

Summary
Cyclooxygenase‐2 (COX‐2) has been recently identified to be involved in the pathogenesis of Alzheimer's disease (AD). Yet, the role of an important COX‐2 metabolic product, prostaglandin (PG) I2, in the pathogenesis of AD remains unknown. Using human‐ and mouse‐derived neuronal cells as well as amyloid precursor protein/presenilin 1 (APP/PS1) transgenic mice as model systems, we elucidated the mechanism of anterior pharynx‐defective (APH)‐1α and pharynx‐defective‐1β induction. In particular, we found that PGI 2 production increased during the course of AD development. Then, PGI 2 accumulation in neuronal cells activates PKA/CREB and JNK/c‐Jun signaling pathways by phosphorylation, which results in APH‐1α/1β expression. As PGI 2 is an important metabolic by‐product of COX‐2, its suppression by NS398 treatment decreases the expression of APH‐1α/1β in neuronal cells and APP/PS1 mice. More importantly, β‐amyloid protein (Aβ) oligomers in the cerebrospinal fluid (CSF) of APP/PS1 mice are critical for stimulating the expression of APH‐1α/1β, which was blocked by NS398 incubation. Finally, the induction of APH‐1α/1β was confirmed in the brains of patients with AD. Thus, these findings not only provide novel insights into the mechanism of PGI 2‐induced AD progression but also are instrumental for improving clinical therapies to combat AD.
Keywords: β‐amyloid protein, anterior pharynx‐defective‐1α/1β, APP/PS1, cyclooxygenase‐2, prostaglandin I2
Introduction
Alzheimer's diseases (AD) is the most common cause of dementia in aged people and is characterized clinically by cognitive decline and pathologically by the accumulation of amyloid β‐protein (Aβ) and hyperphosphorylation of tau in the brain (Hoshino et al., 2007; Arnaud et al., 2009). Here, increases in the expression of several potentially toxic secretases, including BACE‐1, presenilin 1/2 (PS1/2), anterior pharynx‐defective (APH)‐1α/1β, nicastrin, and PEN2, result in the formation of Aβ plaques, synapse dysfunction or loss, neuronal loss, and diffuse brain atrophy, thereby leading to the decline of cognitive abilities (De Strooper, 2003). The γ‐secretases, such as APH‐1α and APH‐1β, are required for notch pathway signaling, for γ‐secretase cleavage of β‐APP, and for Aβ protein accumulation in C. elegans (Francis et al., 2002). Indeed, APH‐1 usually interacts with PEN‐2, nicastrin, and PS to generate an active form of the γ‐secretase complex, which is responsible for the cleavage of β‐APP and the deposition of Aβ (De Strooper, 2003). Once APH‐1 was found in C. elegans (Francis et al., 2002), the APH‐1 complex was then confirmed in several experimental models (Gu et al., 2003; Luo et al., 2003; Hansson et al., 2004). However, the regulatory mechanism of APH‐1α and APH‐1β are often overlooked during the course of AD progression.
To reveal the possible mechanism of APH‐1α and APH‐1β regulation, it is necessary to identify the molecular pathways that are responsible for the deposition of Aβ. Epidemiological and clinical data suggest that nonsteroidal anti‐inflammatory drugs (NSAIDs) are beneficial in the treatment and prevention of AD (Imbimbo et al., 2010). The protective effects of NSAIDs in AD are due to their anti‐inflammatory properties that inhibit cyclooxygenase‐2 (COX‐2) (McGeer, 2000; van Gool et al., 2003). As an important factor in inflammatory reactions of peripheral tissues, COX‐2 has a potential role in the pathogenesis of AD. This has been populously investigated through studies of its metabolic products, the prostaglandins (PGs), including PGE2, PGD2 [and its dehydration end product 15‐deoxy‐Δ12,14‐PGJ2 (15d‐PGJ2)], PGI2, PGF2α, and TXA2 (Akarasereenont et al., 1999). For example, PGE2 treatment increases the ratio of Aβ1–42/Aβ1–40 production in SH‐SY5Y cells and APP23 transgenic mice (Hoshino et al., 2007, 2009). In line with these observations, PGE2 was further verified to stimulate the production of Aβ1–42 alone in C57BL/6 mice (Echeverria et al., 2005). In addition to PGE2, PGD2 shows positive effects on stimulating the production of Aβ1–42 in primary mouse microglia and neuronal cells (Bate et al., 2006). As expected, 15d‐PGJ2 is also able to increase fibrillar Aβ in rat cortical neurons (Takata et al., 2003; Yamamoto et al., 2011) while PGF2α has been shown to be involved in Aβ production in microglia cells (Zhuang et al., 2013). However, the effects of PGI2 on the production of Aβ are not well studied.
In this study, an intracellular signaling pathway by which PGI2 regulates the expression of APH‐1α/1β has been proposed to contribute to the deposition of Aβ. Specifically, PGI2 treatment increases the expression of APH‐1α/1β via the PKA/CREB and JNK/c‐Jun activation pathways in SH‐SY5Y cells, which facilitates the synthesis of Aβ in neuron cells. More importantly, in AD, the Aβ oligomers were localized to the CSF microenvironment, which contributes to the expression of APH‐1α/1β. Reconstructing the signaling network that regulates PGI2‐mediated APH‐1α/1β expression in neuron cells will facilitate the development of strategies to combat AD.
Results
APH‐1α/1β is highly induced in APP/PS1 transgenic mice
Due to previous reports of a pivotal role of APH‐1α/1β in the pathogenesis of AD (Mrak & Griffin, 2001; De Strooper, 2003), we evaluated the expression levels of APH‐1α/1β in AD brains. As shown in Fig. 1a, the immunoreactivity of APH‐1α/1β and Aβ was highly induced at the patients with AD. In accordance with this observation, the mRNA and protein expression levels of APH‐1α/1β were elevated at the patients with AD (Fig. 1b). Due to the limited accessibility of human AD samples, we performed similar experiments in APP/PS1 mice. The results demonstrated that APH‐1α/1β immunostaining was markedly increased in the cerebral cortex as well as in the dentate gyrus region of the hippocampus of APP/PS1 transgenic mice at 6 months of age, when compared to the WT C57BL/6 mice (Fig. 1d). In accordance with our immunostaining observations, the mRNA and protein expression of APH‐1α/1β was upregulated in the cerebral cortex and hippocampus of APP/PS1 mice (Fig. 1c,e). When considered together, these results clearly demonstrate that APH‐1α/1β levels were increased in APP/PS1 transgenic mice. These data agree with previous reports (De Strooper, 2003), suggesting that APH‐1α/1β are possibly involved in aggravating AD.
Figure 1.
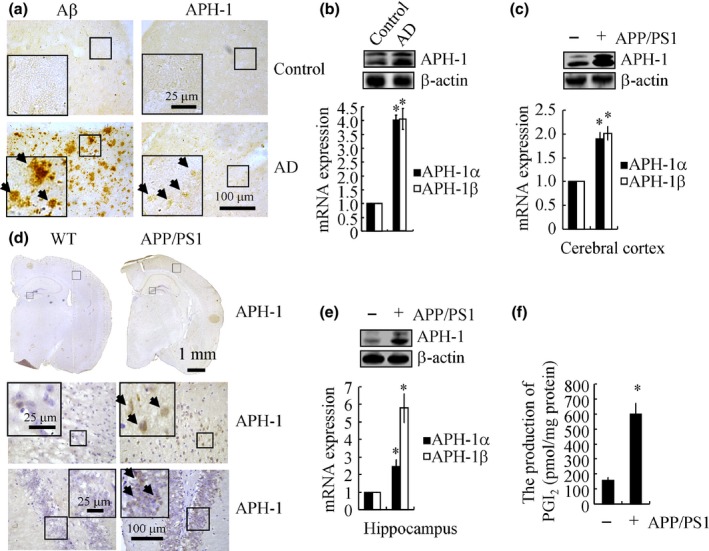
The expression of APH‐1α/1β is markedly increased in patients with AD and APP/PS1 transgenic mice at 6 months of age when compared with control subjects. The tissue blocks of human brains of AD were collected by the New York Brain Bank at Columbia University and Fengtian Hospital of China. Free‐floating slices (40 μm) were prepared using a cryostat (a, b). In selected experiments, the brains of APP/PS1 transgenic mice at 6 months of age were collected following anesthesia and perfusion (c–f). The immunoreactivity of APH‐1 or Aβ1–42 was determined by IHC using an anti‐APH‐1 or Aβ1–42 antibody (a, d). APH‐1α/1β mRNA and protein levels were determined by qRT–PCR and immunoblotting (n = 3 for human sample, n = 6 for mouse samples) (b, c, e). The production of PGI 2 in the brains of APP/PS1 transgenic or of WT mice was determined by PGI 2 determination kits (f). The data represent the means ± S. E. of at least three independent experiments. *P < 0.05 with respect to the WT or normal human controls.
