

Abstract
Reactive multi-target ‘fragment drugs’ represent critical components of current tuberculosis regimens. These compounds, such as pyrazinamide, are old synthetic antimycobacterials that are activated inside Mycobacterium tuberculosis bacilli and are smaller than the usual drug-like, single-target molecules. Based on the success of small ‘dirty’ drugs in the chemotherapy of tuberculosis, we suggested previously that fragment-based whole cell screens should be introduced in our current antimycobacterial drug discovery efforts. Here, we carried out such a screen and characterized bactericidal activity, selectivity and spectrum of hits we obtained. A library of 1725 fragments was tested at a single concentration for growth inhibitory activity against M. bovis BCG as screening strain and 38 of 116 primary hits were confirmed in dose response analyses to be active against virulent M. tuberculosis. Bacterial kill experiments showed that most hits displayed bactericidal activity at their minimal inhibitory concentration. Cytotoxicity assays established that a large proportion of hits displayed a favorable selectivity index for mammalian cells. Importantly, one third of M. tuberculosis active fragments were also active against M. abscessus and M. avium, two emerging non-tuberculous mycobacterial (NTM) pathogens, opening the opportunity to develop broad spectrum antimycobacterials. Activity determination against Gram positive (Staphylococcus aureus) and Gram negative (Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa) bacteria, as well as fungi (Candida albicans, Cryptococcus neoformans) showed only a small overlap indicating a generally narrow spectrum of these novel antimicrobial hits for mycobacteria. In conclusion, we carried out the first fragment-based whole cell screen against bacteria and identified a substantial number of hits with excellent physicochemical properties and dual activity against M. tuberculosis and NTM pathogens. These hits will now be evaluated in animal models of mycobacterial infection to determine whether any of them can be moved forward as a new antimycobacterial fragment drug candidate.
Keywords: fragments, poly-pharmacology, tuberculosis, NTM, M. abscessus, M. avium
Introduction
Mycobacterium tuberculosis remains the most deadly bacterial pathogen globally and acquired resistance to current antimycobacterials threatens global tuberculosis control efforts (WHO, 2015). In addition to the tuberculosis pandemic, new mycobacterial diseases are emerging. The number of infections with intrinsically resistant environmental NTM such as M. abscessus and M. avium are increasing in both developing and developed countries. While prolonged chemotherapies are required for treating M. avium infections, lung disease caused by M. abscessus is considered incurable (Griffith et al., 2007; Nessar et al., 2012; Aksamit et al., 2014; Park and Olivier, 2015). Hence, there is an urgent medical need for new drugs with new mechanisms of action – and with a broad antimycobacterial spectrum of activity.
Current drug discovery strategies are largely based on Paul Ehrlich’s magic bullet ‘one drug–one target’ concept. After a decade of high throughput screening against isolated biochemical targets the antimycobacterial discovery field largely moved back to phenotypic whole cell screens to identify compounds with antimicrobial activity first, then deconvolute the target via selection of spontaneous resistance mutants and whole genome sequencing. This strategic shift is due to the large scale failure of target-based approaches. It turned out that the double membrane-containing mycobacterial cell envelope is a formidable permeability barrier. Enzyme inhibitors identified on isolated targets often do not penetrate this mycobacterial fortress (Payne et al., 2007; Barry et al., 2009; Gwynn et al., 2010). The low permeability of the mycobacterial cell envelope also affects the productivity of phenotypic whole cell screens which use the same standard pharmaceutical chemical libraries employed for target-based screens. Screening those drug-like, rule-of-five compliant (Lipinski, 2000) archives has resulted in very low hit rates (Barry et al., 2009; Campbell, 2011).
Interestingly, critical components of anti-tuberculosis regimens, including the key sterilizing first line drug pyrazinamide, are ‘dirty fragments’: they hit multiple targets, i.e., they are ‘dirty,’ and their molecular weights are in the range of 100–300 g/mol, i.e., they are ‘fragments.’ In addition, these drugs are metabolized inside the tubercle bacillus. Only then, after being ‘activated,’ they exert their antimicrobial activity (Gopal and Dick, 2014). This type of mechanism of action (poly-pharmacology), and physicochemical properties (‘extra’ small and reactive), are at odds with main stream antibacterial drug discovery: attractive leads for medicinal chemistry should inhibit a single target, have a decent size to bind a target with high affinity, and should not be reactive to avoid side effects (Dartois and Barry, 2010).
Fragments came to fame in modern drug discovery due to a novel, ‘fragment-based’ screening approach against molecular targets. This lead finding method makes use of very small, rule-of-3 compliant (Congreve et al., 2003) molecules, with weak binding affinities. Biophysical and structural biology methods are employed to identify such weak binders to a particular target and their modes of binding. The low affinity binders are then extended or combined to develop tight, target-specific, high affinity (nM) compounds. Fragments usually represent attractive starting points for medicinal chemistry as they possess favorable physicochemical and hence pharmacokinetic properties: they are relatively small (<300 g/mol) and rather hydrophilic (clogP < 3) and thus combine attractive water solubility with good absorption and tissue distribution (Scott et al., 2012; Hopkins et al., 2014).
