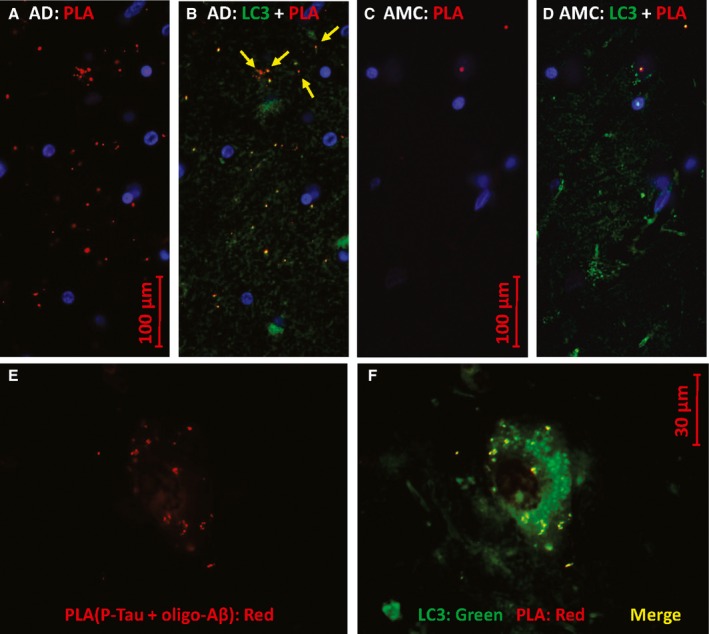
P‐tau co‐localizes with Aβ1–42 oligomers. Antibodies to P‐tau [Thr205] and oligomeric Aβ1–42 (A–D) produce red PLA product if they are within 40 nm. Counterstaining with antibody to LC3B/ATG8, a marker of autophagosomes (B, D, F), appears as green. In E and F, PLA product (red) indicates P‐tau contiguous to Aβ1–42 oligomers. AD hippocampus sections are shown in A, B, E, and F, while C and D show AMC hippocampus.
Similarly, P‐tau colocalizes with sequestosome‐1/p62 in AD hippocampus (Fig. 4A–C) and with Aβ1–42 which appears to be intracellular and located in neurites (Fig. 4D–F) of cells also immunostained for neurofilament heavy chain (NHC, green). The green counterstain in Fig. 5 was antibody to LC3B/ATG8, a microtubule‐associated light‐chain protein with an ubiquitin‐like domain, widely used to mark autophagosomes. Abundant PLA signal, indicating P‐tau contiguous to Aβ1–42, was seen in LC3‐positive puncta of AD hippocampus (panels A, B, E, F) but only rarely in AMC (panels C, D). The antibody for these PLA reactions is specific to Aβ1–42 residues 1–4 and does not bind to APP (Youmans et al., 2012); it preferentially labels neurotoxic oligomers of Aβ1–42 (Tai et al., 2013). These data imply that P‐tau complexes with Aβ1–42 form inside AD neurons and are sequestered in a subset of autophagosomes. Apparent exceptions (e.g., red features at the top of Fig. 5B) all contain yellow subregions indicating co‐localization.
Aggregate proteins enriched in AD play functional roles in aggregation and cytotoxicity
We recently reported that several aggregated proteins identified in a C. elegans model of Huntington disease (strain AM141, expressing polyglutamine [Q40] fused in‐frame to YFP, yellow fluorescent protein) contribute mechanistically to accumulation of aggregates by obstructing their proteolytic clearance.
In a similar fashion, we here used C. elegans amyloidopathy models to test the effects of RNA‐interference (RNAi) knockdowns targeting 21 candidate genes that encode proteins orthologous to constituents substantially more abundant in insoluble aggregates from AD than AMC hippocampus, when isolated by affinity to tau‐ or Aβ‐specific antibodies (Tables 1 and 2). These orthologs are shown in bold, blue font, in the first column of Table 1. Knockdown results for muscle or neuronal expression of human Aβ1‐42 are presented in Fig. 6B, paralleling a diagrammatic representation of aggregate‐specific shifts in protein abundance (Fig. 6A). A strain expressing the human Aβ1‐42 transgene in muscle showed extensive paralysis, retaining only ~40% motility at 1.5 d after induction. Paralysis was attenuated by 15 of the knockdowns (71%) with at least nominal significance (vs. 1 expected by chance at P < 0.05; see blue bars, Fig. 6B). The extent of rescue ranged from 28% to 71% (mean ± SD: 53 ± 14%). A second strain, with pan‐neuronal expression of Aβ1‐42, lost ~90% of chemotaxis to n‐butanol by 2–3 days postinduction. Knockdown of 12 genes (57%) significantly protected from loss of chemotaxis (at P < 0.05; for 6 of those genes, P < 0.005), indicated by red bars in Fig. 6B. The extent of rescue was 30–57% (mean ± SD: 42 ± 9%). In each case, to be as conservative as possible, significance is based on t‐test comparisons of biological replicates only (i.e., N = the number of repeats). Seven of the 21 tested genes produced significant results in both assays. Although most genes that produced a significant impact did so in only one assay, this is not surprising as AD‐aggregate proteins (and their C. elegans orthologs) are likely to be expressed at quite different levels in different cell types.
Figure 6.
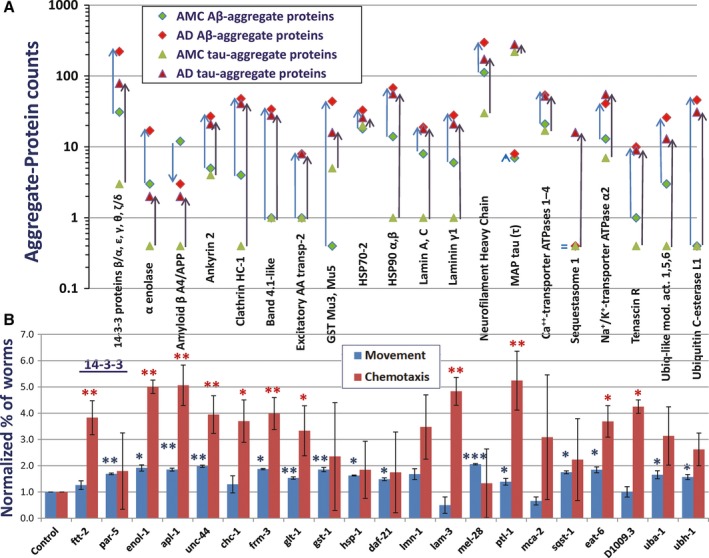
Knockdown of AD‐enriched aggregate proteins rescues aggregation‐induced traits. (A) Proteins shown are overrepresented (in LC‐MS spectral counts) in aggregates isolated from AD relative to NC, except microtubule‐associated protein tau, which is hyperphosphorylated but not more abundant in AD (Table 1). Blue arrows connecting diamond symbols show increased abundance of proteins in tau‐IP aggregates; black arrows connecting triangles show increased abundance in Aβ‐IP aggregates. (B) Of 21 protein types more abundant in AD than in NC aggregates, knockdown of 14 (67%) of their nematode orthologs significantly rescued paralysis of a C. elegans strain expressing human Aβ1‐42 in muscle (blue bars), or rescued chemotaxis disrupted by neuronal expression of Aβ1‐42 (red bars). Significance was assessed by 1‐tailed t‐tests (as lower cytotoxicity was predicted) comparing fractions of unparalyzed or chemo‐attracted worms in three independent assays per group (*P < 0.05; **P < 0.005; ***P < 0.0005). Significance by chi‐squared tests within assays was P < 5 × 10−5 to 10−16.