Elevation of PGI2 accelerates the synthesis of APH‐1α/1β in APP/PS1 transgenic mice
We next sought to elucidate the mechanism by which APH‐1α/1β are upregulated in an AD mouse model. Because evidence suggests that PGI2 is a potential mediator of neuroinflammation (Ford‐Hutchinson et al., 1978; Honda et al., 2005; Pulichino et al., 2006), we sought to determine the concentration of PGI2 in APP/PS1 transgenic mice at 6 months of age. As shown in Fig. 1f, the synthesis of PGI2 was markedly increased in APP/PS1 transgenic mice. To know the roles of PGI2 in AD development, we first screened the effects of PGI2 on the expression of α‐, β‐ or γ‐secretases. The results demonstrated that PGI2 treatment concurrently downregulated the expression of ADAM‐10 and upregulated the expression of BACE‐1, PS2, and APH‐1α/1β in n2a cells (Table 1). To further understand the possible role of PGI2 in AD, we then injected (i.c.v.) PGI2 (2 μg/5 μL) or incubated brain slices from WT C57BL/6 mice with PGI2 (10 μm) for 24 h. IHC staining indicated that APH‐1α/1β expression was induced in both the cerebral cortex and the DG region of APP/PS1 transgenic mice (Fig. 2a,b). In addition, these observations were verified by qRT–PCR and Western blots (Fig. 2c,d). To identify the role of PGI2 in inducing the expression of APH‐1α/1β, we conducted live animal and two‐photon imaging experiments, as described in the ‘Materials and Methods’. The results revealed that PGI2 (2 μg/5 μL) injection (i.c.v.) into the ventricles of WT C57BL/6 mice increased the luciferase activity of the APH‐1α/1β promoters at 12 h following injection. This activity was maximal at 24 h following injection (Fig. 2e). Of note, PBS (−) injection (i.c.v.) does not alter the activity of APH‐1α and APH‐1β promoters (data not shown). Two‐photon imaging results reinforced the notion that PGI2 (2 μg/5 μL) injection (i.c.v.) increased the immunofluorescence of APH‐1α/1β in the cerebral cortex of WT C57BL/6 mice (Fig. 2f). Accordingly, we then treated the SH‐SY5Y and n2a neuronal cells with PGI2 (10 μm) for 48 h. Our data demonstrated that PGI2 treatment clearly increases the expression of APH‐1α/1β in human or mouse neurons (Fig. 2g,h). These results were supported by the immunostaining experiments, and the images obtained using Leica confocal microscopy (Fig. 2i,j). Collectively, our data clearly support the fact that PGI2 elevation increases the synthesis of APH‐1α/1β in vitro and in vivo.
Table 1.
The effects of PGI2 on the expression of α‐, β‐, or γ‐secretases in n2a cells
| Gene Name | Control | PGI2 (10 μm) |
|---|---|---|
| ADAM‐10 | 1 | 0.59 |
| BACE‐1 | 1 | 4.59 |
| PS1 | 1 | 0.99 |
| PS2 | 1 | 1.92 |
| APH‐lα | 1 | 3.25 |
| APH‐lβ | 1 | 1.99 |
| Nicastrin | 1 | 0.94 |
| PEN2 | 1 | 1.08 |
Figure 2.
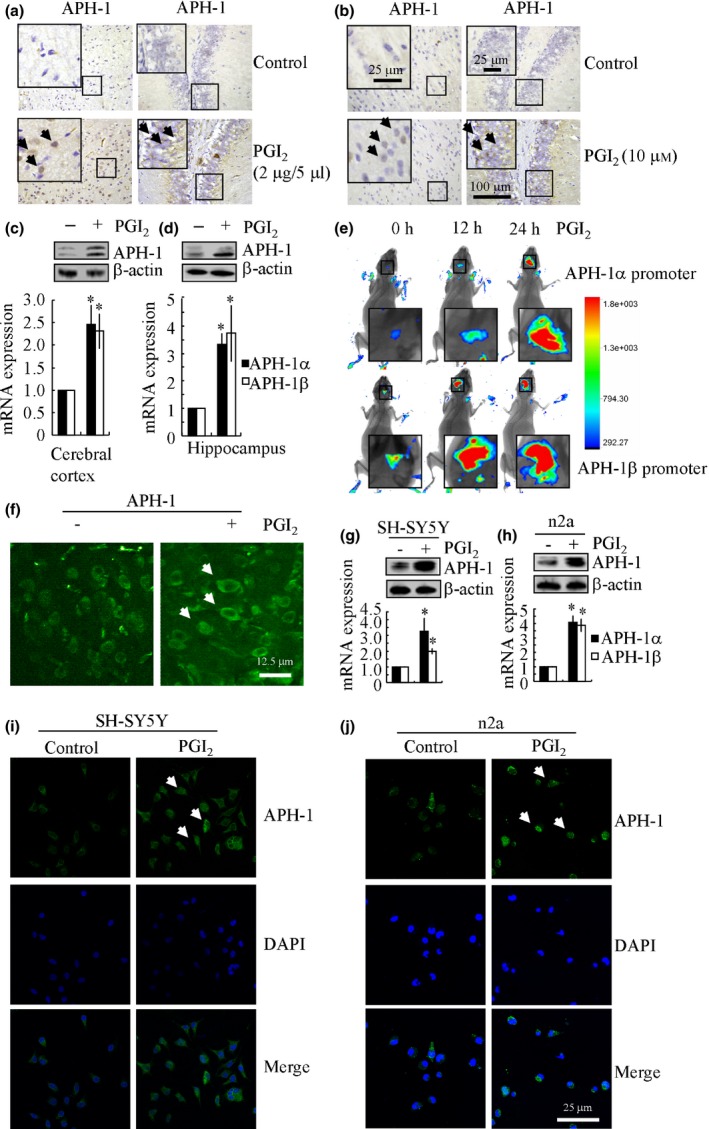
Involvement of PGI 2 in inducing the expression of APH‐1α/1β. (a, c, d) The WT C57BL/6 mice at 6 months of age were injected (i.c.v.) with PGI 2 (2 μg/5 μL), and the brains were then collected and sectioned after 24 h. The immunoreactivity of APH‐1 was determined by immunohistochemistry using an anti‐APH‐1 antibody (a). APH‐1α/1β mRNA and protein levels were determined by qRT–PCR and Western blots, respectively (n = 8) (c, d). (b) The brains of WT C57BL/6 mice at 6 months of age were harvested and freshly sectioned (400 μm) before treatment with PGI 2 (10 μm) for 24 h. The slices were immunostained with APH‐1 antibody. (e) PGI 2 (2 μg/5 μL) or vehicle (PBS) was injected (i.c.v.) into one side of a cerebral ventricle in the absence or presence of preseeded n2a cells that were transfected with APH‐1α/β promoters in the other side of cerebral ventricle (n = 6). Luciferase activities from the different groups of mice were measured using a live animal imaging system. (f) The left cerebral ventricle was injected with PGI 2 (2 μg/5 μL) or vehicle (PBS) solution before staining with an APH‐1 antibody and scanning using two‐photon microscopy (n = 6). (g–j) SH‐SY5Y or n2a cells were treated with PGI 2 (10 μm) for 48 h. APH‐1α/1β mRNA and protein levels were determined by qRT–PCR and Western blots, respectively (g, h). The immunofluorescence of APH‐1 was determined by immunohistochemistry using a primary rabbit APH‐1 antibody and secondary Alexa Fluor 488‐labeled goat anti‐rabbit IgG (i, j). The data represent the means ± SE of three independent experiments. *P < 0.05 with respect to the vehicle‐treated control.