Fragment drugs such as pyrazinamide which are also pro-drugs, are among the most efficacious tuberculosis drugs. Thus hitting multiple targets appears to be an attractive property for antimycobacterials. Furthermore, fragments which are small enough to bind multiple sites are more likely to be accepted as substrate analogs as well and metabolized by the bacterium, again a property that seems to make good antimicrobials for tuberculosis. Being small and moderately lipophilic is critical in achieving in vivo exposure and tissue penetration, usually a major hurdle during lead optimization (Dartois and Barry, 2013; Dartois, 2014). In the case of mycobacteria, these physicochemical properties likely also have a positive impact on cellular pharmacokinetics: fragments might more easily penetrate the double membrane mycobacterial cell envelope as porins, the channels spanning the outer membrane, prefer small hydrophilic molecules. Thus, fragments may have multiple advantages over larger molecules: favorable absorption and systemic pharmacokinetic properties, favorable tissue distribution and better bacterial uptake (Gopal and Dick, 2014).
Based on these considerations we decided to screen a fragment library, typically only used for structure guided lead finding, for hits showing whole cell activity against the tubercle bacillus. We identified 38 fragments showing growth inhibitory activity against M. tuberculosis. We characterized the hit molecules regarding their bactericidal and cytotoxic activity. Furthermore, the hits were profiled for their antimycobacterial, anti-Gram positive/negative and their antifungal spectrum.
Materials and Methods
Assembly of Fragment Library
Fragments were selected based on a computational filtration of 90,000 commercially available molecules from libraries supplied by ChemBridge, Life Chemicals, Enamine, Bionet/Key Organics and Maybridge. The rule-of-3 was applied (Congreve et al., 2003) with additional selections up to molecular weight of 350 for dense functionalities such as sulphonamides. The principle of positive selection of desired hydrogen bonding groups and computational clustering described by Baurin et al. (2004) were also applied to ensure diverse proportional clusters representing a range of polar and solubilising groups. Then a categorization of candidate fragments was made with 0, 1, 2, or 3 H-bond donors, 0, 1, or 2 positively charged nitrogen groups, and up to 1 negatively charged group, the latter being a smaller set. Specific undesired groups were removed such as non-aromatic N–N bonded compounds (e.g., hydrazines), primary anilines, ureas/thioureas, adamantanes, bromo or iodo containing compounds and more than 2 chloro groups. From the approximately 6,000 remaining fragments, further prioritization was carried out with a focus on structures which were not completely flat and had less commonly encountered combinations of functionalities. This resulted in a library of 1725 selected fragment compounds used for whole cell screening described in this report. All fragments are commercially available and the corresponding compound codes of the hits identified in this work are given in Supplementary Table S1.
Bacterial Strains, Cells, Culture Conditions and Chemicals
Mycobacterium tuberculosis H37Rv (ATCC 27294), M. bovis BCG (ATCC 35734), M. abscessus Bamboo (smooth morphotype) and M. avium 11 liquid cultures were grown in Middlebrook 7H9 broth (BD Difco) supplemented with 0.5% albumin, 0.2% glucose, 0.085% sodium chloride, 0.5% glycerol, 0.05% Tween80 as described previously (Lakshminarayana et al., 2015). Escherichia coli (ATCC 25922), Klebsiella pneumoniae (ATCC 700603, MDR), Acinetobacter baumannii (ATCC 19606), Pseudomonas aeruginosa (ATCC 27853), Staphylococcus aureus (ATCC 43300, MRSA), were cultured in cation-adjusted Mueller Hinton broth at 37°C overnight. Candida albicans (ATCC 90028), Cryptococcus neoformans (ATCC 208821) were cultured for 3 days on Yeast Extract-Peptone Dextrose agar at 30°C. A yeast suspension of 106 to 5 × 106 cells/mL (as determined by OD530, absorbance at 530 nm) was prepared from five colonies and subsequently used for screening. HepG2 (ATCC HB.8065) and A549 (ATCC CCL-185) cells were cultured at 37°C with 5% CO2 atmosphere in DMEM media (Gibco) complemented with 10% FBS heat-inactivated (Gibco), penicillin (100 U/mL, Gibco) and streptomycin (100 μg/mL, Gibco). Red blood cells were obtained from Interstate Blood Bank, Inc. Laboratory, USA. Fragments were obtained from various chemical providers shown in Supplementary Table S1. All experiments on M. tuberculosis were conducted in a BSL-3 core facility following biosafety level 3 procedures.