Discussion
Distinct aggregates isolated by Aβ or tau‐IP are both highly enriched for AD‐specific proteins
Aggregates isolated by Aβ IP (thought to reflect initiation or ‘seeding’ by Aβ oligomers) differ markedly with respect to several protein constituents from parallel preparations isolated by tau‐IP (Table 1). This confirms that amyloid plaque aggregates were successfully resolved from neurofibrillary tangles. Considering only aggregates from control hippocampus (yellow highlighting in Table 1), 14‐3‐3 proteins, heparin sulfate, HSP90, lamin A/C, and laminins were detected >10 times as often in Aβ‐IP (75 hits) as in tau‐IP aggregates (7 hits). Conversely, ApoD and clusterin taken together were 45 times less abundant (3 vs. 135 hits) when associated with Aβ than when associated with tau. The same points apply to aggregates from AD hippocampus (blue highlighting), but different sets of proteins were most differential between aggregate types: α‐enolase and talins 1 and 2 are >10 times as abundant (56 vs. 5 hits) when associated with Aβ as when linked to tau, while sequestosome‐1 is only found in tau aggregates, and polyubiquitin (UBB) is 3.3 times as abundant there.
The ability to distinguish immunologically between aggregates cannot be taken for granted, given that Aβ aggregates contain some tau, and tau‐affinity aggregates (at least from AD tissue) contain a little Aβ [or APP, which may also be recognized by most antibodies to Aβ (Youmans et al., 2012)]. Nevertheless, the amount of ‘crossover’ (based on data in Table 1) is relatively small: Tau was identified in Aβ aggregates at only 3% of the level in tau‐affinity aggregates (15 vs. 499 hits), whereas Aβ was also quite scarce in tau tangles compared to Aβ aggregates (2 vs. 15 hits).
It is noteworthy that 90% (90/100) of the proteins comprising these aggregates were enriched (or in a few cases depleted) in AD relative to NC, concordantly for both tau‐IP and Aβ‐IP aggregates. Most shifts (74%) were significant in both, even after adjusting for total protein (higher in AD aggregates). Thus, the ‘aggregate proteome’ is remarkably distinctive for AD and could be useful diagnostically, in addition to shedding light on mechanisms of protein accrual.
Total aggregates increase with age in normal C. elegans (David et al., 2010; Ayyadevara et al., 2014) and in several tissues of normal mice (Ayyadevara et al., 2016 and unpublished data), indicating that protein aggregation is a quite broadly conserved feature of aging. In view of our evidence for common processes underlying the growth of both Aβ and tau aggregates, as discussed above, it is surprising that ‘total aggregates’ (sarcosyl‐insoluble material without IP) differed far less between AD and NC than did either type of neuropathological aggregate. That is, although total aggregates did differ in composition between these two sources (115 proteins differed with chi‐squared P < 0.05 vs. 40 expected by chance), the ratios were far less extreme than those in Aβ and tau aggregates (Table 1). This implies that aggregates are heterogeneous and that those containing either tau or Aβ are more specific to AD than aggregates overall—many of which are likely to reflect age‐dependent proteostasis failure unrelated to the specific pathophysiology of AD (Morimoto & Cuervo, 2014).
Aggregate proteomics are more informative for AD than total proteomics
AD‐specific proteins comprised a much larger fraction of total proteins in immuno‐isolated aggregates (our data) than in total protein prepared from affected frontal cortex without isolation of aggregates (Emory ADRC Brain Bank, https://www.synapse.org/#!Synapse:syn2580853/wiki/). In a proteomic comparison that identified 8027 proteins from a total of 16 pools representing affected tissue from 7 neurological diseases (plus controls), the mean ± SD number of proteins identified per pool was 3508 ± 163. Of those proteins, only 28 (0.8%) and 34 (1.0%) differed with nominal significance (P ≤ 0.05) between AD and ‘AD/PD’ (PD with some AD traits), respectively, and the common control pools. These results imply that aggregate proteomes are much less complex (comprise fewer proteins) than total tissue proteomes and yet have a far higher fraction of identified proteins that differ significantly between AD and controls. Moreover, although >85% of the proteins identified in aggregates were also identified in the Emory protein set, very few proteins appeared significant in both assays. Of 136 proteins in Tables 1 and 2, only four differed significantly between AD and NC total protein pools in the Emory study: APP, tau, NADH dehydrogenase (NDUFA4), and secernin‐1 (SCRN1). Conversely, proteins that differed significantly between AD/PD and NC in the Emory study included only 6 proteins that also appeared significant in our comparisons: tau, 14‐3‐3γ, HSPβ‐1, peroxiredoxin‐2, required for meiotic nuclear division protein 1 (RMND1), and pyridoxal phosphate phosphatase (PDXP). Even considering all seven neuropathology pools, only 17 proteins were significant in both the Emory study and our own. In contrast, our tables include 15 of the 26 proteins (58%) identified as potentially differential in amyloid plaque isolated by laser‐capture microdissection from two AD cases (Liao et al., 2004). The above results are consistent with our hypothesis that neurotoxic aggregates have a protein composition quite distinct from the affected tissues in which they reside.
Post‐translational modifications contribute to AD‐specific aggregates
Table 1 summarizes proteomic data for oxidations (summing over mono‐, di‐, and trioxidation), phosphorylations and acetylations. These PTMs are elevated 2.8‐fold to 3.5‐fold in AD relative to NC (1.7‐ to 2.1‐fold even after adjustment for increased aggregation in AD), only in Aβ‐ or tau‐IP aggregates but not in total aggregates. These changes add charged, predominantly nucleophilic sites that may block normal protein interactions, and could destabilize hydrophobic regions otherwise buried in the protein interior. Subsequent misfolding exposes hydrophobic residues that can then coalesce with hydrophobic ‘extrusions’ from other proteins. Some PTMs, in particular oxidations, may arise from elevated ROS levels typical of AD neuroinflammation (Liu et al., 2011; Heneka et al., 2015). It is revealing that AD does not affect total aggregates in the same way, implying that the underlying molecular dysfunction does not generalize to types of aggregates that accompany normal aging. Moreover, total aggregates provide a sort of negative control which serves to exclude most sources of artefactual modification, such as oxidations occurring in lysates during aggregate isolation. While such PTMs might occur or alter after lysis, additional ad hoc assumptions are needed to explain how they could distinguish between aggregate types.
In addition to these PTMs, specifically sought during peptide identification under Mascot, we also detected higher levels of polyubiquitin (UBB)—presumably reflecting post‐translational tagging of proteins—in aggregates from AD than from NC. UBB was twice as abundant in AD‐derived aggregates isolated by Aβ‐IP and 3.7× more abundant in AD aggregates isolate by tau‐IP (Table 1). Other approaches imply that ubiquitinated proteins accrue in AD due to compromised proteasomal activity (Cecarini et al., 2007); higher prevalence of this deficit in neurons than astrocytes (Orre et al., 2013) may contribute to the abundance of neuronal proteins in our results.
