

Learning mechanisms in brain reward pathways are hijacked after experience with drugs of abuse, shaping drug-related memories that underlie compulsive drug-taking behavior. Ventral tegmental area (VTA) dopamine neuronal dysfunction is an initial step for the development of addiction. Drugs of abuse may silence genes in the VTA, while compounds called histone deacetylase (HDAC) inhibitors may oppose this. We demonstrate that acute morphine-induced synaptic plasticities in the VTA are reversible by an HDAC inhibitor.
Keywords: ventral tegmental area, synaptic plasticity, electrophysiology, synaptic transmission, histone deacetylase
Abstract
Dopamine (DA) dysfunction originating from the ventral tegmental area (VTA) occurs as a result of synaptic abnormalities following consumption of drugs of abuse and underlies behavioral plasticity associated with drug abuse. Drugs of abuse can cause changes in gene expression through epigenetic mechanisms in the brain that underlie some of the lasting neuroplasticity and behavior associated with addiction. Here we investigated the function of histone acetylation and histone deacetylase (HDAC)2 in the VTA in recovery of morphine-induced synaptic modifications following a single in vivo exposure to morphine. Using a combination of immunohistochemistry, Western blot, and whole cell patch-clamp recording in rat midbrain slices, we show that morphine increased HDAC2 activity in VTA DA neurons and reduced histone H3 acetylation at lysine 9 (Ac-H3K9) in the VTA 24 h after the injection. Morphine-induced synaptic changes at glutamatergic synapses involved endocannabinoid signaling to reduce GABAergic synaptic strength onto VTA DA neurons. Both plasticities were recovered by in vitro incubation of midbrain slices with a class I-specific HDAC inhibitor (HDACi), CI-994, through an increase in acetylation of histone H3K9. Interestingly, HDACi incubation also increased levels of Ac-H3K9 and triggered GABAergic and glutamatergic plasticities in DA neurons of saline-treated rats. Our results suggest that acute morphine-induced changes in VTA DA activity and synaptic transmission engage HDAC2 activity locally in the VTA to maintain synaptic modifications through histone hypoacetylation.
NEW & NOTEWORTHY
Learning mechanisms in brain reward pathways are hijacked after experience with drugs of abuse, shaping drug-related memories that underlie compulsive drug-taking behavior. Ventral tegmental area (VTA) dopamine neuronal dysfunction is an initial step for the development of addiction. Drugs of abuse may silence genes in the VTA, while compounds called histone deacetylase (HDAC) inhibitors may oppose this. We demonstrate that acute morphine-induced synaptic plasticities in the VTA are reversible by an HDAC inhibitor.
although opioids are among the most effective pain relievers, their potential for abuse is undeniable. Certain brain areas are known loci for drug-induced changes in activity, gene expression, and synaptic transmission (McClung et al. 2005). These include the nucleus accumbens (NAc), prefrontal cortex, and ventral tegmental area (VTA), all of which are key parts of the brain's reward circuitry. The VTA is of particular interest as a locus for drug reward since enhanced dopamine (DA) transmission from the VTA is an early neuroadaptation in response to a single exposure to drugs of abuse and triggers subsequent synaptic changes in projection areas of the VTA, such as the NAc and prefrontal cortex (Hyman et al. 2006; Kauer and Malenka 2007). Drug-evoked synaptic plasticity at glutamatergic and GABAergic synapses onto VTA DA neurons significantly contributes to drug-induced changes in DA cell excitability and DA signaling. Long-term potentiation (LTP) of glutamatergic synapses onto VTA DA neurons can be triggered 24 h after a single in vivo exposure to opioids and other drugs promoting DA cell excitability and behavioral sensitization (Brown et al. 2010; Saal et al. 2003; Ungless et al. 2001). Moreover, a single exposure to a drug of abuse such as morphine has been shown to block LTP and may occlude long-term depression (LTD) at GABAergic synapses onto VTA DA neurons, further enhancing DA signaling from the VTA (Dacher and Nugent 2011; Nugent et al. 2007).