Antimicrobial and Antifungal Assays
Antimycobacterial screens were carried out as follows. All mycobacterial precultures were harvested at mid-log phase and diluted to an OD600 (absorbance at 600 nm) of 0.05 in complete 7H9 medium. M. bovis BCG bacterial suspensions were then dispensed into 96-well plates (200 μL/well) with a single concentration of 500 μM of fragment per well in the primary screen. Plates were incubated for 5 days at 37°C under shaking (100 rpm). Compounds causing growth inhibition >80% as compared to untreated control in the same plate were considered positive hits. Cells were manually resuspended and OD was measured at 600 nm on M200Pro plate reader (Tecan) (Murugasu-Oei and Dick, 2000; Gopal and Dick, 2015; Lakshminarayana et al., 2015). Hits that were re-confirmed in the same assay from re-ordered fresh powder stock were then tested against M. tuberculosis in a dose-response format (twofold serial dilution from a maximum concentration of 500 μM). Active hits were subsequently tested against M. abscessus and M. avium in a similar manner. M. abscessus and M. avium containing 96-well plates were incubated for 3 and 5 days respectively at 37°C under shaking (100 rpm). Ten micromolar ciprofloxacin was used as a positive control for M. tuberculosis and M. bovis BCG. Hundred micromolor kanamycin was used as a positive control for M. abscessus and M. avium. Bactericidal activity against M. bovis BCG was determined by exposing approximately 106 CFU to MIC90 concentration for 5 days. CFU were then recovered on agar plates for enumeration and compared to CFU recovered from inoculum as described previously (Murugasu-Oei and Dick, 2000; Lakshminarayana et al., 2015). Fold-kill was calculated as the reduction in CFU in treated samples as compared to inoculum. All experiments were carried out two times independently and mean values are shown. Standard deviations were smaller than 20%.
Antimicrobial screening against gram positive and negative bacteria as well as fungi was performed by the Community for Antimicrobial Drug Discovery, University of Queensland, funded by the Wellcome Trust (UK) and The University of Queensland (Australia). A sample of an overnight culture of the respective test bacterium was diluted 40-fold in fresh broth and incubated at 37°C for 1.5–3 h. The resultant mid-log phase cultures were diluted (CFU/mL measured by OD600), then 45 μL was added to each well of the compound containing plates, giving a cell density of 5 × 105 CFU/mL. The ten best compounds were tested in a twofold serial dilution format at a maximum concentration of 100 μM. All the plates were covered and incubated at 37°C for 18 h without shaking. Inhibition of bacterial growth was determined measuring absorbance at 600 nm (OD600), using a Tecan M1000 Pro monochromator plate reader. The percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (bacteria without inhibitors) on the same plate as references. Colistin and Vancomycin were used as positive bacterial inhibitor standards for Gram negative and Gram-positive bacteria, respectively, at four concentrations including two above and two below the MIC of the respective pathogens. All experiments were carried out two times independently and mean values are shown. Standard deviations were smaller than 20%.
Fungal stock suspensions described above were diluted with Yeast Nitrogen Base broth to a final concentration of 2.5 × 103 CFU/mL. Then, 45 μL of the fungi suspension was added to each well of the compound-containing plates. The ten best compounds were tested in a twofold serial dilution format at a maximum concentration of 100 μM. Plates were covered and incubated at 35°C for 24 h without shaking. Growth inhibition of C. albicans was determined measuring absorbance at 530 nm (OD530), while the growth inhibition of C. neoformans was determined measuring the difference in absorbance between 600 and 570 nm (OD600-570), after the addition of resazurin (0.001% final concentration) and incubation at 35°C for additional 2 h. The absorbance was measured using a Biotek Synergy HTX plate reader. The percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (fungi without inhibitors) on the same plate. Fluconazole was used as a positive fungal inhibitor standard for C. albicans and C. neoformans at four concentrations including two above and two below the MIC. All experiments were carried out two times independently and mean values are shown. Standard deviations were smaller than 20%.
Cytotoxicity Assays
The MTS [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reduction assay (Promega) was used according to manufacturer’s guidelines to assess HepG2 or A549 cell viability after exposure to a range of compound concentrations (twofold serial dilution) (Riss et al., 2004). Absorbance was read with a Tecan M200Pro plate reader at 570 nm. Hundred micromolor of carbonyl cyanide m-chlorophenylhydrazone was used as a cytotoxic positive control. Hemolytic activity was evaluated by exposing 106 red blood cells to two-fold serial dilutions of compounds in phosphate-buffered saline for 24 h. Red blood cells were subsequently centrifuged at 1000 g for 10 min and the supernatant was collected and homogenized by vortexing. Absorbance was read at 540 nm. A Triton X-100 treated control (2% Triton) was used to define 100% of hemoglobin release (Dausset and Contu, 1967). All experiments were carried out two times independently and mean values are shown. Standard deviations were smaller than 20%.