Analyses of individual tau‐IP samples confirm AD‐specific protein enrichment in aggregates
We analyzed tau‐IP aggregates from individual samples of caudal hippocampus in order to estimate the intra‐ and intergroup variance and to evaluate the dependence of aggregate protein levels on age, the extent of AD histopathology, and PMI. We found by multivariate regression that age and PMI affected about as many proteins as would be expected by chance (6 and 8%, respectively, of the total tested), whereas the level of AD pathology was significantly associated with the abundance of 13 proteins (26%) in tau‐IP aggregates. Five of these proteins had also differed significantly between AD and AMC in our initial intergroup comparison, but three others had attained significance only in the second group comparison and/or the pooled‐sample analysis. Half of the proteins (13/26) that were significant in either comparison of individual samples, had also reached significance in the pools, but the remainder were insufficiently abundant. The sensitivity of proteomic detection is inevitably limiting, so that any given comparison will yield many false negatives—proteins that are either not detected or are observed at levels too low to meet statistical thresholds.
In situ data support proteomics results
We used standard immunohistochemical procedures and also PLA, a newer method to sensitively and precisely detect sites of close protein:protein contiguity, to provide independent evidence bearing on several key proteomic observations. Multifluor images suggest heterogeneous aggregate structures specific to AD (Fig. 2A–C), while also supporting co‐localization of P‐tau and dynactin. Proximity ligation amplification (Fig. 2E–H) quite specifically indicated structures in AD hippocampus wherein tau and dynactin lie within 40 nm of each other and thus are likely to interact directly. Similarly, tau or P‐tau was shown to coexist in close proximity to 14‐3‐3 proteins, α‐internexin, sequestosome‐1/p62, and Aβ1–42 (including its neurotoxic oligomers (Tai et al., 2013)) (Figs 3, 4, 5).
Although amyloid beta peptides Aβ1–42 and Aβ1–40 are best known as components of extracellular plaque, they are also found within neurons (Youmans et al., 2012) where neurofibrillary tangles reside. We consistently identified Aβ1–42 or its precursor APP in tau‐IP fractions (Table 2, line 1). In all sections of AD hippocampus examined (but never in AMC), we observed abundant PLA signal for tau in close proximity to Aβ1–42 oligomers (Figs 4 and 5) in puncta that counter‐stained for the autophagosome marker LC3B/ATG8 (Fig. 5), consistent with their co‐aggregation in AD neurons. Direct interaction between tau and Aβ in aggregates would have important implications for AD etiology, perhaps rendering moot any debate as to which of these molecules is the primary driver of AD pathology.
The 14‐3‐3 proteins recruit misfolded proteins and associated chaperones to dynein motors for transport to aggresomes (Jia et al., 2014). Zeta and eta isoforms of 14‐3‐3 were detected in AD/tau and PD aggregates, respectively (Qureshi et al., 2013; Plotegher et al., 2014). All five 14‐3‐3 isoforms detected here were enriched in AD aggregates, sevenfold after Aβ1–42 IP and 26‐fold after tau‐IP (Tables 1 and 2), while dynein H and L chains were exclusively associated with AD aggregates of both kinds (Table 1). The prominence of aggregate proteins involved in retrograde axonal transport agrees well with the prevalence of retromer‐associated genes in AD genomewide association studies (Vardarajan et al., 2012). Dynactins, which are also subunits of the dynein motor complex, are less abundant but also specific to both AD IP fractions (Table 1). α‐Internexin, a neural intermediate filament protein implicated in both AD (Dickson et al., 2005) and ALS (Page et al., 2011), was consistently enriched in AD tau‐IP aggregates (Table 2). Sequestosome‐1, which helps to recruit misfolded proteins and their aggregates to either proteasomes or autophagosomes (Wooten et al., 2006), was highly enriched in tau‐IP aggregates exclusively when derived from AD (Tables 1 and 2). In that context, it is not surprising that tau/Aβ1–4 PLA signal also colocalizes with punctate immunostain for LC3B/ATG8, a marker of autophagosome membranes (Fig. 5). While these in situ data clearly represent just a small sampling of the protein interactions predicted by co‐isolation of the above proteins in total tau‐IP experiments (Tables 1 and 2), it is encouraging that the results of these quite distinct procedures are consistent with one another and extend prior inference of autophagy impairment in AD as well as other neurodegenerative diseases (Cardenas et al., 2012).
Functional assays imply a nonrandom aggregation process
RNAi knockdowns are functional assays that seek morphologic or behavioral changes arising from disruption of specific proteins and pathways. Knockdown data imply that aggregate components are mechanistically involved in the accrual process, at least in nematodes and very likely also in humans, given the high conservation of pathways across vast evolutionary distances. Thus, they may furnish valuable therapeutic targets for AD and other progressive diseases in which aggregation plays a causal role.
Our data also imply that aggregate growth is not an entirely random process, but rather involves something akin to a pathway: protein accrual occurring in an ordered sequence, either obligatory or preferred. This follows from the remarkable observation that aggregation can be greatly attenuated by any of several interventions. Some RNA interference targets reduce aggregates to the extent that they contribute to them, consistent with roles as structural components of aggregates, whereas others exert effects so potent as to suggest that their disruption must block key steps in a sequential accrual process (Ayyadevara et al., 2014). If these latter proteins accrued in a stochastic manner, it should not be possible for combined contributions to exceed 100%. However, relatively large effects of RNAi were observed in C. elegans aggregation models after RNAi targeted nematode orthologs of 14‐3‐3 proteins, α‐enolase, amyloid precursor‐like protein, ankyrin‐2, clathrin heavy chain, band 4.1‐like protein, excitatory amino acid transporter‐2, GST‐mu, HSPs 70 and 90, lamin, laminins, tau, neurofilament heavy chains, sequestosome‐1, Na+/K+‐transporter ATPase α2, tenascin‐R, ubiquitin‐like modifier activators, and ubiquitin C‐terminal esterase L1 (Fig. 6B). These RNAi treatments were tested for efficacy in two nematode models. In the first model, 15 of the 21 RNAi treatments significantly restored the loss of motility due to Aβ expression in body‐wall muscle. The mean ± SD rescue was 53 ± 14% of the untreated deficit (controls fed RNAi vector only), and the sum of rescue effects was 8 times the maximal impairment! In the second model, 12 RNAi treatments significantly restored the loss of normal chemotaxis that follows pan‐neuronal Aβ expression. In this case, rescue averaged 42 ± 9% of the untreated deficit, for a sum of rescue effects exceeding 5 times the maximal deficiency. These data are inconsistent with models in which aggregate constituents coalesce in a random manner, but instead imply a preferred sequence, that is, an ordered series of accretion steps.
Roles of aggregate proteins enriched or depleted in AD
AD‐enriched aggregates include proteins associated with heat‐shock and unfolded protein responses, inflammation, and/or innate immunity—categories previously shown to be associated with aging (Finch et al., 2010; van Oosten‐Hawle & Morimoto, 2014). Examples include ApoE, complement C3, chaperones (e.g., HSP70.2), plectin, ATP‐dependent RNA helicases, BM‐specific heparin sulfate, filamins A and C, lamin A/C, laminin, talin, tenascins, and vinculin.