PGI2 inhibition impaired the expression of APH‐1α/1β following intranasal administration of NS398 in APP/PS1 transgenic mice
Because PGI2 is an important metabolic product of COX‐2, we treated APP/PS1 transgenic mice with a COX‐2‐specific inhibitor, NS398 (50 μg kg−1 day−1), for 5 months. Our results revealed that PGI2 was significantly suppressed by NS398 (50 μg kg−1 day−1) administration in APP/PS1 transgenic mice (Fig. 3a). More interestingly, the inhibition of PGI2 by NS398 resulted in a decrease in the levels of APH‐1α/1β in APP/PS1 transgenic mice by IHC staining (Fig. 3c). Similar results were verified by qRT–PCR or Western blot experiments (Fig. 3e,f). Of note, NS398 treatment does not affect the body weight of mice and induce wound healing to the mice as previously indicated (Wang et al., 2011a). We then injected APP/PS1 transgenic mice with NS398 (2 μg/5 μL) for 24 h. The results demonstrated that the injection (i.c.v.) of NS398 (2 μg/5 μL) shows a suppressive effect on the production of PGI2 in APP/PS1 transgenic mice (Fig. 3b). Of note, our results reinforce the hypothesis that NS398 treatment (2 μg/5 μL) suppressed the expression of APH‐1α/1β (Fig. 3d,g,h) by inhibiting the production of PGI2. In addition, the production of sAPPα was restored to the control level (i.e. the basal level of WT mice), whereas the increased production of sAPPβ was attenuated to the basal level of WT mice following intranasal administration of NS398 treatment for 5 months in APP/PS1 mice (Fig. 3i,j). Intranasal administration of NS398 also decreased the production of Aβ1–42 in APP/PS1 mice (Fig. 3i,j), which potentially decelerates the pathogenesis of AD. To further verify the role of NS398 in suppressing the expression of APH‐1α/1β, we performed intracerebroventricular injections of the inhibitor. Similar to nasal administration, NS398 injection (i.c.v., 2 μg/5 μL) reversed the concurrent downregulation of sAPPα and upregulation of sAPPβ in APP/PS1 mice (Fig. 3k,l). Therefore, our results concretely support the hypothesis that NS398 has the ability to suppress the expression of APH‐1α/1β by decreasing the production of PGI2 in APP/PS1 transgenic mice.
Figure 3.
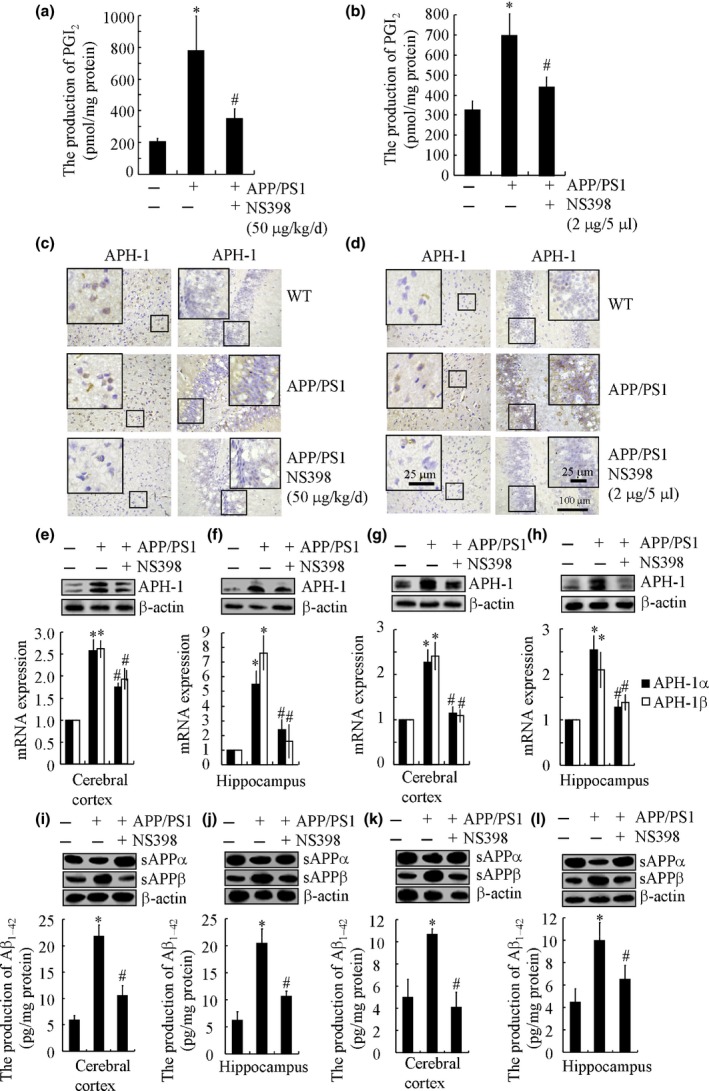
NS398 administration decreases the expression of APH‐1α/1β via inhibiting the production of PGI 2 in APP/PS1 transgenic mice. The APP/PS1 transgenic mice at 1 month of age received NS398 (50 μg kg−1 day−1) for 5 months before harvesting brain slices (n = 3) (a, c, e, f, i, j). In selected experiments, the APP/PS1 transgenic mice at the 6 months of age were injected (i.c.v.) with NS398 (2 μg/5 μL). The brains were then collected and sectioned after 24 h (n = 8) (b, d, g, h, k, l). The production of PGI 2 was determined by PGI 2 enzyme immunoassay kits (a, b). The immunoreactivity of APH‐1 was determined by immunohistochemistry using an anti‐APH‐1 antibody (c, d). APH‐1α/1β mRNA and protein expression levels were determined by qRT–PCR and Western blots, respectively (e–h). The production of sAPPα and sAPPβ was determined by Western blots (i–l). The production of Aβ1–42 was determined by Aβ1–42 ELISA kits (i‐l). The data represent the means ± SE of three independent experiments. *P < 0.05 with respect to WT mice. # P < 0.05 compared to APP/PS1 transgenic mice.
Critical role of PKA/CREB and JNK/c‐Jun signaling pathways in mediating PGI2‐induced APH‐1α/1β expression in n2a cells
We next aimed to elucidate the signaling pathways of APH‐1α/1β synthesis in PGI2‐treated n2a cells. First, 48 h of PGI2 (10 μm) treatment activated PKA/CREB and JNK/c‐Jun signaling pathways by the phosphorylation of CREB and c‐Jun (Fig. 4a–c), which resulted in the synthesis of APH‐1α and APH‐1β in n2a cells (Fig. 4a,c). To further elucidate the role of PKA/CREB and JNK/c‐Jun signaling pathways in regulating the expression of APH‐1α/1β, we treated n2a cells with the PKA pharmacological inhibitor H89 (1 μm) or JNK‐specific inhibitor SP600125 (10 μm). Treatment of n2a cells with H89 (1 μm) or SP600125 (10 μm) not only suppressed the phosphorylation of CREB and c‐Jun (Fig. 4a–c) but also reversed the synthesis of APH‐1α/1β in PGI2‐treated n2a cells (Fig. 4a,c).
Figure 4.