Physico-Chemical Characterization of Hits
Molecular properties of the fragment hits were calculated using Molinspiration software1. Hydrogen bond donors are defined as the total number of OH + NH groups. Hydrogen bond acceptors are defined as the O + N count. Calculated LogP (cLogP) was determined using the miLogP2.2 (November 2005) software from Molinspiration. Topological Polar Surface Area (TPSA) (Ertl et al., 2000) is defined as the sum of the surfaces of the polar atoms in a molecule. Number of rotatable bonds (NROT) is defined as the count of any single non-ring bond, bounded to a non-hydrogen atom; NROT does not include amide bonds which are considered non-rotatable.
Results
Identification of Hits Active against M. tuberculosis from a Fragment Library
A library of 1725 fragments was carefully selected out of a collection of 90,000 commercially available fragments based on their physicochemical and chemical properties (see Material and Methods). We first screened the library against our biosafety level 2 screening strain M. bovis BCG as a surrogate for M. tuberculosis in order to identify hits with whole-cell activity.
The screen was carried out using a single concentration of 500 μM of the test compounds. Growth inhibition of at least 80% was defined as cut-off which resulted in 184 primary hits (11% hit rate). These hits were re-purchased as powders and the compounds were re-tested in the same single point 500 μM assay resulting in 116 confirmed hits (7% hit rate). For these 116 hits, growth inhibition dose response was measured against M. tuberculosis and compounds with an MIC50 < 500 μM were defined as whole cell actives, resulting in 38 hits (2% hit rate). A cut-off of 500 μM was derived from the MIC50 of the anti-tuberculosis fragment drug pyrazinamide which was selected as a benchmark (Via et al., 2015). The screening cascade and hit confirmation is depicted in Figure 1. Figure 2 and Table 1 show the structures and the MIC50 of the most potent M. tuberculosis active hits. Table 2 describes in detail their physicochemical properties. The structures and activities of all 38 hits are shown in Supplementary Table S1.
FIGURE 1.
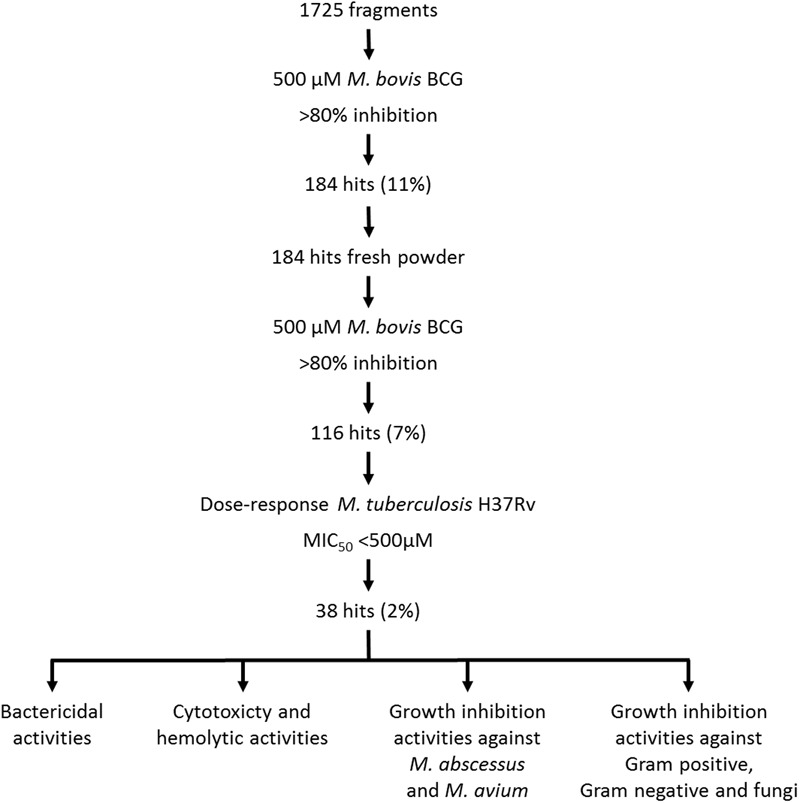
Screening cascade and hit characterization. (Upper part) Screening cascade with associated number of hits. (Bottom part) Various hits characterization activities. For details see Results.
FIGURE 2.
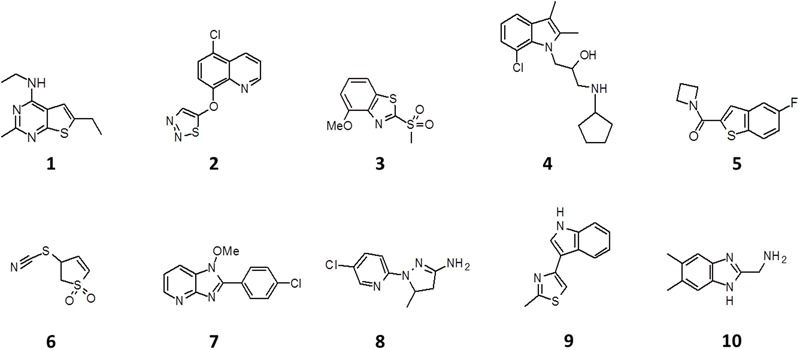
Structure of the 10 most potent M. tuberculosis actives. See Table 1 for antibacterial, antifungal and cytotoxic activities.