Many of the proteins that were here characterized as differentially enriched or depleted in aggregates from AD, relative to NC tissue, have been previously implicated in neurodegenerative diseases and/or protein aggregation. For example, ubiquitin carboxy‐terminal hydrolase L1 (which hydrolyzes ubiquitin added to C‐termini of proteins) is mutated in strict association with PD in one pedigree, and a common polymorphism is associated with PD risk (Leroy et al., 1998). Moreover, it has also been implicated in Aβ neurotoxicity in AD (Gong et al., 2006). Carbonyl reductase is elevated in hippocampi of 3xTg‐AD mice, which have markedly elevated levels of protein carbonylation (Shen et al., 2015). Clusterin (ApoJ) polymorphism is associated with AD (Lambert et al., 2009), and its circulating levels predict AD progression (Thambisetty et al., 2010). Other apolipoproteins are also strongly linked to AD risk: ApoD catalyzes reduction of peroxidized lipids and its levels increase with age and AD, while ApoE has allele‐specific effects on Aβ levels and AD risk (Tai et al., 2013; Dassati et al., 2014).
We recently showed that neuronal levels of the motor protein dynactin are affected by age, AD, and ApoE genotype (Aboud et al., 2015). Dynactin activates dynein chains to couple organelles to microtubules for fast axonal retrograde transport (Waterman‐Storer et al., 1997). Suppression of dynein impairs clearance of α‐synuclein aggregates via the autophagosome/lysosome pathway (Li et al., 2013). Excitatory amino acid transporter 2 deficiency has been implicated in AD, HD, and ALS‐PD complex (Yi & Hazell, 2006). Filamin C mutations are associated with CVD and with protein aggregation in cardiac cells (Kley et al., 2013). Astrocytic GFAP expression is associated with aging and AD (Middeldorp & Hol, 2011); dominant GFAP mutations in Alexander disease cause protein aggregates that contain GFAP, vimentin, plectin, ubiquitin, and small chaperones such as α‐crystallin (Hagemann et al., 2009). The 14‐3‐3 proteins, expressed ubiquitously but most abundant in brain, bind as dimers to >200 known target proteins. They were first identified in Lewy bodies of PD, where they are thought to bind α‐synuclein (Plotegher et al., 2014). However, they also bind tau in AD neurofibrillary tangles (Qureshi et al., 2013; Jia et al., 2014), and mutant huntingtin (86Q) in an HD model (Jia et al., 2014), and they have been implicated in aggresome formation (Jia et al., 2014). Mu‐class GST variants are risk factors for PD (Wahner et al., 2007) as well as AD (de Mendonca et al., 2014). Heat‐shock protein defects have been implicated in PD (Yang et al., 2015) and AD (Ou et al., 2014), and HspB1 mutations are associated with familial motor neuron diseases (Muranova et al., 2015). Hsp90 and its co‐chaperones regulate tau and Aβ processing (Blair et al., 2014), and Hsp90 may specifically protect TDP‐43 from ROS‐induced aggregation (Chang et al., 2013a). The mitochondrial HSP70, also called ‘stress‐70’ or ‘mortalin’, is implicated in PD and in vitro longevity (Wadhwa et al., 2015). Lamin A mutants induce nuclear protein aggregation (Hubner et al., 2006). Laminins complex with Aβ, preventing its fibril formation (Henriques et al., 2014). Band 4.1‐like protein 1 binds and stabilizes dopamine receptors (Binda et al., 2002).
Tau is hyperphosphorylated in AD (6–12 sites/molecule, vs. 2–4 normally) and consequently forms oligomers with normal tau, which cohere to MAPs 1A, 1B, and 2 (Iqbal et al., 2013). Neurofilament light chain is a CSF biomarker of late‐onset AD (Skillback et al., 2014) and PD (Herbert et al., 2015). Peptidyl‐prolyl cis/trans isomerase A regulates protein folding at proline residues and affects tau aggregation (Blair et al., 2015). Peroxiredoxins 1 and 2 protect against huntingtin neurotoxicity (Pitts et al., 2012) and dopaminergic neurodegeneration (Hu et al., 2011). Phosphoglycerate mutase activity in brain decreases with aging and especially with AD (Meier‐Ruge et al., 1984), perhaps explained by its AD‐specific accumulation in aggregates. Secernin‐1 was identified as an early marker of neurodegeneration in a transgenic mouse overexpressing human tau (Chang et al., 2013b). Natural variation in sequestosome‐1, which escorts aggregates to autophagosomes, is associated with early‐onset AD (Cuyvers et al., 2015). Spectrin has been utilized as a model of spontaneous amyloid formation (Castello et al., 2015), and the spectrin β4 locus is one of three genes strongly hypermethylated in AD (Sanchez‐Mut et al., 2013). Phosphorylation of synapsin‐1 triggers neurotransmitter release but is blocked by binding huntingtin protein in HD (Xu et al., 2013). Syntaxin‐1 is a synaptic SNARE protein that forms age‐ and PD‐dependent co‐aggregates with α‐synuclein (Garcia‐Reitbock et al., 2010). Tenascin‐R co‐aggregates with APP at nodes of Ranvier on myelinated axons (Xu et al., 2014). Knockdown of UBA1 or talin‐2 increased aggregation of a polyglutamine array in C. elegans and of transgenic huntingtin (Htt‐Q74‐GFP) in cultured human HEK293 cells (Teuling et al., 2011). Frame‐shifted misreading of ubiquitin‐B inhibits proteasomes in transgenic mice and phenotypically mimics AD (Irmler et al., 2012). Vinculin is one of three extracellular matrix protein hubs in a protein‐interaction network of ‘age‐related disease proteins’ (Wolfson et al., 2009). Ubiquitin hybrid (fusion) genes, Uba52 and Uba80, encode ubiquitin fused to ribosomal proteins and are upregulated during tumor‐cell apoptosis (Han et al., 2012).
Surprisingly, pyruvate kinase suppresses Aβ fibril formation in vitro, as effectively as any known chaperone (Luo et al., 2014). Mitochondrial aconitase activity is reduced almost twofold in lymphocytes from AD relative to normal controls (Mangialasche et al., 2015). ATP synthase α is found on the cell surface of neurons, where it binds oligomeric Aβ to initiate neurotoxicity (Xing et al., 2013). Adenylate kinase overexpression is a common feature of AD, PD, ALS, and epilepsy (Boison & Aronica, 2015). Phosphatidylethanolamine‐binding protein is ~20% more abundant in hippocampus of AD than NC, where it is thought to inhibit proteasomes (Chen et al., 2006). Microglial γ‐enolase is neuroprotective in a mouse model of AD (Hafner et al., 2013). Astrocyte protein PEA‐15 protects neuronal cells in vitro against MPTP‐induced dopaminergic cell death (a model of PD) and raises dopamine levels in mouse striatum in vivo (Ahn et al., 2014). Aspartate aminotransferase and aldolase are major targets for deglycation repair by DJ‐1, which is highly induced in many cases of PD (Richarme et al., 2015). Neurofilament medium polypeptide is one of three plasma proteins strongly associated with ALS (Haggmark et al., 2014). Cytochrome c oxidase mutations and polymorphisms are associated with multiple neurodegenerative diseases, including AD (Loera‐Castaneda et al., 2014).
Conclusions
Aggregates are characteristic of most or all neurodegenerative diseases and confirmed diagnosis typically depends on immunohistochemical detection of just one or a few of the best known proteins in foci typical of each pathology. However, it has become increasingly clear that there are many more proteins coalesced in aggregates, than have been identified (let alone pursued) to date. Moreover, the specific catalogue of aggregated proteins in each disease is far from random, and based on evidence presented here, probably reflects an ordered process of protein accrual. Similar aggregates form in nematode models of human neurodegenerative diseases, expressing a human transgene to produce a protein thought to initiate or ‘seed’ aggregation. These models can provide insights into the underlying processes that predispose to, and contribute to, aggregation. Further, they provide a facile platform on which to test the functional roles of each protein component.