Neuroplastic changes in response to drugs of abuse also include drug-induced transcriptional changes. Drugs of abuse such as morphine most likely enact these transcriptional changes via epigenetic mechanisms. In the VTA, such modifications are necessary for reward learning (Day et al. 2013). Epigenetic mechanisms such as histone lysine-tail acetylation are able to encode environmental stimuli into a change in mRNA expression levels by chromatin remodeling, promoting gene transcription. Such chromatin modification has been shown to alter gene expression in neurons and neuroplasticity underlying memory formation (Abel and Zukin 2008; Haggarty and Tsai 2011). Acute and chronic administration of psychostimulants and morphine elicits histone modifications such as histone acetylation at a specific subset of learning-associated genes in the brain reward circuitry including the VTA that could facilitate and consolidate lasting neural and behavioral plasticity induced by drug abuse. Moreover, increasing histone acetylation through pre- or coadministration of histone deacetylase (HDAC) inhibitors (HDACis) in conjunction with abused drugs promotes or opposes drug-induced synaptic and behavioral abnormalities, suggesting that this epigenetic mark might be required to potentiate or prevent an addicted state (Jing et al. 2011; Kalda et al. 2007; Kennedy et al. 2013; Kumar et al. 2005; Levine et al. 2005; Renthal et al. 2007, 2009; Rogge and Wood 2013; Sanchis-Segura et al. 2009; Sun et al. 2012; Wang et al. 2010). This opens the possibility of targeting epigenetic mechanisms during initial learning or memory retrieval of drug-associated cues as a novel pharmacological therapy for extinction of drug-seeking behaviors. It is worthwhile to mention that in all studies investigating the effects of HDAC inhibition on addictive behaviors, HDACis have been used as pretreatment drugs or in conjunction with drugs of abuse; therefore it is unknown whether HDAC inhibition per se is sufficient to reverse a drug-induced synaptic plasticity after a drug exposure. Stress and drugs of abuse tap into a common synaptic mechanism to alter DA signaling (Niehaus et al. 2010; Nugent et al. 2007; Saal et al. 2003). Recently, we demonstrated that a severe early-life stress (i.e., 24-h maternal deprivation) induces synaptic abnormalities within the VTA that are reversible by HDAC inhibition, suggesting that early-life stress-induced neuroplasticity in the VTA involves HDACs to trigger long-lasting GABAergic and DA dysfunction (Authement et al. 2015). Here we tested whether HDAC inhibition locally in the VTA is sufficient to reverse epigenetic modifications and synaptic plasticities associated with a single in vivo injection of morphine 24 h after exposure.
MATERIALS AND METHODS
All experiments were carried out in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Uniformed Services University Institutional Animal Care and Use Committee. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Morphine treatment.
Sprague-Dawley male rats (P14–P21) received either one intraperitoneal injection of morphine sulfate (10 mg/kg) dissolved in 0.9% saline or injection of comparable volumes of saline 24 h prior to death for electrophysiological recordings or immunohistochemical studies. Only one cell per animal (saline or morphine) was recorded; therefore all reported n values represent the number of animals recorded.
Immunofluorescence and image analysis.
Morphine- and saline-treated rats were anesthetized with an intraperitoneal injection containing ketamine (85 mg/kg) and xylazine (10 mg/kg) and perfused through the aorta with heparinized 1× phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA; USB, Cleveland, OH). The brains were dissected and placed in 4% PFA for 24 h and then cryoprotected by submersion in 20% sucrose for 3 days, frozen on dry ice, and stored at −70°C until sectioned. Sections of the VTA were cut with a cryostat (Leica CM1900) and mounted on slides. Serial coronal sections (20 μm) of the midbrain containing the VTA (from −4.92 to −6.72 mm caudal to bregma; Paxinos and Watson 2007) were fixed in 4% PFA for 5 min, washed in 1× PBS, and then blocked in 10% normal horse serum (NHS) containing 0.3% Triton X-100 in 1× PBS for 1 h. Sections were incubated in rabbit anti-tyrosine hydroxylase (TH) (1:1,000; Calbiochem, San Diego, CA) and mouse anti-HDAC2 (1:1,000; Abcam, Cambridge, MA) in carrier solution (5% NHS in 0.1% Triton X-100 in 1× PBS) overnight at room temperature. After rinsing in 1× PBS, sections were incubated for 2 h in Alexa Fluor 488-labeled goat anti-rabbit IgG and Alexa Fluor 568-labeled goat anti-mouse IgG (both diluted 1:200). Finally, sections were rinsed in 1× PBS, dried, and coverslipped with ProLong mounting medium containing DAPI to permit visualization of nuclei. Background staining was assessed by omission of primary antibody in the immunolabeling procedure (negative control). VTA tissue sections of rats with previously established presence of TH/HDAC2-immunoreactive neurons were processed as positive control tissue. Images in Fig. 1 were captured with a Zeiss Confocal Inverted Microscope System (Carl Zeiss) ×40/1.4 NA oil immersion objective. At three AP locations (−5.4, −5.7, and −6.0 relative to bregma) six TH-positive neurons were identified within the VTA. From each TH-positive neuron two HDAC2 density readings (3 μm × 3 μm) were taken from the somatic region (clearly labeled with TH) and the nuclear region (clearly labeled with DAPI). Three background density readings were taken from an area clearly not labeled with HDAC2. All density readings were normalized to background.