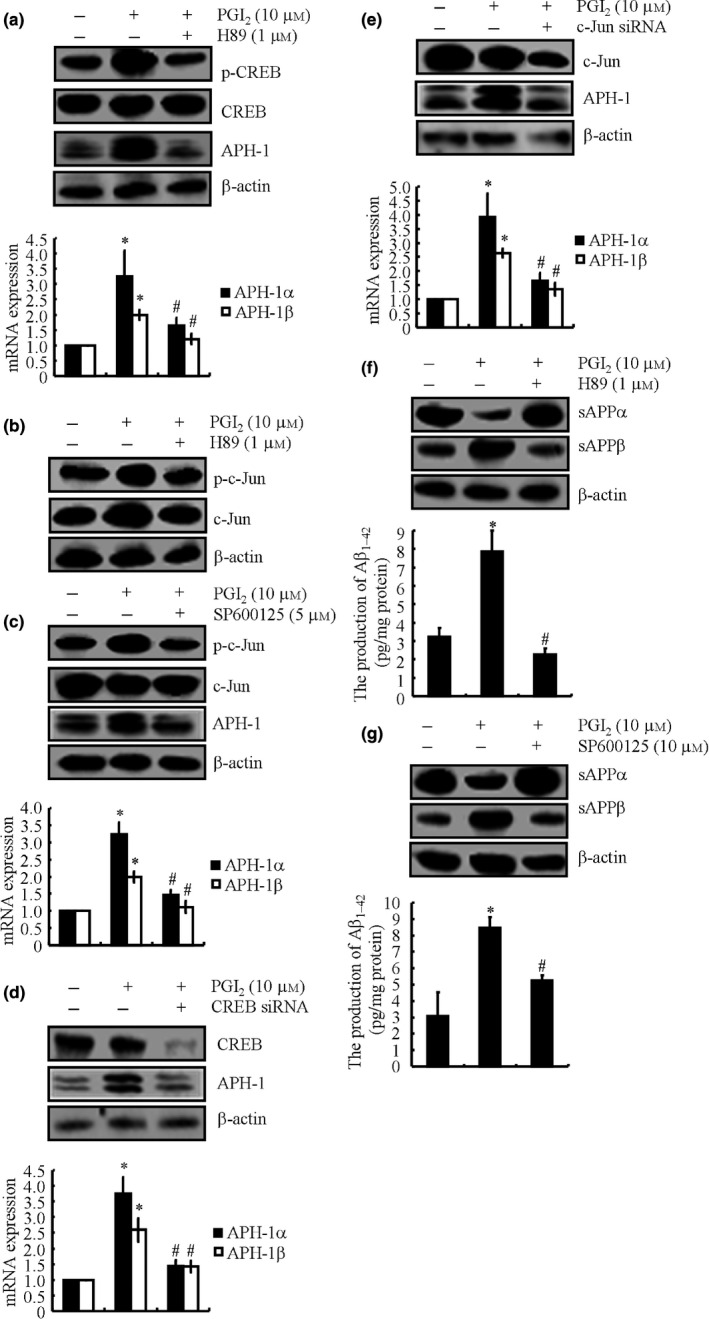
PGI 2 elevation stimulates the expression of APH‐1α/1β via the PKA/CREB and JNK/c‐Jun signaling pathways in cultured neuronal cells. n2a cells were treated with PGI 2 (10 μm) in the absence or presence of H89 (1 μm) (a, b, f) or SP600125 (5 μm) (c, g) cells for 48 h. In distinct experiments, n2a cells were transfected with CREB (d) or c‐Jun siRNA (e) before treating the cells with PGI 2 (10 μm) for 48 h. APH‐1α/1β mRNA and protein levels were determined by qRT–PCR and Western blots, respectively (a, c–e). Phosphorylated CREB and c‐Jun as well as total CREB and c‐Jun were detected by immunoblotting using specific antibodies (a–e). The production of sAPPα and sAPPβ was determined by Western blots (f, g). The production of Aβ1–42 was determined by Aβ1–42 ELISA kits (f, g). The data represent the means ± SE of three independent experiments. *P < 0.05 with respect to the vehicle‐treated or vector‐transfected control. # P < 0.05 compared to PGI 2‐treated alone.
To verify these observations and to account for the nonspecificity of the pharmacological inhibitors, we transfected n2a cells with siRNAs that were specific for interfering with the expression of CREB or c‐Jun prior to incubating them with PGI2 (10 μm). As shown in Fig. 4d and e, CREB and c‐Jun knockdown efficiently decreased the protein levels of CREB and c‐Jun. As a consequence, the knockdown of CREB or c‐Jun inhibited the effects of PGI2 on inducing the synthesis of APH‐1α/1β in n2a cells (Fig. 4d,e). In addition, inhibiting the signaling pathways of PKA/CREB and JNK/c‐Jun concurrently results in the restoration of the production of sAPPα and a decrease in the production of sAPPβ to the basal level in PGI2‐treated n2a cells (Fig. 4f,g). More importantly, inhibiting the activity of the PKA/CREB or the JNK/c‐Jun signaling pathways resulted in the attenuation of Aβ1–42 formation in PGI2‐activated n2a cells (Fig. 4f,g). Therefore, these observations support the hypothesis that PKA/CREB and JNK/c‐Jun signaling pathways are important in mediating PGI2‐induced APH‐1α/1β expression, which results in Aβ1–42 deposition in neuron cells.
Aβ oligomers in the CSF of APP/PS1 mice have the ability to stimulate the expression of APH‐1α/1β
Because NS398 incubation decreases the expression of APH‐1α/1β and Aβ1–42 deposition by decreasing the production of PGI2 in vitro and in vivo, we next examined the potential contribution of Aβ1–42 to the pathogenesis of AD. We conducted experiments to determine whether Aβ1–42 is confined to microenvironments that are related to AD in APP/PS1 transgenic mice. In brief, the cerebrospinal fluid (CSF) of APP/PS1 transgenic mice was injected into WT C57BL/6 mice in the absence or presence of Aβ antibody for 2 weeks prior to sacrifice. When compared to control animals, the expression of APH‐1α/1β in both the cerebral cortex and hippocampus was markedly increased by the injection (i.c.v.) of APP/PS1 CSF (Fig. 5a–c). In addition, the elevated induction of APH‐1α/1β was suppressed by the injection (i.c.v.) of the Aβ antibody (1 μg/5 μL) (Fig. 5a–c). Therefore, these observations demonstrate that the confinement of the secreted form of Aβ1–42 to AD‐related microenvironments might induce the expression of APH‐1α/1β in a PGI2‐dependent manner.
Figure 5.
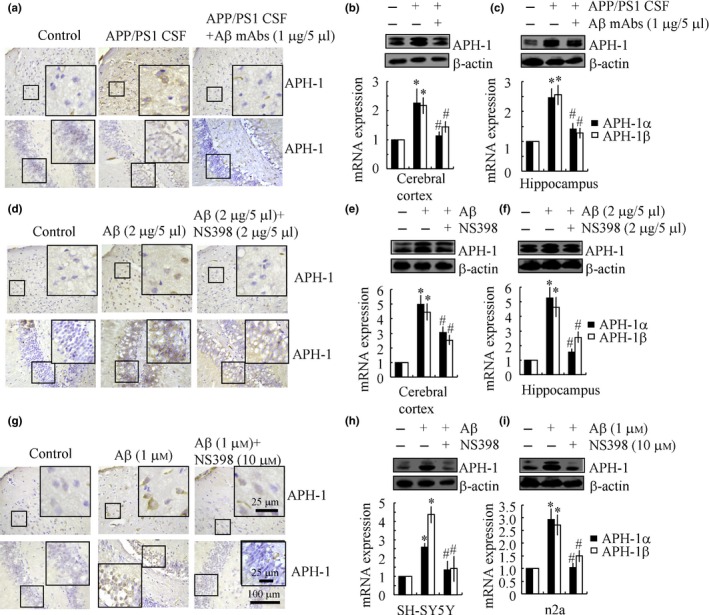
NS398 treatment diminished the effects of Aβ oligomers on inducing the expression of APH‐1α/1β. Cerebrospinal fluid (CSF) was obtained from APP/PS1 transgenic mice, which was then injected (i.c.v.), in the absence or presence of Aβ antibody (1 μg/5 μL), into C57BL/6 mice for 2 weeks before sacrifice (a–c). In selected experiments, the WT C57BL/6 mice at 6 months of age were injected (i.c.v.) with Aβ oligomers (2 μg/5 μL) in the absence or presence of NS398 (2 μg/5 μL). The brains were then collected and sectioned after 24 h (d–f). In separate experiments, the brains of WT C57BL/6 mice at 6 months of age were harvested and freshly sectioned (400 μm) before treatment with Aβ (1 μμ) in the absence or presence of NS398 (10 μm) for 24 h (g). In distinct experiments, SH‐SY5Y or n2a cells were treated with Aβ (1 μm) in the absence or presence of NS398 (10 μm) for 24 h (h, i). The immunoreactivity of APH‐1 was determined by IHC using an anti‐APH‐1 antibody (a, d, g). APH‐1α/1β mRNA and protein expression was determined by qRT–PCR and Western blots, respectively (n = 8) (b, c, e, f, h, i). The data represent the means ± SE of three independent experiments. *P < 0.05 with respect to vehicle‐treated controls. # P < 0.05 compared to APP/PS1 CSF‐ or Aβ‐treated alone.