In the current study, we have learned from this approach that Alzheimer disease aggregates, purified by affinity to antibodies against total tau or Aβ1–42/APP, are distinguished by several proteins that are specific to one type of aggregate, but nevertheless have in common the great majority of contributing proteins. PTMs are markedly increased in tau‐ and Aβ‐IP (but not total) aggregates when they were derived from AD, consistent with greater recruitment of modified proteins into AD‐specific aggregates, although not into other aggregates. Through a better understanding of the sequences of events that are shared by diverse aggregates, we may learn features that will constitute generic targets for small‐molecule interventions. At the same time, disease‐specific features may unveil nuances by which different seed proteins provoke aggregation and neurotoxicity in affected areas.
A remarkable conclusion from the current data is that aggregates, especially those specific to AD brain, contain many proteins that are far more abundant in AD than in control aggregates—the majority of which play functional roles in the accrual process, and/or were previously implicated in neurodegenerative diseases and other pathologies. This is such a frequent outcome that it is difficult to explain how so many candidate proteins could contribute mechanistically to a single disease. Many of these proteins were also reported to play roles in aging, but one feature they all share is the propensity for aggregation. We therefore propose that aggregation in general, and disease‐specific aggregates in particular, provide insights into proteostasis failure and its consequences, which in turn underlies age‐ and disease‐specific pathologies.
Experimental procedures
Proteomics methods
Tissues from caudal hippocampus, flash frozen and stored at −80 °C, were pulverized in a mortar and pestle cooled on dry ice as described (Ayyadevara et al., 2014). After a brief low‐speed spin (5 min at ~2200 g), protein was assayed (Bradford; Bio‐Rad) in supernatants and equal protein contents were pooled or analyzed individually as indicated. Samples were incubated with DYNAL Protein‐G magnetic beads coated with antibody against total tau (ab80579, Abcam) or Aβ1–17 (ab11132, Abcam). Bound aggregates rinsed 3×, were eluted, and brought to 0.1 m HEPES buffer with 1% v/v sarcosyl, 5‐mm EDTA, and protease inhibitors. After centrifugation 30 min at 100 000 g, pellets were resuspended in Laemmli buffer containing 2% w/v SDS and 0.5% v/v ß‐mercaptoethanol and heated 5 min at 95 °C. Proteins from each immuno‐pulldown (IP) and from total aggregates without IP (equal fractions based on initial protein content rather than IP recovery) were resolved on 1% SDS‐acrylamide gels, which were Coomassie stained, sliced, and excised for tryptic digestion in situ (Promega, Madison WI). Eluted peptides were analyzed by LC‐MS/MS as described (Byrum et al., 2013; Ayyadevara et al., 2014).
Proteins were identified by spectral match in the UniProtKB/SwissProt database. For tau‐IP2, electrophoresis was replaced by a nanoparticle‐adsorption protocol (Luchini et al., 2008). Details are provided as Data S1.
Statistics
Significance statistics for protein raw counts (Table 1) were calculated using 2‐tailed chi‐squared tests with Yates’ correction; note that Fisher's exact test is inappropriate for 2 × 2 tables with large cell entries. For each protein, a 2 × 2 table contains AD and NC spectral counts in the top row, and the total for all proteins in the lower row, from which we assess significant enrichment in either AD or NC tissues. This is a very conservative approach, as it ‘corrects’ the biologically important observation that AD aggregates have more protein than NC aggregates. To determine the number of significant proteins expected by chance, we excluded proteins with insufficient counts in both cells to possibly attain significance. The number remaining (proteins adequately represented to achieve significance) was then multiplied by the alpha value, 0.05 (the threshold for nominal significance, without correction for multiple endpoints) to calculate the number expected by chance. In Table 1, the minimal count numbers required to achieve chi‐squared significance were 4, 10, 4, 10, 6, and 7, for spectral‐count columns numbered 1–6, respectively.
Significance statistics for Table 2 were calculated by unpaired, 2‐tailed t‐tests. As above, to calculate the expected numbers of nominally significant proteins (at P < 0.05), an adjustment was made for proteins that were identified in too few samples to possibly attain significance. The cutoff was arbitrarily set at a minimum of 3 detections in either the AD or NC group, for each protein. That is, a protein was considered capable of achieving significance if it was identified in 3 or more individual samples in either the AD or NC group.
Immunohistochemistry
Formalin‐fixed paraffin‐embedded hippocampal sections of human brain were cut in seven‐micrometer sections. The sections were heated in a 60 °C incubator for one hour, and then, paraffin was dissolved in xylene. Tissue was rehydrated through graded alcohol to water. Slides were immersed in citrate buffer (for antigen retrieval) as it cooled from boiling, over a span of 30 min. After blocking (60 min.) in serum‐free protein block (Dako, Carpinteria CA), slides were incubated overnight at 4 °C in a humidified container with one or more of the following antibodies: Dynactin/p50, 1:500 (Novus, Littleton CO; NBP2‐16115); AT8 (tau‐pSer202, pThr205), 1:100 (Thermo, Waltham MA; MN1020); LC3B, 1:50 (Abcam Cambridge, U.K.; ab65054); pan‐14‐3‐3, 1:50 (Santa Cruz Dallas TX; sc629); SQSTM1, 1:50 (Santa Cruz sc10117); tau‐pThr205, 1:50 (Santa Cruz sc101817); tau5, 1:50 (Abcam ab80579); Neurofilament, 1:1000 (Millipore, Billerica MA; AB5539); Alpha‐Internexin, 1:250 (Abcam ab40758), and MOAB‐2, 1:200 (kind gift from Leon Tai, Univ. of Illinois, Chicago).
Slides were incubated 1 h in the appropriate secondary antibodies (Invitrogen, Carlsbad CA): AF488 (A21202), AF594 (A21207), and AF647 (A21447 and A31573), followed by 2 min in 0.1% Sudan Black B in 70% ethanol to quench autofluorescence. Finally, DAPI (Thermo 62247), diluted 1:1000, was applied for 5 min to visualize nuclei. Slides were mounted under Prolong Gold medium, and the edges sealed with clear nail polish.
Proximity ligation amplification
Sections used for Duolink PLA (Sigma‐Aldrich, St. Louis MO) were treated as IHC sections (see above) until 4 °C incubation with primary antibodies. After this step, slides were treated according to the manufacturer's protocol. In brief, slides were washed in successive PLA wash buffers, and incubated 1 h with oligonucleotide‐conjugated secondary antibodies, washed, and then treated with ligase, 30 min at 37 °C. Slides were again washed as above, covered in amplification solution containing polymerase and fluorescent probes, and incubated at 37 °C for 100 min. Slides were washed, blocked with 0.1% Sudan Black B in 70% ethanol to reduce autofluorescence, and mounted in DuoLink Mounting Medium with DAPI.