Table 1.
Antibacterial, antifungal and cytotoxic activities of the 10 most potent M. tuberculosis actives.
|
M. tuberculosis |
M. bovis BCG |
M. abscessus |
M. avium |
S. aureus |
E. coli |
K. pneumoniae |
A. baumannii |
P. aeruginosa |
C. albicans |
C. neoformans |
HepG2 |
A549 |
RBC |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | MIC50 | MIC50 | Fold kill at MIC90a | MIC50 | Growth Inhibition Spectrum (MIC80)b | CC50c | SI | CC50c | SI | HC50c | SI | |||||||
| 1 | 40 | 5 | 100 | >500 | >500 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | i | >24 | i | >24 | i | >24 |
| 2 | 43 | 56 | 100 | >500 | 130 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 200 | 5 | i | >7 | 250 | 6 |
| 3 | 52 | 20 | nd | >500 | >500 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | nd | nd | nd | nd | i | >13 |
| 4 | 55 | 56 | 100 | 125 | 53 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 45 | 1 | 60 | 1 | 125 | 2 |
| 5 | 60 | 68 | 10 | >500 | >500 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | i | >41 | i | >41 | i | >41 |
| 6 | 92 | 53 | 100 | >500 | >500 | 100 | >100 | >100 | 100 | >100 | 100 | >100 | 75 | 1 | 1000 | 11 | 2200 | 24 |
| 7 | 104 | 19 | 10 | 150 | 145 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 850 | 8 | 1800 | 17 | i | >23 |
| 8 | 123 | 13 | 100 | >500 | 275 | >100 | 100 | >100 | 100 | >100 | 100 | >100 | 900 | 7 | i | >20 | i | >20 |
| 9 | 126 | 39 | 10 | >500 | >500 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 500 | 4 | i | >20 | i | >18 |
| 10 | 131 | 53 | 10 | >500 | >500 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 900 | 7 | 1900 | 15 | 2100 | 16 |
Activities of hits against mycobacteria, Gram positive and negative bacteria, fungi and mammalian cells are shown. Concentrations are reported in micromolar (μM). MIC50, MIC80, MIC90, minimum inhibitory concentration that inhibits 50, 80, and 90% of bacterial growth, respectively. MIC90 were in general 1–5 × MIC50. CC50, HC50, cytotoxic and hemolytic concentrations that kill or lyse 50% of cells as compared to untreated control, respectively. SI, selectivity index as defined by CC50 or HC50/MIC50. RBC, red blood cells. nd, not determined. afold-kill as compared to untreated control; ball top 10 hits showed an MIC80 ≥ 32 μg/mL (or approximately 100 μM), the highest concentration tested; ci, inactive denotes no cytotoxic or hemolytic activity at five times the MIC90; All experiments were carried out two times independently and mean values are shown. Standard deviations were smaller than 20%.
Table 2.
Physico-chemical properties of the 10 most potent M. tuberculosis actives.
| Fragments # | MW | HBD1 | HBA2 | clogP3 | TPSA4 | NROT5 |
|---|---|---|---|---|---|---|
| 1 | 221.3 | 1 | 3 | 2.54 | 37.8 | 3 |
| 2 | 263.7 | 0 | 4 | 3.30 | 47.9 | 2 |
| 3 | 243.3 | 0 | 4 | 1.98 | 56.3 | 2 |
| 4 | 320.9 | 2 | 3 | 4 | 37.2 | 5 |
| 5 | 235.3 | 0 | 2 | 1.88 | 20.3 | 1 |
| 6 | 175.2 | 0 | 3 | 0.04 | 57.9 | 1 |
| 7 | 259.7 | 0 | 4 | 3.35 | 40.0 | 2 |
| 8 | 210.7 | 2 | 4 | 1.76 | 54.5 | 1 |
| 9 | 214.3 | 1 | 2 | 2.41 | 28.7 | 1 |
| 10 | 175.2 | 3 | 3 | 1.52 | 54.7 | 1 |
MW, molecular weight; HBD, Hydrogen Bond Donor; HBA, Hydrogen Bond Acceptor; 1Properties calculated using Molinspiration software (www.molinspiration.com); HBD, OH + NH count. 2HBA, O + N count. cLogP, partition coefficient between n-octanol and water log; 3cLogP, calculated LogP (miLogP2.2 – November 2005). 4TPSA, Topological Polar Surface Area. 5NROT, number of rotatable bonds.