To confirm these observations, we performed experiments to directly evaluate the involvement of Aβ1–42 oligomers in the progression of AD. Aβ oligomer (2 μg/5 μL) injection (i.c.v.) clearly increases the expression of APH‐1α/1β in both the cerebral cortex and hippocampus of WT C57BL/6 mice (Fig. 5d–f). The upregulation of APH‐1α/1β was further suppressed by NS398 (2 μg/5 μL) injection (i.c.v.) in Aβ1–42‐stimulated C57BL/6 mice (Fig. 5d–f). These in vivo observations were reinforced by in vitro experiments that demonstrated that Aβ (1 μm) treatment stimulates the expression of APH‐1α/1β in neuron cells by organotypic slice or cell culture (Fig. 5g–i). Thus, Aβ1–42 oligomers are critical for worsening AD.
Discussion
β‐amyloid protein (Aβ) deposition and hyperphosphorylation of tau are pathological characteristics of AD (1). As the role of PGI2 in AD development is presently unknown, we designed a study to identify the aggravating effects of PGI2 on AD. The major findings of this study are as follows: (i) APH‐1α/1β expression was markedly upregulated during the course of AD development; (ii) the accumulation of PGI2 in neuron cells induced the mRNA and protein expression of APH‐1α/1β in APP/PS1 mice; (iii) the PKA/CREB and JNK/c‐Jun signaling pathways are critical for mediating the effects of PGI2 on stimulating the expression of APH‐1α/1β, which is critical for γ‐cleavage of β‐APP and producing Aβ1–42; and (iv) Aβ1–42 oligomers in the CSF of APP/PS1 mice are responsible for augmenting the activity APH‐1α/1β, which potentially aggravates the pathogenesis of AD (Fig. 6).
Figure 6.
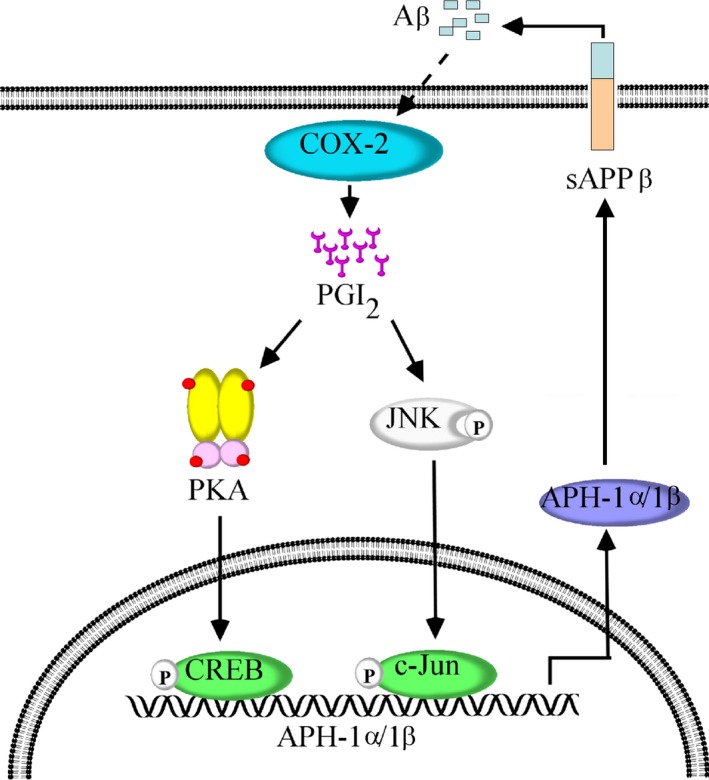
Proposed cascade of the signaling events regulating the pathogenesis of AD by PGI 2. In detail, elevated levels of PGI 2 in APP/PS1 transgenic mice will enhance the expression of APP‐1α/1β via the PKA/CREB and JNK/c‐Jun pathways in neuron cells of APP/PS1 transgenic mice, which in turn aggravates the pathogenesis of AD. Moreover, the highly secreted Aβ oligomers from neuron cells are able to reciprocally regulate the expression of APH‐1α/1β, which further aggravate the pathogenesis of AD in vivo. PGI 2, a metabolic product of COX‐2, inhibition by NS398 administration reversed the effects of APP/PS1 overexpression in stimulating the expression of APH‐1α/1β, which potentially contributes to improvement in study ability and decline in cognitive ability in APP/PS1 transgenic mice.
Substantial evidence indicates that prostaglandins, such as PGE2 and 15d‐PGJ2, are important for Aβ deposition and tau tangling, which contribute to the role of COX‐2 in the pathophysiology of AD (Hoshino et al., 2007; Arnaud et al., 2009). However, knowledge concerning the specific function of PGI2 in the human brain is limited. Indeed, the assumption that PGI2 is synthesized in brain tissue in vivo is based on observations in primary culture of astrocytes and meningeal cells (Murphy et al., 1985) as well as of the expression of PGI2 receptor (PI) in the rodent brain (Oida et al., 1995; Takechi et al., 1996). Siegle et al. (Siegle et al., 2000) supported this hypothesis by demonstrating, via IHC and in situ hybridization, that glia and neuron cells in the human brain express PGI2 synthase (PGIS). Moreover, the expression of PGIS in the human brain was supported by the detection of 6‐keto‐PGF1α, a stable degradation product of PGI2, in human brain homogenates by enzyme immunoassay kits (Siegle et al., 2000). Our data agree with this prior work (Siegle et al., 2000) by demonstrating the presence of PGI2 in the brains of C57BL/6 mice. PGI2 production was elevated in the brain of APP/PS1 transgenic mice when compared with that of the WT control. PGI2 synthesis may result from COX‐2 upregulation in APP/PS1 transgenic mice (data not shown). Yosojima et al. (Yasojima et al., 1999) reported that COX‐2 was substantially upregulated in the affected areas of AD brains. In addition, COX‐2 is responsible for the systemic synthesis of PGI2 (McAdam et al., 1999). Therefore, PGI2 synthesis was markedly increased during the course of AD progression.
PGI2 elevation was initially found to be involved in the actions of inflammation (Ford‐Hutchinson et al., 1978). In addition, treatment with PGI2 analogs, including iloprost and treprostinil, suppressed TNF‐α expression in human myeloid dendritic cells (Kuo et al., 2012). Schuh et al. (2014) also reported that the early induction of PGI2 at the site of traumatic injury resulted in the aggregation of IL‐1β‐expressing macrophages as a critical reason for neuropathic pain. Although the effects of these cytokines on Aβ production are still in debating, these prior works have indicated that PGI2 might play its roles in AD via inducing the production of cytokines. Apart from the inflammatory effects of PGI2, the ability of neuron cells to express elevated amounts of PGI2 in APP/PS1 transgenic mice suggests that PGI2 may be important to the pathogenesis of AD. Because there is no report that demonstrates the functional significance of PGI2 in regulating Aβ deposition, we assayed for the synthesis of α‐, β‐, and γ‐secretases in PGI2‐treated neuronal cells. The results demonstrated that the expression of BACE‐1 and APH‐1α/1β was upregulated while ADAM‐10 expression was downregulated in PGI2‐treated n2a cells. Therefore, PGI2 elevation could possibly accelerate the deposition of Aβ1–42 by decreasing the expression of α‐secretase and increasing the expression of β‐ and γ‐secretases. As the roles of ADAM‐10 and BACE‐1 in Aβ deposition have been thoroughly investigated (Niemitz, 2013), we sought to determine the effects of PGI2 in stimulating the expression of APH‐1α/1β. Although there are no reports that demonstrate the effects of PGI2 in inducing the expression of APH‐1α/1β, APH‐1α/1β are required for notch pathway signaling, for γ‐secretase cleavage of β‐APP, and for Aβ protein accumulation in C. elegans (Francis et al., 2002). Indeed, APH‐1 often combines with PEN‐2, nicastrin, and PS to generate an active form of γ‐secretase complex, which is responsible for the cleavage of β‐APP and for the deposition of Aβ (De Strooper, 2003). Once APH‐1 was found in C. elegans (Francis et al., 2002), the APH‐1 complex was confirmed in several experimental models (Gu et al., 2003; Luo et al., 2003; Hansson et al., 2004). Along these lines, APH‐1α/1β might also be involved in regulating the deposition of Aβ1–42 in response to PGI2 stimulation.