Immunofluorescence images were captured at identical exposure settings and with identical fluor‐specific filters, using a Nikon Eclipse E600 microscope (Melville, NY, USA) equipped with a Coolsnap ES monochrome camera (Photometrics, Tucson, AZ, USA). Images were taken through either 20× or 40× objective lenses, and reproduced at 120×, with the exception of Fig. 5E–F (100× objective, reproduced at 400×). Tissue samples for proteomic analyses were flash frozen in liquid nitrogen after dissection, placed on dry ice and stored at −80 °C. Formalin‐fixed paraffin‐embedded (FFPE) samples for immunohistochemistry were prepared in a standard histological processor.
Funding
This work was supported by grants to RJSR (VA Merit, VA Senior Research Career Scientist Award), SA (subaward of NIH/NIA grant P30 AG028718 [J. Wei, P.I.]), SWB (NIH R03 AG043784 and P01 AG012411; Sturgis Charitable & Educational Trust); and WSTG (NIH P01AG012411; Windgate Charitable Foundation).
Conflict of interests
The authors have no conflict of interests.
Author contributions
SA and RJSR planned the proteomic experiments and functional assays in C. elegans; SA, RA, and MB performed all nematode studies and aggregate preparations; SWB and WSTG provided AD and NC hippocampal samples and thin sections, along with metadata; AJT, EP, and WZ performed proteomic analyses; PAP performed all immunohistochemistry and PLA assays; and RJSR wrote the paper with input from all authors.
Supporting information
Figure S1. Enrichment of proteins and post‐translational modifications in pooled hippocampal tissue from AD relative to normal controls.
Figure S2. Hippocampal tau‐IP aggregate proteins, AD vs. AMC.
Table S1. Subject Data.
Data S1. Full proteomics data.
Acknowledgments
We thank Richard Jones for assistance with tissue sample selection and metadata retrieval, Leon Tai (Univ. of Illinois, Chicago) for kindly providing MOAB‐2 antibody, and the Univ. of Arkansas for Medical Sciences (UAMS) Proteomics Core for protein analyses.
Contributor Information
Srinivas Ayyadevara, Email: AyyadevaraSrinivas@uams.edu.
Robert J. Shmookler Reis, Email: rjsr@uams.edu
References
- Abdel‐Salam OM (2014) The paths to neurodegeneration in genetic Parkinson's disease. CNS Neurol. Disord. Drug Targets 13, 1485–1512. [DOI] [PubMed] [Google Scholar]
- Aboud O, Parcon PA, DeWall KM, Liu L, Mrak RE, Griffin WS (2015) Aging, Alzheimer's, and APOE genotype influence the expression and neuronal distribution patterns of microtubule motor protein dynactin‐P50. Front Cell Neurosci. 9, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn EH, Kim DW, Shin MJ, Kim HR, Kim SM, Woo SJ, Eom SA, Jo HS, Kim DS, Cho SW, Park J, Eum WS, Choi SY (2014) PEP‐1‐PEA‐15 protects against toxin‐induced neuronal damage in a mouse model of Parkinson's disease. Biochim. Biophys. Acta 1840, 1686–1700. [DOI] [PubMed] [Google Scholar]
- Ayyadevara S, Balasubramaniam M, Gao Y, Yu LR, Alla R, Shmookler Reis RJ (2014) Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome‐blocking complexes. Aging Cell 14, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyadevara S, Mercanti F, Wang XW, Mackintosh SG, Tackett AJ, Prayaga VS, Romeo F, Shmookler Reis RJ, Mehta JL (2016) Age‐ and hypertension‐associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 67, 1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ (2015) Linking RNA dysfunction and neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 12, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda AV, Kabbani N, Lin R, Levenson R (2002) D2 and D3 dopamine receptor cell surface localization mediated by interaction with protein 4.1N. Mol. Pharmacol. 62, 507–513. [DOI] [PubMed] [Google Scholar]
- Blair LJ, Sabbagh JJ, Dickey CA (2014) Targeting Hsp90 and its co‐chaperones to treat Alzheimer's disease. Expert Opin. Ther. Targets 18, 1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair LJ, Baker JD, Sabbagh JJ, Dickey CA (2015) The emerging role of peptidyl‐prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer's disease. J. Neurochem. 133, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Aronica E (2015) Comorbidities in Neurology: is adenosine the common link? Neuropharmacology 97, 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd‐Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM Jr, Klein JB, Ferguson J, Link CD, Butterfield DA (2006) Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1‐42): implications for Alzheimer's disease. Neurobiol. Aging 27, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del TK (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Di DF, Swomley AM, Head E, Perluigi M (2014) Redox proteomics analysis to decipher the neurobiology of Alzheimer‐like neurodegeneration: overlaps in Down's syndrome and Alzheimer's disease brain. Biochem. J. 463, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum SD, Larson SK, Avaritt NL, Moreland LE, Mackintosh SG, Cheung WL, Tackett AJ (2013) Quantitative proteomics identifies activation of hallmark pathways of cancer in patient melanoma. J. Proteomics Bioinform. 6, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas AM, Ardiles AO, Barraza N, Baez‐Matus X, Caviedes P (2012) Role of tau protein in neuronal damage in Alzheimer's disease and Down syndrome. Arch. Med. Res. 43, 645–654. [DOI] [PubMed] [Google Scholar]
- Castello F, Casares S, Ruedas‐Rama MJ, Orte A (2015) The first step of amyloidogenic aggregation. J. Phys. Chem. B 119, 8260–8267. [DOI] [PubMed] [Google Scholar]
- Cecarini V, Ding Q, Keller JN (2007) Oxidative inactivation of the proteasome in Alzheimer's disease. Free Radic. Res. 41, 673–680. [DOI] [PubMed] [Google Scholar]
- Chang HY, Hou SC, Way TD, Wong CH, Wang IF (2013a) Heat‐shock protein dysregulation is associated with functional and pathological TDP‐43 aggregation. Nat. Commun. 4, 2757. [DOI] [PubMed] [Google Scholar]
- Chang SH, Jung IS, Han GY, Kim NH, Kim HJ, Kim CW (2013b) Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer's disease. Biochem. Biophys. Res. Commun. 430, 670–675. [DOI] [PubMed] [Google Scholar]
- Chen Q, Wang S, Thompson SN, Hall ED, Guttmann RP (2006) Identification and characterization of PEBP as a calpain substrate. J. Neurochem. 99, 1133–1141. [DOI] [PubMed] [Google Scholar]
- Cuyvers E, van der Zee J, Bettens K, Engelborghs S, Vandenbulcke M, Robberecht C, Dillen L, Merlin C, Geerts N, Graff C, Thonberg H, Chiang HH, Pastor P, Ortega‐Cubero S, Pastor MA, Diehl‐Schmid J, Alexopoulos P, Benussi L, Ghidoni R, Binetti G, Nacmias B, Sorbi S, Sanchez‐Valle R, Lladó A, Gelpi E, Almeida MR, Santana I, Clarimon J, Lleó A, Fortea J, de Mendonça A, Martins M, Borroni B, Padovani A, Matěj R, Rohan Z, Ruiz A, Frisoni GB, Fabrizi GM, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Sleegers K; BELNEU Consortium, EU EOD Consortium (2015) Genetic variability in SQSTM1 and risk of early‐onset Alzheimer dementia: a European early‐onset dementia consortium study. Neurobiol. Aging 36, 2005–2022. [DOI] [PubMed] [Google Scholar]
- Dassati S, Waldner A, Schweigreiter R (2014) Apolipoprotein D takes center stage in the stress response of the aging and degenerative brain. Neurobiol. Aging 35, 1632–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C (2010) Widespread protein aggregation as an inherent part of aging in C. elegans . PLoS Biol. 8, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M (2014) Redox proteomics analysis of HNE‐modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 71, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson TC, Chuckowree JA, Chuah MI, West AK, Vickers JC (2005) alpha‐Internexin immunoreactivity reflects variable neuronal vulnerability in Alzheimer's disease and supports the role of the beta‐amyloid plaques in inducing neuronal injury. Neurobiol. Dis. 18, 286–295. [DOI] [PubMed] [Google Scholar]