Fig. 1.

Morphine increases HDAC2 expression in VTA DA neurons. A: examples of brain sections stained with antibodies to TH (green), HDAC2 (red), and DAPI (blue) with merged panels, which show the expression of HDAC2 in TH neurons in the VTA of saline (top)- and morphine (bottom)-treated rats. Scale bar, 20 μm. B: averaged levels of nuclear and somatic HDAC2 expression at 3 AP levels from saline- and morphine-treated rats. In this and all subsequent figures, numbers in parentheses indicate the number of animals examined. *Statistical significance.
Slice preparation for electrophysiology and Western blot.
Saline- or morphine-treated rats were anesthetized with isoflurane and immediately decapitated. The brains were quickly dissected and placed into ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM) 126 NaCl, 21.4 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.00 MgSO4, 11.1 glucose, and 0.4 ascorbic acid, saturated with 95% O2-5% CO2. Horizontal midbrain slices containing the VTA were cut at 250 μm (for electrophysiology) and incubated in ACSF at 34°C for at least 1 h. Slices were then transferred to a recording chamber and perfused with ascorbic acid-free ACSF at 28°C. Midbrain slices were cut at 400 μm for Western blot experiments.
For HDACi treatment, slices were incubated in the presence of CI-994 dissolved in DMSO and diluted to the final concentration in ACSF. The final concentration of CI-994 (20 μM) was selected for brain slice incubation because it is approximately the concentration measured in the brain after a systemic, intraperitoneal administration of 30 mg/kg CI-994 (Graff et al. 2014). Controls were incubated in the same dilution of DMSO (1:1,000), which did not affect synaptic transmission. These slices were allowed to incubate for 2–4 h before transfer to the recording chamber or dissection of the VTA for Western blots. In some of our control recordings, slices were only incubated in DMSO-free ACSF and the data for both DMSO and DMSO-free groups were pooled together as controls in the graphs. For chemically induced GABAergic LTD experiments, drugs were bath applied after 10 min of stable baseline recordings of evoked inhibitory postsynaptic currents (IPSCs) and remained in the bath throughout the experiment. The CB1 receptor agonist R-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN 55212-2) was dissolved in DMSO (1:1,000 dilution) and diluted to 2 μM in ACSF. Stock solutions for the group I metabotropic glutamate receptor (mGluR) agonist dihydroxyphenylglycine (DHPG, 50 μM) was made by dissolving in deionized water. All drugs were purchased from Sigma, Tocris, or Calbiochem.
Western blot.
The VTA was dissected bilaterally from horizontal midbrain slices (400 μM) of morphine- or saline-treated rats. For HDACi treatment, slices were incubated in ACSF in the presence of CI-994 dissolved in DMSO or the same dilution of vehicle (DMSO) for 2–4 h before being snap frozen in liquid nitrogen and stored at −80°C. Tissues were thawed, washed in ice-cold PBS, and lysed in RIPA buffer containing protease inhibitors (Sigma). Samples were then sonicated, incubated on ice for 30 min, and centrifuged at 10,000g for 20 min at 4°C. Protein concentration in the supernatant was determined with a Pierce BCA Protein Assay Kit (Life Technologies). Equal amounts of protein (20 μg) were combined with loading buffer, boiled for 5 min, and loaded onto 4–20% precast polyacrylamide gel (Bio-Rad Laboratories). Separated proteins were transferred onto nitrocellulose membranes, blocked with casein-based blocking reagent (I-Block, Life Technologies) for 60 min at room temperature, and then incubated overnight at 4°C with rabbit anti-acetyl-histone H3K9 (Ac-H3K9, 1:1,000; Cell Signaling, no. 9649), or mouse anti-β-actin (1:10,000; Abcam, no. ab6276). After incubation, the membranes were washed with PBS-T and exposed to the appropriate horseradish peroxidase-linked secondary antibody (Cell Signaling). Blots were developed with Clarity Western ECL Substrate (Bio-Rad Laboratories) and detected with a Fuji LAS-3000 image acquisition system (Fuji, Stamford, CT).
Electrophysiology.