Bactericidal Activities and Cytotoxicity of Hits
To determine the bactericidal activities of the hits we exposed tubercle bacilli to MIC90 concentrations of the respective compounds and counted CFU after 5 days of treatment. Table 1; Supplementary Table S1 shows that more than half of the hits reduced the number of CFU at least 100-fold showing that most fragment hits displayed strong cidal activity. To evaluate in vitro tolerability of the 38 M. tuberculosis actives we measured their cytotoxicity against two mammalian cell lines as well as their membrane toxicity using a red blood cell lysis assay. Table 1; Supplementary Table S1 show that half of the hits showed acceptable cytotoxic and hemolytic activity with a selectivity index of more than 5.
Activity of M. tuberculosis Actives against M. abscessus and M. avium
To establish the spectrum of activity of the 38 M. tuberculosis active hits against other mycobacteria we measured their growth inhibition activity against the fast growing M. abscessus and the slow growing M. avium. Table 1; Supplementary Table S1 shows that two thirds of the M. tuberculosis actives were also active against M. avium, while one third showed activity against M. abscessus. Figure 3 shows that the 13 M. abscessus active hits in fact represent a subset of the M. avium actives.
FIGURE 3.
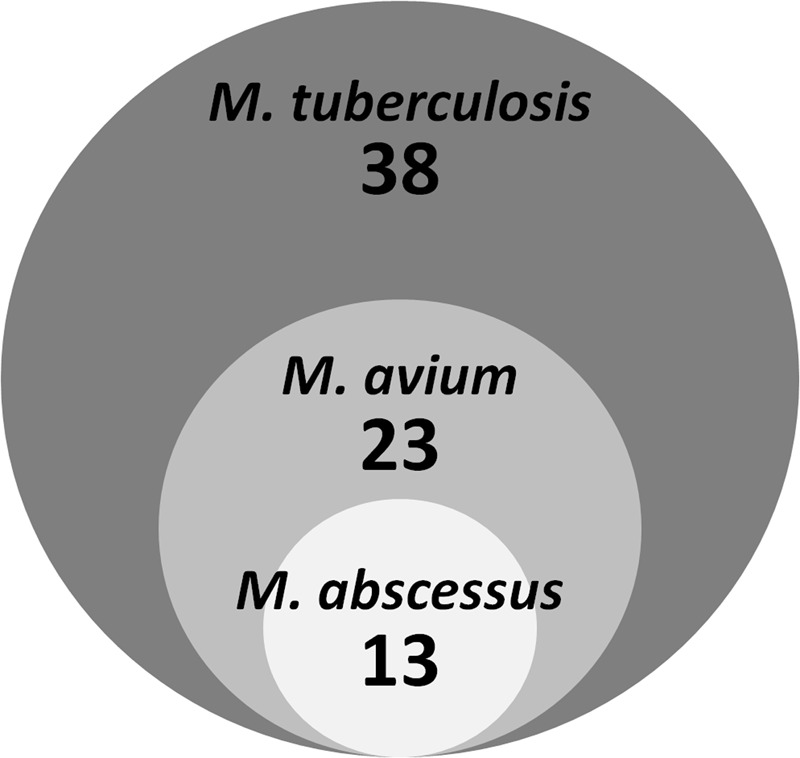
Activity of M. tuberculosis actives against M. avium and M. abscessus. The diagram shows overlapping activity of 38 M. tuberculosis hits against two non-tuberculous mycobacterial species.
Activity of M. tuberculosis Actives against Gram Positive and Gram Negative Pathogens and Fungi
To examine whether the M. tuberculosis actives have a mostly Mycobacterium-specific range of activity or tend to display broad spectrum activity we determined their growth inhibition against Gram positive (Staphylococcus aureus) and negative (Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa) bacteria, as well as fungi (Candida albicans, Cryptococcus neoformans). Table 1; Supplementary Table S1 show that only a small overlap was observed indicating a generally narrow spectrum of the fragment hits for mycobacteria.
Discussion
Based on the success of pyrazinamide and other ‘dirty’ fragment drugs in the treatment of tuberculosis patients, we previously proposed that fragment-based whole cell screens, currently not part of mainstream drug discovery efforts, should be included in antimycobacterial discovery activities (Gopal and Dick, 2014). Here we carried out such a screen of 1725 compounds and identified 38 rule-of-3 compliant fragments (hit rate 2%) with growth inhibitory activity against M. tuberculosis. When compared to the hit rate of standard pharmaceutical libraries (<0.5%; Campbell, 2011) this higher hit rate is consistent with our expectation that small hydrophilic fragment compounds should have favorable bacterial cell penetration properties compared to larger and more hydrophobic molecules (Gopal and Dick, 2014). Interestingly, most hits showed strong mycobactericidal activity while a large fraction retained an acceptable selectivity index for mammalian cells. All hits displayed excellent physic-chemical properties and low molecular weight but no particular structure-activity relationship could be established. General pathogen spectrum profiling of the 38 anti-tuberculosis hits against Gram positive and negative bacteria and against fungi revealed a limited overlap suggesting that most of the hits are rather Mycobacterium-specific.