PGI2 is important for regulating the expression of APH‐1α/1β, which is regulated by the PKA/CREB and JNK/c‐Jun signaling pathways and leads to Aβ1–42 deposition. Consistent with our observations, Su et al. (2003) reported that H89 treatment suppressed the production of Aβ in cells that have been stably transfected with human APP bearing a ‘Swedish mutation’. They further found that the PKA inhibitor abolishes the mature form of intracellular APP and accumulates the immature form (Su et al., 2003). In addition, Marambaud et al. (Marambaud et al., 1999) found that H89 inhibited the production of Aβ1–40 and Aβ1–42 in HEK293 cells that expressed the APP/PS1 genes. However, these studies were not extended to the expression of β‐ or γ‐secretases. Although the PKA inhibitor has shown similar effects in the suppression of the production of Aβ1–42, the role of H89 in Aβ‐induced memory deficit is not conclusively identified (Amini et al., 2015). In addition to the PKA signaling pathway, the JNK/c‐Jun signaling pathways have also been suggested to be involved in Aβ deposition. For example, Jung et al. (2015) reported that the c‐Jun N‐terminal kinase mediates the effects of auraptene on the production of Aβ by activating γ‐secretase. Shen et al. (2008) supported this observation by showing that JNK‐dependent activation of γ‐secretase is responsible for Aβ deposition in H2O2‐stimualted SH‐SY5Y cells. In detail, γ‐secretase as well as presenilin nicastrin is involved in mediating the effects of SP600125 on suppressing the production of Aβ1–42 (Kuo et al., 2008; Rahman et al., 2012). More importantly, the inhibition of c‐Jun N‐terminal kinase activation reverses the AD phenotype in APP/PS1 mice (Zhou et al., 2015). Along these lines, our data further found that the PKA/CREB and JNK/c‐Jun pathways are important for Aβ deposition by activating APH‐1α/1β in PGI2‐stimulated cells and APP/PS1 mice.
We will focus this discussion on the role of Aβ regulation in the pathogenesis of AD. Interestingly, the expression of APH‐1α/1β was upregulated when we injected (i.c.v.) the CSF of APP/PS1 mice into WT mice. This upregulation was attenuated by the addition of Aβ antibodies. These observations clearly indicate the possible role of CSF‐bound Aβ of APP/PS1 mice in upregulating the expression of APH‐1α/1β. However, previous studies have suggested that the CSF‐bound Aβ1–42 level progressively reduced in patients with AD (Mo et al., 2015). These observations indicate that the total level of Aβ1–42 in the CSF of APP/PS1 mice might not be critical for upregulating the expression of APH‐1α/1β. As noted by Lopez‐Gonzalez et al. (2015), the self‐aggregated characteristics of Aβ1–42 result in the Aβ1–42 oligomers being critical for AD initiation. In agreement with this observation, our data demonstrated that Aβ oligomer injection (i.c.v.) has the ability to stimulate the expression of APH‐1α/1β. More interestingly, NS398 blocked the effects of Aβ oligomers on inducing the expression of APH‐1α/1β. These observations clearly indicated the possible roles of Aβ oligomers in activating COX‐2, which potentially further aggravates AD. In agreement with our hypothesis, Kotilinek et al. (2008) also suggested that possible cross talk exists between COX‐2 and Aβ.
In conclusion, we elucidated the signaling pathway by which PGI2 regulates the expression of APH‐1α/1β in neuron cells of APP/PS1 mice. We found that PGI2 treatment upregulates the synthesis of APH‐1α/1β by activating the PKA/CREB and JNK/c‐Jun signaling pathways, which results in Aβ formation in neuronal cells. Aβ injection (i.c.v.) further stimulates the expression of APH‐1α/1β, which potentially contributes to the pathogenesis of AD.
Experimental procedures
Reagents
Unless otherwise specified, all reagents used for the study were described in the supporting information.
Transgenic mice and treatments
The female wild‐type (WT) or APP/PS1 transgenic mice [B6C3‐Tg (APPswe, PSEN1dE9) 85Dbo/J (Stock Number: 004462)] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Genotyping was performed at 3–4 weeks after birth. In selected experiments, mice at the age of 1 month were treated with NS398 (50 μg kg−1 day−1) for 5 months before determining the expression of APH‐1α/1β. Each group contains 3 mice. The brains of animals in different groups were collected after anesthesia and perfusion as previously described (Yu et al., 2015).
Cerebrospinal fluid collection
Cerebral spinal fluid (CSF) was collected according to a published method (Liu et al., 2004) with minor modifications as previously described (Wang et al., 2015; Yu et al., 2015).
Aβ1–42 preparation
The methods for preparing Aβ oligomers or fibrils had been described previously (Dahlgren et al., 2002; Moore et al., 2002; Pan et al., 2011). The detailed information was provided in the supportive information.
Intracerebroventricular injection (i.c.v.)
NS398, PGI2, Aβ, Aβ antibody, or vehicle (PBS) were injected (i.c.v.) into WT or APP/PS1 transgenic mice as previously described (Yu et al., 2015). Each group contains eight mice. In selected experiments, the WT mice were injected (i.c.v.) with the CSF of APP/PS1 mice. At indicated time intervals, the brains were harvested under anesthesia and perfusion as previously described (Yu et al., 2015).
Organotypic slice culture of brain tissue
Brain tissue was freshly collected from WT C57BL/6 or APP/PS1 transgenic mice at 6 months of age. Serial sections (400 μm thick) were cut using a chopper without fixation. Each group contains eight mice. The tissue sections were immediately cultured in DMEM/high‐glucose medium with 10% FBS. In a separate set of experiments, the tissues were grown in serum‐free medium for an additional 24 h before incubation with PGI2 (10 μm) or Aβ oligomers (1 μm) in the absence or presence of NS398 (10 μm), as previously described (Yu et al., 2015). The tissue sections were fixed and immunostained with APH‐1 antibody by IHC staining kit (Invitrogen, Carlsbad, CA, USA).
Luciferase assays and live animal imaging
The live animal imaging was performed as previously described (Wang et al., 2015; Yu et al., 2015). In brief, the n2a cells that were transfected with APH‐1α/β promoter were preseeded on one side of a cerebral ventricle. PGI2 or vehicle (PBS) solutions were then injected (i.c.v.) into the other cerebral ventricle. At different time intervals, the mice were anesthetized and injected (i.c.v.) with luciferin at the side cerebral ventricle, which was preseeded with n2a cells. Each group contains 6 mice. The scan was performed exactly five min after luciferin introduction. All images were analyzed using Bruker in vivo imaging systems (MS FX PRO, Carestream, Billerica, MA, USA).
Two‐photon imaging
In vivo two‐photon recording was performed as previously described (Wang et al., 2015; Yu et al., 2015). In brief, a custom‐built, two‐photon microscope that was based on a chameleon excitation laser operating at 690–1064 nm was used. The laser‐scanning unit was mounted on an upright microscope that was equipped with a water immersion objective (Zeiss; 20×, Beijing, China). The fluorescence was detected using specific antibody staining. The brain slices were stained and scanned before and after the injection (i.c.v.) of PGI2 or vehicle (PBS) solutions. Each group contains 6 mice.
Quantitative real‐time PCR (qRT–PCR)
qRT–PCR assays were performed with the MiniOpticon Real‐Time PCR detection system (Bio‐Rad, Hercules, CA, USA) using total RNA and the GoTaq one‐step Real‐Time PCR kit with SYBR green (Promega, Madison, WI, USA) and the appropriate primers as previously described (Wang et al., 2010, 2011b,c, 2015,b). The GenBank accession number and forward and reverse primers for human or mouse BACE‐1, APH‐1α, APH‐1β, and GAPDH are provided in our previous publications (Wang et al., 2014c; Guan et al., 2015a,b; Yu et al., 2015). Other primers are shown in Table 2, and the gene expression values were normalized to those of GAPDH.
Table 2.