- Dillin A, Cohen E (2011) Ageing and protein aggregation‐mediated disorders: from invertebrates to mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch CE, Morgan TE, Longo VD, de Magalhaes JP (2010) Cell resilience in species life spans: a link to inflammation? Aging Cell 9, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB (2014) What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog. Neurobiol. 117, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della CL, Spano P, Tofaris GK, Goedert M, Spillantini MG (2010) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain 133, 2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O (2006) Ubiquitin hydrolase Uch‐L1 rescues beta‐amyloid‐induced decreases in synaptic function and contextual memory. Cell 126, 775–788. [DOI] [PubMed] [Google Scholar]
- Hafner A, Glavan G, Obermajer N, Zivin M, Schliebs R, Kos J (2013) Neuroprotective role of gamma‐enolase in microglia in a mouse model of Alzheimer's disease is regulated by cathepsin X. Aging Cell 12, 604–614. [DOI] [PubMed] [Google Scholar]
- Hagemann TL, Boelens WC, Wawrousek EF, Messing A (2009) Suppression of GFAP toxicity by alphaB‐crystallin in mouse models of Alexander disease. Hum. Mol. Genet. 18, 1190–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggmark A, Mikus M, Mohsenchian A, Hong MG, Forsstrom B, Gajewska B, Baranczyk‐Kuzma A, Uhlen M, Schwenk JM, Kuzma‐Kozakiewicz M, Nilsson P (2014) Plasma profiling reveals three proteins associated to amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 1, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XJ, Lee MJ, Yu GR, Lee ZW, Bae JY, Bae YC, Kang SH, Kim DG (2012) Altered dynamics of ubiquitin hybrid proteins during tumor cell apoptosis. Cell Death Dis. 3, e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques AG, Oliveira JM, Gomes B, Ruivo R, da Cruz e Silva EF, da Cruz e Silva OA (2014) Complexing Abeta prevents the cellular anomalies induced by the Peptide alone. J. Mol. Neurosci. 53, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert MK, Aerts MB, Beenes M, Norgren N, Esselink RA, Bloem BR, Kuiperij HB, Verbeek MM (2015) CSF neurofilament light chain but not FLT3 ligand discriminates Parkinsonian disorders. Front Neurol. 6, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J, Hastings T, Greenamyre JT, Chen J (2011) Peroxiredoxin‐2 protects against 6‐hydroxydopamine‐induced dopaminergic neurodegeneration via attenuation of the apoptosis signal‐regulating kinase (ASK1) signaling cascade. J. Neurosci. 31, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner S, Eam JE, Hubner A, Jans DA (2006) Laminopathy‐inducing lamin A mutants can induce redistribution of lamin binding proteins into nuclear aggregates. Exp. Cell Res. 312, 171–183. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Gong CX, Liu F (2013) Hyperphosphorylation‐induced tau oligomers. Front Neurol. 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M, Gentier RJ, Dennissen FJ, Schulz H, Bolle I, Holter SM, et al (2012) Long‐term proteasomal inhibition in transgenic mice by UBB(+1) expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol. 124, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA (2012) Interaction between pathogenic proteins in neurodegenerative disorders. J. Cell Mol. Med. 16, 1166–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Wu Y, Zhou Y (2014) 14‐3‐3 and aggresome formation: implications in neurodegenerative diseases. Prion 8, 173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kley RA, van der Ven PF, Olive M, Hohfeld J, Goldfarb LG, Furst DO, Vorgerd M (2013) Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy 9, 422–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41, 1094–1099. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al (1998) The ubiquitin pathway in Parkinson's disease. Nature 395, 451–452. [DOI] [PubMed] [Google Scholar]
- Li D, Shi JJ, Mao CJ, Liu S, Wang JD, Chen J, Wang F, Yang YP, Hu WD, Hu LF, Liu CF (2013) Alteration of dynein function affects alpha‐synuclein degradation via the autophagosome‐lysosome pathway. Int. J. Mol. Sci. 14, 24242–24254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI, Peng J (2004) Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 279, 37061–37068. [DOI] [PubMed] [Google Scholar]
- Lio D, Licastro F, Scola L, Chiappelli M, Grimaldi LM, Crivello A, Colonna‐Romano G, Candore G, Franceschi C, Caruso C (2003) Interleukin‐10 promoter polymorphism in sporadic Alzheimer's disease. Genes Immun. 4, 234–238. [DOI] [PubMed] [Google Scholar]
- Liu Q, Xie F, Alvarado‐Diaz A, Smith MA, Moreira PI, Zhu X, Perry G (2011) Neurofilamentopathy in neurodegenerative diseases. Open Neurol J 5, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loera‐Castaneda V, Sandoval‐Ramirez L, Pacheco Moises FP, Macias‐Islas MA, Alatorre Jimenez MA, Gonzalez‐Renovato ED, et al (2014) Novel point mutations and A8027G polymorphism in mitochondrial‐DNA‐encoded cytochrome c oxidase II gene in Mexican patients with probable Alzheimer disease. Int. J. Alzheimers. Dis. 2014, 794530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchini A, Geho DH, Bishop B, Tran D, Xia C, Dufour RL, Jones CD, Espina V, Patanarut A, Zhou W, Ross MM, Tessitore A, Petricoin EF III, Liotta LA (2008) Smart hydrogel particles: biomarker harvesting: one‐step affinity purification, size exclusion, and protection against degradation. Nano Lett. 8, 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Warmlander SK, Graslund A, Abrahams JP (2014) Non‐chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid beta peptide. J. Biol. Chem. 289, 27766–27775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangialasche F, Baglioni M, Cecchetti R, Kivipelto M, Ruggiero C, Piobbico D, et al (2015) Lymphocytic mitochondrial aconitase activity is reduced in Alzheimer's disease and mild cognitive impairment. J. Alzheimers Dis. 44, 649–660. [DOI] [PubMed] [Google Scholar]
- Meier‐Ruge W, Iwangoff P, Reichlmeier K (1984) Neurochemical enzyme changes in Alzheimer's and Pick's disease. Arch. Gerontol. Geriatr. 3, 161–165. [DOI] [PubMed] [Google Scholar]
- de Mendonca E, Salazar Alcala E, Fernandez‐Mestre M (2014) Role of genes GSTM1, GSTT1, and MnSOD in the development of late‐onset Alzheimer disease and their relationship with APOE*4. Neurologia S0213–4853, 235–237. [DOI] [PubMed] [Google Scholar]
- Middeldorp J, Hol EM (2011) GFAP in health and disease. Prog. Neurobiol. 93, 421–443. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Cuervo AM (2014) Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl 1), S33–S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranova LK, Weeks SD, Strelkov SV, Gusev NB (2015) Characterization of mutants of human small heat shock protein HspB1 carrying replacements in the N‐terminal domain and associated with hereditary motor neuron diseases. PLoS ONE 10, e0126248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten‐Hawle P, Morimoto RI (2014) Organismal proteostasis: role of cell‐nonautonomous regulation and transcellular chaperone signaling. Genes Dev. 28, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Dooves S, Kooijman L, Chan ET, Kirk CJ, et al (2013) Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 136, 1415–1431. [DOI] [PubMed] [Google Scholar]