Importantly, a substantial proportion of M. tuberculosis hits also exhibited activity against NTM pathogens, specifically M. abscessus and M. avium. New drugs to treat lung disease caused by NTM represent an new urgent medical need as M. avium causes a large number of infections requiring prolonged treatments, and M. abscessus infections, although smaller in number, are considered incurable (Griffith, 2011). The identification of hits active against both M. tuberculosis and NTM opens the opportunity to progress anti M. tuberculosis hits as broad spectrum antimycobacterials.
The identified antimycobacterial fragment hits will now be moved forward following two different strategies. Firstly, we will ask whether we can ‘identify the next pyrazinamide,’ i.e., a new antimycobacterial fragment drug. To explore this possibility we will directly move hits into efficacy studies using mouse models of mycobacterial infection, after selecting a subset of compounds showing acceptable in vivo tolerability and pharmacokinetics. If metabolic stability turns out to be an issue, minor chemical modifications will be carried out to determine whether this property can be improved without affecting potency. The objective of this first strategy is to determine whether any of the fragment hits can be moved more or less directly, i.e., without a major lead optimization program, to preclinical development as a fragment drug (Dick et al., 2007). In a second strategy we ask whether we can use any of these rule-of-3 compliant fragment hits to generate a more standard drug-like molecule, i.e., a rule-of-5 compliant, high affinity, high potency, single target compound. Due to their fragment nature, all hits have excellent physico-chemical properties and hence represent attractive starting points for lead optimization campaigns.
In parallel to the two pronged drug discovery program, the mechanism of action of the fragment hits will be determined. It appears likely, due to their nature as promiscuous binders, that the new antimycobacterials have multiple targets, i.e., that they act as ‘dirty’ drugs. Furthermore, due to their potential to act as enzyme substrates, and similarly to other anti-TB fragment drugs such as isoniazid (INH), ethionamide (ETH), para-amino salicylic acid (PAS) or pyrazinamide (PZA), it is likely that a subset of the 38 fragment hits will be metabolized and ‘activated’ by the bacilli, i.e., that they act as reactive ‘dirty’ drugs (Gopal and Dick, 2014). Dissecting the mechanisms of action of existing tuberculosis fragment drugs resulted in the identification of several novel targets and novel concepts of antimicrobial action (Gopal and Dick, 2014). Hence we expect that the mechanism of action studies of this collection of novel, mostly bactericidal fragments may uncover novel antimicrobial concepts.
Conclusion
We carried out the first fragment-based whole cell screen against mycobacteria and identified a substantial number of hits with excellent physicochemical properties which represent a rich source for antimycobacterial drug discovery and chemical biology projects.
Author Contributions
WM and TD conceived and designed the project. WM, JJL, SYY, and PMR carried out the experiments. WM, BWD, and TD wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research is supported by the Singapore Ministry of Health’s National Medical Research Council under its TCR Flagship grant NMRC/TCR/011-NUHS/2014 and Centre Grant ‘MINE,’ Research Core #4 NMRC/CG/013/2013 to TD and is part of Singapore Programme of Research Investigating New Approaches to Treatment of Tuberculosis (SPRINT-TB; http://www.sprinttb.org) managed by Kristina Rutkute and led by Nick Paton. BD and PR’s work is supported by the National University of Singapore’s Drug Development Unit grant R-711-000-022-133. We thank Sabai Phyu and Martin Gengenbacher, Yong Loo Lin School of Medicine BSL3 core facility, for support. Screening of the M. tuberculosis actives against Gram positive/negative bacteria and fungi was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (UK) and The University of Queensland (Australia), and we would like to thank Johannes Zuegg, Mathilde Desselle, and Alysha Elliott for their contribution.