The primer sequences for α‐, β‐, or γ‐secretases
| Gene symbol | GenBank number | Sequences |
|---|---|---|
| ADAM‐10 | NM_007399 |
F‐ATTGCTGCTTCGATGCCAAC R‐GCACCGCATGAAAACATCAC |
| PS1 | NM_008943 |
F‐GCTTGTAGGCGCCTTTAGTG R‐CATCTGGGCATTCTGGAAGT |
| PS2 | NM_011183 |
F‐AAGAACGGGCAGCTCATCTA R‐TCCAGACAGCCAGGAAGAGT |
| NCT | NM_021607 |
F‐TGTGCAGTGCCCAAATGATG R‐GGCCACATTCCAGAAAAAGGAC |
| PEN2 | NM_025498 |
F‐ACTGAAAACTGCGGCATCTC R‐ATTGGGGCAGATGGGAAATG |
Immunostaining
Human SH‐SY5Y and mouse n2a cells were immunostained as previously described (Wang et al., 2011c). In brief, cells were permeabilized with 0.1% Triton X‐100 for 1 min at 4 °C, fixed with 4% paraformaldehyde for 10 min at 37 °C, washed with PBS (−), and incubated in buffer containing 1% BSA/PBS (−) for 10 min at room temperature. Cells were then incubated with a rabbit antibody to APH‐1 for 60 min at room temperature, washed with 1% BSA/PBS (−), and incubated in buffer containing Alexa Fluor 488‐labeled goat anti‐rabbit IgG for 60 min at room temperature. The cells were then washed five times with 1% BSA/PBS (−) before incubation in DAPI solution for five min. Finally, the cells were washed five times with 1% BSA/PBS (−) and once with deionized water before observation under confocal microscopy (Leica, TCS‐SP8, Liaoning, Shenyang, China).
Human brain samples
Human brain samples were obtained from New York Brain Bank, serial numbers P535‐00 (normal), T4339, T4304, and 235‐95 (patients with AD). Another two normal brain samples were obtained from Fengtian Hospital of China (the patients are 59‐ and 63‐year‐old men who were diagnosed as cerebral edema, and the normal tissues are collected surrounding the tissues of cerebral edema).
The information for immunohistochemistry (IHC), cell culture, Western blot analysis, measurement of the Aβ1–42 or PGI2 concentration in the culture medium or the brain of mice, Transfection and Animal committee, was described in the supporting information.
Statistical analysis
All data are represented as the mean ± SE of at least three independent experiments. The statistical significance of the differences between the means was determined using a Student's t‐test or a one‐way ANOVA, where appropriate. If the means were found to be significantly different, multiple pairwise comparisons were performed using the Tukey's test (Wang et al., 2015,c; Guan et al., 2015a,b; Yu et al., 2015).
Funding
No funding information provided.
Conflict of interest
The authors declare no competing financial interests.
Author contributions
P. W. and P.P.G conceived and performed all of the experiments, participated in the design of the study, and wrote the manuscript. J.W.G., L.L.C., G.B.X., X.Y, and Y.W. carried out some of the experiments. P.W. and Z.Y.W. conceived the experiments, interpreted the data, and wrote the manuscript.
Supporting information
Data S1 Experimental procedures.
Acknowledgments
This work was supported in part or in whole by the National Natural Science Foundation of China (CN) (31571064, 81500934, 31300777 and 31371091), the Fundamental Research Funds of China (N142004002, N130120002, N120520001, N120320001, N141008001/7 and L1520001), the National Natural Science Foundation of Liaoning, China (CN) (2015020662), and the Liaoning Provincial Talent Support Program (LJQ2013029).
References
- Akarasereenont P, Techatrisak K, Chotewuttakorn S, Thaworn A (1999) The induction of cyclooxygenase‐2 in IL‐1beta‐treated endothelial cells is inhibited by prostaglandin E2 through cAMP. Mediators Inflamm. 8, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini E, Nassireslami E, Payandemehr B, Khodagholi F, Foolad F, Khalaj S, Hamedani MP, Azimi L, Sharifzadeh M (2015) Paradoxical role of PKA inhibitor on amyloidbeta‐induced memory deficit. Physiol. Behav. 149, 76–85. [DOI] [PubMed] [Google Scholar]
- Arnaud LT, Myeku N, Figueiredo‐Pereira ME (2009) Proteasome‐caspase‐cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J. Neurochem. 110, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, Kempster S, Williams A (2006) Prostaglandin D2 mediates neuronal damage by amyloid‐beta or prions which activates microglial cells. Neuropharmacology 50, 229–237. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ (2002) Oligomeric and fibrillar species of amyloid‐beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph‐1, Pen‐2, and Nicastrin with Presenilin generate an active gamma‐Secretase complex. Neuron 38, 9–12. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Clerman A, Dore S (2005) Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta‐amyloid exposure. Eur. J. Neurosci. 22, 2199–2206. [DOI] [PubMed] [Google Scholar]
- Ford‐Hutchinson AW, Walker JR, Davidson EM, Smith MJ (1978) PGI2: a potential mediator of inflammation. Prostaglandins 16, 253–258. [DOI] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D (2002) aph‐1 and pen‐2 are required for Notch pathway signaling, gamma‐secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97. [DOI] [PubMed] [Google Scholar]
- van Gool WA, Aisen PS, Eikelenboom P (2003) Anti‐inflammatory therapy in Alzheimer's disease: is hope still alive? J. Neurol. 250, 788–792. [DOI] [PubMed] [Google Scholar]
- Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, Li W, Ruan X, Luthra A, Mount HT, Tandon A, Fraser PE, St George‐Hyslop P (2003) APH‐1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin nicastrin complexes. J. Biol. Chem. 278, 7374–7380. [DOI] [PubMed] [Google Scholar]
- Guan PP, Guo JW, Yu X, Wang Y, Wang T, Konstantopoulos K, Wang ZY, Wang P (2015a) The role of cyclooxygenase‐2, interleukin‐1beta and fibroblast growth factor‐2 in the activation of matrix metalloproteinase‐1 in sheared‐chondrocytes and articular cartilage. Sci. Rep. 5, 10412. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Guan PP, Yu X, Guo JJ, Wang Y, Wang T, Li JY, Konstantopoulos K, Wang ZY, Wang P (2015b) By activating matrix metalloproteinase‐7, shear stress promotes chondrosarcoma cell motility, invasion and lung colonization. Oncotarget 6, 9140–9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M (2004) Nicastrin, presenilin, APH‐1, and PEN‐2 form active gamma‐secretase complexes in mitochondria. J. Biol. Chem. 279, 51654–51660. [DOI] [PubMed] [Google Scholar]
- Honda M, Kitagawa N, Tsutsumi K, Morikawa M, Nagata I, Kaminogo M (2005) Magnetic resonance angiography evaluation of external carotid artery tributaries in moyamoya disease. Surg. Neurol. 64, 325–330. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Homan T, Tanaka K, Sugimoto Y, Araki W, Narita M, Narumiya S, Suzuki T, Mizushima T (2007) Involvement of prostaglandin E2 in production of amyloid‐beta peptides both in vitro and in vivo. J. Biol. Chem. 282, 32676–32688. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Namba T, Takehara M, Nakaya T, Sugimoto Y, Araki W, Narumiya S, Suzuki T, Mizushima T (2009) Prostaglandin E2 stimulates the production of amyloid‐beta peptides through internalization of the EP4 receptor. J. Biol. Chem. 284, 18493–18502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Solfrizzi V, Panza F (2010) Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Front. Aging Neurosci. 2, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CG, Uhm KO, Horike H, Kim MJ, Misumi S, Ishida A, Ueda Y, Choi EK, Kim YS, Michikawa M, Hida H (2015) Auraptene increases the production of amyloid‐beta via c‐Jun N‐terminal kinase‐dependent activation of gamma‐secretase. J. Alzheimers Dis. 43, 1215–1228. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH (2008) Cyclooxygenase‐2 inhibition improves amyloid‐beta‐mediated suppression of memory and synaptic plasticity. Brain 131, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo LH, Hu MK, Hsu WM, Tung YT, Wang BJ, Tsai WW, Yen CT, Liao YF (2008) Tumor necrosis factor‐alpha‐elicited stimulation of gamma‐secretase is mediated by c‐Jun N‐terminal kinase‐dependent phosphorylation of presenilin and nicastrin. Mol. Biol. Cell 19, 4201–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CH, Lin CH, Yang SN, Huang MY, Chen HL, Kuo PL, Hsu YL, Huang SK, Jong YJ, Wei WJ, Chen YP, Hung CH (2012) Effect of prostaglandin I2 analogs on cytokine expression in human myeloid dendritic cells via epigenetic regulation. Mol. Med. 18, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Herukka SK, Minkeviciene R, van Groen T, Tanila H (2004) Longitudinal observation on CSF Abeta42 levels in young to middle‐aged amyloid precursor protein/presenilin‐1 doubly transgenic mice. Neurobiol. Dis. 17, 516–523. [DOI] [PubMed] [Google Scholar]