Whole cell recordings were performed on midbrain slices with a patch amplifier (Multiclamp 700B) under infrared-differential interference contrast microscopy. Data acquisition and analysis were carried out with Digidata 1440A, pCLAMP 10 (Molecular Devices, Union City, CA), and Mini Analysis 6.0.3 (Synaptosoft). Signals were filtered at 3 kHz and digitized at 10 kHz. The recording ACSF was the same as the cutting solution except that it was ascorbic acid free. The appearance of an Ih current (≥50 pA) in response to stepping cells from −50 mV to −100 mV was used to identify VTA DA neurons as previously described (Dacher et al. 2013). Paired AMPA receptor (AMPAR)-mediated excitatory postsynaptic currents (EPSCs) were stimulated with a bipolar stainless steel stimulating electrode placed 200–500 mm rostral to the recording site in the VTA at 0.1 Hz (100 μs). Combined EPSCs were evoked and recorded in ACSF perfusion containing picrotoxin (100 μM) while the cell was voltage clamped at +40 mV. Patch pipettes were filled with (in mM) 117 Cs-gluconate, 2.8 NaCl, 5 MgCl2, 2 ATP-Na+, 0.3 GTP-Na+, 0.6 EGTA, and 20 mM HEPES, with intracellular spermine (10 μM) (pH adjusted to 7.28 with CsOH, osmolarity adjusted to 275–280 mosM). d-APV (50 μM) was added to the perfusion to block NMDA receptor (NMDAR)-mediated currents and isolate AMPAR-mediated currents. Subtraction of AMPAR-mediated currents from combined excitatory currents allowed us to measure AMPA-to-NMDA ratios. Pharmacologically isolated AMPAR-mediated evoked EPSCs were also recorded to measure AMPAR rectification. These recordings were performed in the presence of picrotoxin (100 μM), d-APV (50 μM), and intracellular spermine (10 μM) included in Cs-gluconate-based internal solution. AMPAR EPSCs were recorded at several holding potentials ranging from −65 to +40 mV. AMPAR rectification was determined by dividing peak AMPAR EPSC amplitudes recorded at −65 mV by those recorded at +40 mV. Paired GABAA receptor (GABAAR)-mediated IPSCs were similarly evoked (at 0.1 Hz, duration 100 μs, 50 ms interstimulation interval), isolated, and recorded in ACSF containing 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μM) and strychnine (1 μM). The internal solution was similar to miniature IPSC (mIPSC) recordings as stated below. Cells were voltage clamped at −70 mV for these recordings.
Whole cell recordings of GABAAR-mediated mIPSCs were performed in ACSF perfused with DNQX (10 μM), strychnine (1 μM), and tetrodotoxin (TTX, 1 μM). The patch pipettes (3–6 MΩ) were filled with (in mM) 125 KCl, 2.8 NaCl, 2 MgCl2, 2 ATP-Na+, 0.3 GTP-Na+, 0.6 EGTA, and 10 HEPES (pH adjusted to 7.28 with KOH, osmolarity adjusted to 275–280 mosM). Similarly, AMPAR-mediated miniature EPSCs (mEPSCs) were isolated in ACSF perfused with picrotoxin (100 μM), d-APV (50 μM), and TTX (1 μM). Patch pipettes for mEPSC recordings were filled with (in mM) 117 Cs-gluconate, 2.8 NaCl, 5 MgCl2, 2 ATP-Na+, 0.3 GTP-Na+, 0.6 EGTA, and 20 HEPES. VTA neurons were voltage-clamped at −70 mV and recorded over 10 sweeps, each lasting 50 s. The cell input resistance and series resistance were monitored through all experiments, and if these values changed by >10% data were not included.
Data analysis.
Values are presented as means ± SE. Statistical significance was determined with unpaired Student's t-test and one-way or two-way ANOVA with Bonferroni post hoc analysis with significance level of P < 0.05. The peak values of the evoked paired EPSCs or IPSCs were measured relative to the same baseline. A stable baseline value was considered in each sweep of paired pulses starting at 20–50 ms right before the emergence of the EPSC or IPSC with pCLAMP 10 software. Paired-pulse ratio (PPR, 50-ms interstimulus interval) was measured over 5-min epochs of 30 EPSCs or IPSCs each. The average value for the amplitude of all 30 EPSC2/IPSC2 was divided by the average value for the amplitude of the corresponding 30 EPSC1/IPSC1 and reported as the mean PPR for that epoch. Unpaired Student's t-test was used to determine statistical significance for baseline PPRs of EPSCs or IPSCs between saline and morphine animals. Levels of drug-induced depression in response to WIN55212-2 and DHPG are reported as averaged IPSC amplitudes for 5 min just before bath application compared with averaged IPSC amplitudes during the 5-min period from 35 to 40 min after the application. The magnitude of chemical LTD in Fig. 4C is expressed as percentage of synaptic change. Statistical significance was determined with repeated-measures ANOVA with significance level of P < 0.05 for chemical LTD (WIN55212-2 and DHPG) experiments. Mini Analysis software was used to detect and measure mIPSCs and mEPSCs with preset detection parameters of mIPSCs and mEPSCs and an amplitude cutoff of 5 pA. The Kolmogorov-Smirnov (KS) test was performed for the statistical analyses of cumulative probability plots of mEPSCs and mIPSCs.