Abbreviations
- CFU
colony forming units
- NTM
non-tuberculous mycobacteria
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01392
References
- Aksamit T. R., Philley J. V., Griffith D. E. (2014). Nontuberculous mycobacterial (NTM) lung disease: the top ten essentials. Respir. Med. 108 417–425. 10.1016/j.rmed.2013.09.014 [DOI] [PubMed] [Google Scholar]
- Barry C. E., III, Boshoff H. I., Dartois V., Dick T., Ehrt S., Flynn J., et al. (2009). The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7 845–855. 10.1038/nrmicro2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurin N., Aboul-Ela F., Barril X., Davis B., Drysdale M., Dymock B., et al. (2004). Design and characterization of libraries of molecular fragments for use in NMR screening against protein targets. J. Chem. Inf. Comput. Sci. 44 2157–2166. 10.1021/ci049806z [DOI] [PubMed] [Google Scholar]
- Campbell J. (2011). High-throughput assessment of bacterial growth inhibition by optical density measurements. Curr. Protoc. Chem. Biol. 3:100115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M., Carr R., Murray C., Jhoti H. (2003). A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 8 876–877. 10.1016/S1359-6446(03)02831-9 [DOI] [PubMed] [Google Scholar]
- Dartois V. (2014). The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12 159–167. 10.1038/nrmicro3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V., Barry C. E. (2010). Clinical pharmacology and lesion penetrating properties of second- and third-line antituberculous agents used in the management of multidrug-resistant (MDR) and extensively-drug resistant (XDR) tuberculosis. Curr. Clin. Pharmacol. 5 96–114. 10.2174/157488410791110797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V., Barry C. E., III. (2013). A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorg. Med. Chem. Lett. 23 4741–4750. 10.1016/j.bmcl.2013.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dausset J., Contu L. (1967). Drug-induced hemolysis. Annu. Rev. Med. 18 55–70. 10.1146/annurev.me.18.020167.000415 [DOI] [PubMed] [Google Scholar]
- Dick T., Dartois V., Keller T., Matter A. (2007). “TB drug discovery from target identification to proof of concept studies,” in Handbook of Tuberculosis: Clinics, Diagnostics, Therapy and Epidemiology eds Kaufmann S. H. E., van Helden P. (Weinheim: Wiley-VCH; ) 139–159. [Google Scholar]
- Ertl P., Rohde B., Selzer P. (2000). Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43 3714–3717. 10.1021/jm000942e [DOI] [PubMed] [Google Scholar]
- Gopal P., Dick T. (2014). Reactive dirty fragments: implications for tuberculosis drug discovery. Curr. Opin. Microbiol. 21 7–12. 10.1016/j.mib.2014.06.015 [DOI] [PubMed] [Google Scholar]
- Gopal P., Dick T. (2015). The new tuberculosis drug Perchlozone® shows cross-resistance with thiacetazone. Int. J. Antimicrob. Agents 45 430–433. 10.1016/j.ijantimicag.2014.12.026 [DOI] [PubMed] [Google Scholar]
- Griffith D. E. (2011). The talking Mycobacterium abscessus blues. Clin. Infect. Dis. 52 572–574. 10.1093/cid/ciq252 [DOI] [PubMed] [Google Scholar]
- Griffith D. E., Aksamit T., Brown-Elliott B. A., Catanzaro A., Daley C., Gordin F., et al. (2007). An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 175 367–416. 10.1164/rccm.200604-571ST [DOI] [PubMed] [Google Scholar]
- Gwynn M. N., Portnoy A., Rittenhouse S. F., Payne D. J. (2010). Challenges of antibacterial discovery revisited. Ann. N. Y. Acad. Sci. 1213 5–19. 10.1111/j.1749-6632.2010.05828.x [DOI] [PubMed] [Google Scholar]
- Hopkins A. L., Keseru G. M., Leeson P. D., Rees D. C., Reynolds C. H. (2014). The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13 105–121. 10.1038/nrd4163 [DOI] [PubMed] [Google Scholar]
- Lakshminarayana S. B., Huat T. B., Ho P. C., Manjunatha U. H., Dartois V., Dick T., et al. (2015). Comprehensive physicochemical, pharmacokinetic and activity profiling of anti-TB agents. J. Antimicrob. Chemother. 70 857–867. 10.1093/jac/dku457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. (2000). Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44 235–249. 10.1016/S1056-8719(00)00107-6 [DOI] [PubMed] [Google Scholar]
- Murugasu-Oei B., Dick T. (2000). Bactericidal activity of nitrofurans against growing and dormant Mycobacterium bovis BCG. J. Antimicrob. Chemother. 46 917–919. 10.1093/jac/46.6.917 [DOI] [PubMed] [Google Scholar]
- Nessar R., Cambau E., Reyrat J. M., Murray A., Gicquel B. (2012). Mycobacterium abscessus: a new antibiotic nightmare. J. Antimicrob. Chemother. 67 810–818. 10.1093/jac/dkr578 [DOI] [PubMed] [Google Scholar]
- Park I. K., Olivier K. N. (2015). Nontuberculous mycobacteria in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin. Respir. Crit. Care Med. 36 217–224. 10.1055/s-0035-1546751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J., Gwynn M. N., Holmes D. J., Pompliano D. L. (2007). Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6 29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- Riss T. L., Moravec R. A., Niles A. L., Benink H. A., Worzella T. J., Minor L., et al. (2004). “Cell viability assays,” in Assay Guidance Manual eds Sittampalam G. S., Coussens N. P., Nelson H., Arkin M., Auld D., Austin C., et al. (Rockville, MD: Bethesda; ). [Google Scholar]
- Scott D. E., Coyne A. G., Hudson S. A., Abell C. (2012). Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51 4990–5003. 10.1021/bi3005126 [DOI] [PubMed] [Google Scholar]
- Via L. E., Savic R., Weiner D. M., Zimmerman M. D., Prideaux B., Irwin S. M., et al. (2015). Host-mediated bioactivation of pyrazinamide: implications for efficacy, resistance, and therapeutic alternatives. ACS Infect. Dis. 1 203–214. 10.1021/id500028m [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2015). Global Tuberculosis Report 2015. Geneva: World Health Organization. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.