- Ou JR, Tan MS, Xie AM, Yu JT, Tan L (2014) Heat shock protein 90 in Alzheimer's disease. Biomed. Res. Int. 2014, 796869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page T, Gitcho MA, Mosaheb S, Carter D, Chakraverty S, Perry RH, Bigio EH, et al (2011) FUS immunogold labeling TEM analysis of the neuronal cytoplasmic inclusions of neuronal intermediate filament inclusion disease: a frontotemporal lobar degeneration with FUS proteinopathy. J. Mol. Neurosci. 45, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Jung HY, Kim TD, Park JH, Yang CH, Kim J (2002) Distinct roles of the N‐terminal‐binding domain and the C‐terminal‐solubilizing domain of alpha‐synuclein, a molecular chaperone. J. Biol. Chem. 277, 28512–28520. [DOI] [PubMed] [Google Scholar]
- Pitts A, Dailey K, Newington JT, Chien A, Arseneault R, Cann T, Thompson LM, Cumming RC (2012) Dithiol‐based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J. Biol. Chem. 287, 22717–22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Kumar D, Tessari I, Brucale M, Munari F, Tosatto L, et al (2014) The chaperone‐like protein 14‐3‐3eta interacts with human alpha‐synuclein aggregation intermediates rerouting the amyloidogenic pathway and reducing alpha‐synuclein cellular toxicity. Hum. Mol. Genet. 23, 5615–5629. [DOI] [PubMed] [Google Scholar]
- Qureshi HY, Li T, MacDonald R, Cho CM, Leclerc N, Paudel HK (2013) Interaction of 14‐3‐3zeta with microtubule‐associated protein tau within Alzheimer's disease neurofibrillary tangles. Biochemistry 52, 6445–6455. [DOI] [PubMed] [Google Scholar]
- Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A (2015) Parkinsonism‐associated protein DJ‐1/Park7 is a major protein deglycase that repairs methylglyoxal‐ and glyoxal‐glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 290, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Kageyama Y, Iijima M, Sesaki H (2015) PARK2/Parkin becomes critical when DNM1L/Drp1 is absent. Autophagy 11, 573–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrabhatla P, Jaffe H, Pant HC (2011) Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer's NFTs. FASEB J. 25, 3896–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A, et al (2013) DNA methylation map of mouse and human brain identifies target genes in Alzheimer's disease. Brain 136, 3018–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Chen C, Yang A, Chen Y, Liu Q, Ni J (2015) Redox proteomics identification of specifically carbonylated proteins in the hippocampi of triple transgenic Alzheimer's disease mice at its earliest pathological stage. J. Proteomics. 123, 101–113. [DOI] [PubMed] [Google Scholar]
- Skillback T, Farahmand B, Bartlett JW, Rosen C, Mattsson N, Nagga K, Kilander L, Religa D, Wimo A, Winblad B, Rosengren L, Schott JM, Blennow K, Eriksdotter M, Zetterberg H (2014) CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology 83, 1945–1953. [DOI] [PubMed] [Google Scholar]
- Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, et al (2013) Levels of soluble apolipoprotein E/amyloid‐beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 288, 5914–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuling E, Bourgonje A, Veenje S, Thijssen K, de Boer J, van der Velde J, Swertz M, Nollen E (2011) Modifiers of mutant huntingtin aggregation: functional conservation of C. elegans‐modifiers of polyglutamine aggregation. PLoS Curr. 3, RRN1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al (2010) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch. Gen. Psychiatry 67, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardarajan BN, Bruesegem SY, Harbour ME, Inzelberg R, Friedland R, St George‐Hyslop P, Seaman MN, Farrer LA (2012) Identification of Alzheimer disease‐associated variants in genes that regulate retromer function. Neurobiol. Aging 33, 2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan J, Haapasalo A, Bottcher C, Miettinen R, Kurkinen KM, Lu A, et al (2011) Alzheimer's disease‐associated ubiquilin‐1 regulates presenilin‐1 accumulation and aggresome formation. Traffic 12, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J (2008) Calpain‐mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 38, 78–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Ryu J, Ahn HM, Saxena N, Chaudhary A, Yun CO, Kaul SC (2015) Functional significance of point mutations in stress chaperone mortalin and their relevance to Parkinson disease. J. Biol. Chem. 290, 8447–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahner AD, Glatt CE, Bronstein JM, Ritz B (2007) Glutathione S‐transferase mu, omega, pi, and theta class variants and smoking in Parkinson's disease. Neurosci. Lett. 413, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman‐Storer CM, Karki SB, Kuznetsov SA, Tabb JS, Weiss DG, Langford GM, Holzbaur EL (1997) The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc. Natl Acad. Sci. USA 94, 12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson M, Budovsky A, Tacutu R, Fraifeld V (2009) The signaling hubs at the crossroad of longevity and age‐related disease networks. Int. J. Biochem. Cell Biol. 41, 516–520. [DOI] [PubMed] [Google Scholar]
- Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC (2006) Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62's role in neurodegenerative disease. J. Biomed. Biotechnol. 2006, 62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S, Shen D, Chen C, Wang J, Liu T, Yu Z (2013) Regulation of neuronal toxicity of beta‐amyloid oligomers by surface ATP synthase. Mol. Med. Rep. 8, 1689–1694. [DOI] [PubMed] [Google Scholar]
- Xu Q, Huang S, Song M, Wang CE, Yan S, Liu X, Gaertig MA, Yu SP, Li H, Li S, Li XJ (2013) Synaptic mutant huntingtin inhibits synapsin‐1 phosphorylation and causes neurological symptoms. J. Cell Biol. 202, 1123–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu DE, Zhang WM, Yang ZZ, Zhu HM, Yan K, Li S, Bagnard D, Dawe GS, Ma QH, Xiao ZC (2014) Amyloid precursor protein at node of Ranvier modulates nodal formation. Cell Adh. Migr. 8, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang Y, Zhao S, Zhang Z, Tong X, Wei F, Lu Z (2015) Heat shock protein 70 induction by glutamine increases the α‐synuclein degradation in SH‐SY5Y neuroblastoma cells. Mol. Med. Rep., 12, 5524–5530. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 48, 394–403. [DOI] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Kanekiyo T, Stine WB Jr, Michon SC, Nwabuisi‐Heath E, et al (2012) Intraneuronal Abeta detection in 5xFAD mice by a new Abeta‐specific antibody. Mol. Neurodegener. 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KY, Yang S, Warraich ST, Blair IP (2014) Ubiquilin 2: a component of the ubiquitin‐proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol. 50, 123–126. [DOI] [PubMed] [Google Scholar]
- Zigman WB (2013) Atypical aging in Down syndrome. Dev. Disabil. Res. Rev. 18, 51–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Enrichment of proteins and post‐translational modifications in pooled hippocampal tissue from AD relative to normal controls.
Figure S2. Hippocampal tau‐IP aggregate proteins, AD vs. AMC.
Table S1. Subject Data.
Data S1. Full proteomics data.