- Lopez‐Gonzalez I, Schluter A, Aso E, Garcia‐Esparcia P, Ansoleaga B, LLorens F, Carmona M, Moreno J, Fuso A, Portero‐Otin M, Pamplona R, Pujol A, Ferrer I (2015) Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J. Neuropathol. Exp. Neurol. 74, 319–344. [DOI] [PubMed] [Google Scholar]
- Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H (2003) PEN‐2 and APH‐1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 278, 7850–7854. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Ancolio K, Alves da Costa C, Checler F (1999) Effect of protein kinase A inhibitors on the production of Abeta40 and Abeta42 by human cells expressing normal and Alzheimer's disease‐linked mutated betaAPP and presenilin 1. Br. J. Pharmacol. 126, 1186–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdam BF, Catella‐Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA (1999) Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)‐2: the human pharmacology of a selective inhibitor of COX‐2. Proc. Natl Acad. Sci. U.S.A. 96, 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL (2000) Cyclo‐oxygenase‐2 inhibitors: rationale and therapeutic potential for Alzheimer's disease. Drugs Aging 17, 1–11. [DOI] [PubMed] [Google Scholar]
- Mo JA, Lim JH, Sul AR, Lee M, Youn YC, Kim HJ (2015) Cerebrospinal fluid beta‐amyloid1‐42 levels in the differential diagnosis of Alzheimer's disease–systematic review and meta‐analysis. PLoS One 10, e0116802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW (2002) A CD36‐initiated signaling cascade mediates inflammatory effects of beta‐amyloid. J. Biol. Chem. 277, 47373–47379. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS (2001) Interleukin‐1, neuroinflammation, and Alzheimer's disease. Neurobiol. Aging 22, 903–908. [DOI] [PubMed] [Google Scholar]
- Murphy S, Jeremy J, Pearce B, Dandona P (1985) Eicosanoid synthesis and release from primary cultures of rat central nervous system astrocytes and meningeal cells. Neurosci. Lett. 61, 61–65. [DOI] [PubMed] [Google Scholar]
- Niemitz E (2013) ADAM10 and Alzheimer's disease. Nat. Genet. 45, 1273. [Google Scholar]
- Oida H, Namba T, Sugimoto Y, Ushikubi F, Ohishi H, Ichikawa A, Narumiya S (1995) In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br. J. Pharmacol. 116, 2828–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, Chen XC (2011) Microglial phagocytosis induced by fibrillar beta‐amyloid is attenuated by oligomeric beta‐amyloid: implications for Alzheimer's disease. Mol. Neurodegener. 6, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulichino AM, Rowland S, Wu T, Clark P, Xu D, Mathieu MC, Riendeau D, Audoly LP (2006) Prostacyclin antagonism reduces pain and inflammation in rodent models of hyperalgesia and chronic arthritis. J. Pharmacol. Exp. Ther. 319, 1043–1050. [DOI] [PubMed] [Google Scholar]
- Rahman M, Zhang Z, Mody AA, Su DM, Das HK (2012) Intraperitoneal injection of JNK‐specific inhibitor SP600125 inhibits the expression of presenilin‐1 and Notch signaling in mouse brain without induction of apoptosis. Brain Res. 1448, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh CD, Pierre S, Weigert A, Weichand B, Altenrath K, Schreiber Y, Ferreiros N, Zhang DD, Suo J, Treutlein EM, Henke M, Kunkel H, Grez M, Nusing R, Brune B, Geisslinger G, Scholich K (2014) Prostacyclin mediates neuropathic pain through interleukin 1beta‐expressing resident macrophages. Pain 155, 545–555. [DOI] [PubMed] [Google Scholar]
- Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N (2008) Hydrogen peroxide promotes Abeta production through JNK‐dependent activation of gamma‐secretase. J. Biol. Chem. 283, 17721–17730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegle I, Klein T, Zou MH, Fritz P, Komhoff M (2000) Distribution and cellular localization of prostacyclin synthase in human brain. J. Histochem. Cytochem. 48, 631–641. [DOI] [PubMed] [Google Scholar]
- Su Y, Ryder J, Ni B (2003) Inhibition of Abeta production and APP maturation by a specific PKA inhibitor. FEBS Lett. 546, 407–410. [DOI] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Umeki M, Tsuchiya D, Kakimura J, Taniguchi T, Gebicke‐Haerter PJ, Shimohama S (2003) Possible involvement of small oligomers of amyloid‐beta peptides in 15‐deoxy‐delta 12,14 prostaglandin J2‐sensitive microglial activation. J. Pharmacol. Sci. 91, 330–333. [DOI] [PubMed] [Google Scholar]
- Takechi H, Matsumura K, Watanabe Y, Kato K, Noyori R, Suzuki M (1996) A novel subtype of the prostacyclin receptor expressed in the central nervous system. J. Biol. Chem. 271, 5901–5906. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhu F, Lee NH, Konstantopoulos K (2010) Shear‐induced interleukin‐6 synthesis in chondrocytes: roles of E prostanoid (EP) 2 and EP3 in cAMP/protein kinase A‐ and PI3‐K/Akt‐dependent NF‐kappaB activation. J. Biol. Chem. 285, 24793–24804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Zheng W, Wang T, Xie JW, Wang SL, Zhao BL, Teng WP, Wang ZY (2011a) Huperzine A activates Wnt/beta‐catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 36, 1073–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhu F, Konstantopoulos K (2011b) Interleukin‐6 synthesis in human chondrocytes is regulated via the antagonistic actions of prostaglandin (PG)E2 and 15‐deoxy‐Delta(12,14)‐PGJ2. PLoS One 6, e27630. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang P, Zhu F, Tong Z, Konstantopoulos K (2011c) Response of chondrocytes to shear stress: antagonistic effects of the binding partners Toll‐like receptor 4 and caveolin‐1. FASEB J. 25, 3401–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Chen SH, Hung WC, Paul C, Zhu F, Guan PP, Huso DL, Kontrogianni‐Konstantopoulos A, Konstantopoulos K (2015) Fluid shear promotes chondrosarcoma cell invasion by activating matrix metalloproteinase 12 via IGF‐2 and VEGF signaling pathways. Oncogene 34, 4558–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Guan PP, Wang T, Yu X, Guo JJ, Konstantopoulos K, Wang ZY (2014b) Interleukin‐1beta and cyclic AMP mediate the invasion of sheared chondrosarcoma cells via a matrix metalloproteinase‐1‐dependent mechanism. Biochim. Biophys. Acta 1843, 923–933. [DOI] [PubMed] [Google Scholar]
- Wang P, Guan PP, Wang T, Yu X, Guo JJ, Wang ZY (2014c) Aggravation of Alzheimer's disease due to the COX‐2‐mediated reciprocal regulation of IL‐1beta and Abeta between glial and neuron cells. Aging Cell 13, 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang P, Yu X, Guan PP, Guo JW, Wang Y, Zhang Y, Zhao H, Wang ZY (2015) Magnesium ion influx reduces neuroinflammation in Abeta precursor protein/Presenilin 1 transgenic mice by suppressing the expression of interleukin‐1beta. Cell. Mol. Immunol. doi: 10.1038/cmi.2015.93. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Takase K, Kishino J, Fujita M, Okamura N, Sakaeda T, Fujimoto M, Yagami T (2011) Proteomic identification of protein targets for 15‐deoxy‐Delta(12,14)‐prostaglandin J2 in neuronal plasma membrane. PLoS One 6, e17552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL (1999) Distribution of cyclooxygenase‐1 and cyclooxygenase‐2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 830, 226–236. [DOI] [PubMed] [Google Scholar]
- Yu X, Guan PP, Guo JW, Wang Y, Cao LL, Xu GB, Konstantopoulos K, Wang ZY, Wang P (2015) By suppressing the expression of anterior pharynx‐defective‐1alpha and ‐1beta and inhibiting the aggregation of beta‐amyloid protein, magnesium ions inhibit the cognitive decline of amyloid precursor protein/presenilin 1 transgenic mice. FASEB J. 29, 5044–5058. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang M, Du Y, Zhang W, Bai M, Zhang Z, Li Z, Miao J (2015) Inhibition of c‐Jun N‐terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 77, 637–654. [DOI] [PubMed] [Google Scholar]
- Zhuang J, Zhang H, Zhou R, Chen L, Chen J, Shen X (2013) Regulation of prostaglandin F2alpha against beta amyloid clearance and its inflammation induction through LXR/RXR heterodimer antagonism in microglia. Prostaglandins Other Lipid Mediat. 106, 45–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Experimental procedures.