Fig. 4.

Morphine-induced changes of GABAergic synaptic transmission onto VTA DA neurons are reversed by HDAC inhibition. A: sample GABAAR-mediated mIPSC traces from saline-treated and morphine-treated rats. Calibration bars, 30 pA, 5 s. B: average mIPSC amplitude and frequency from saline-treated and morphine-treated rats with DMSO and CI-994. C, insets: representative traces from saline with bath application of WIN 55212-2 (top) or DHPG (bottom): averaged IPSCs before or the peak response 35 min after WIN 55212-2 or DHPG. Calibration bars, 100 pA, 25 ms. Averaged experiments with bath-applied WIN 55212-2 (2 μM) or DHPG (50 μM) in saline or morphine. WIN 55212-2 induced a significant rundown of IPSCs in saline- but not morphine-treated rats. DHPG induced a significant LTD in both saline- and morphine-treated rats. D–F: cumulative probability plots of amplitude and frequency (interevent interval) for all mIPSCs in saline-treated and morphine-treated rats. CI-994 (20 μM) reversed morphine-induced pre- and postsynaptic LTD. *Statistical significance.
RESULTS
Acute morphine exposure induces epigenetic modifications at the level of histone acetylation through HDAC2.
Exposure to drugs of abuse has been shown to modify histone acetylation in brain reward regions (Robison and Nestler 2011). Given that HDAC2, a member of the class I HDACs, is the most highly expressed HDAC in neurons and plays a critical role in synaptic plasticity and memory (Guan et al. 2009), we hypothesized that acute morphine exposure decreases histone acetylation through an increase in HDAC2 expression in VTA DA neurons. With a double-immunofluorescence staining technique using antibodies against TH (marker for DA neurons) and HDAC2, we found higher levels of both somatic and nuclear HDAC2 immunoreactivity in TH-positive cells of morphine-treated compared with saline-treated rats at three AP levels within the VTA (−5.4, −5.7, and −6 mm caudal to bregma; Paxinos and Watson 2007) (Fig. 1, saline, n = 5; morphine, n = 6; 2-way ANOVA, P < 0.0001). Furthermore, we analyzed HDAC2 expression in DA neurons of substantia nigra pars compacta to show the selectivity of morphine's effect on HDAC2 levels in VTA DA neurons and found that HDAC2 levels were only upregulated in VTA DA neurons (data not shown).
To determine whether increased HDAC2 expression leads to changes in epigenetic modifications, we performed Western blot assays of VTA homogenates isolated from saline- and morphine-treated rats to quantify acetylated histone H3 (using an antibody against Ac-H3K9). There was a significant decrease in levels of H3K9 acetylation in morphine-treated rats (Fig. 2A; saline, n = 3; morphine, n = 3, unpaired Student's t-test, P < 0.0001). Given that our previous work demonstrated the reversal of maternal deprivation-induced synaptic abnormalities of VTA by in vitro CI-994 treatment, we sought to determine whether the changes in Ac-H3K9 were similarly reversible. Our results here confirm that a 2- to 4-h incubation of brain slices with the HDACi does indeed increase histone acetylation. We found that levels of AC-H3K9 were significantly higher in brain slices from morphine-treated rats incubated in CI-994 compared with vehicle (DMSO) (Fig. 2C; morphine + DMSO, n = 5; morphine + CI-994, n = 4, unpaired Student's t-test, P < 0.0001). Interestingly, this treatment also significantly increased Ac-H3K9 levels in saline-treated animals (Fig. 2B; saline + DMSO, n = 6; saline + CI-994, n = 6, unpaired Student's t-test, P < 0.001). Given that acute morphine increased HDAC2 activity and triggered reversible changes in histone acetylation, we next tested whether morphine-induced synaptic modifications in response to a single morphine exposure were also reversible by the HDACi.
Fig. 2.
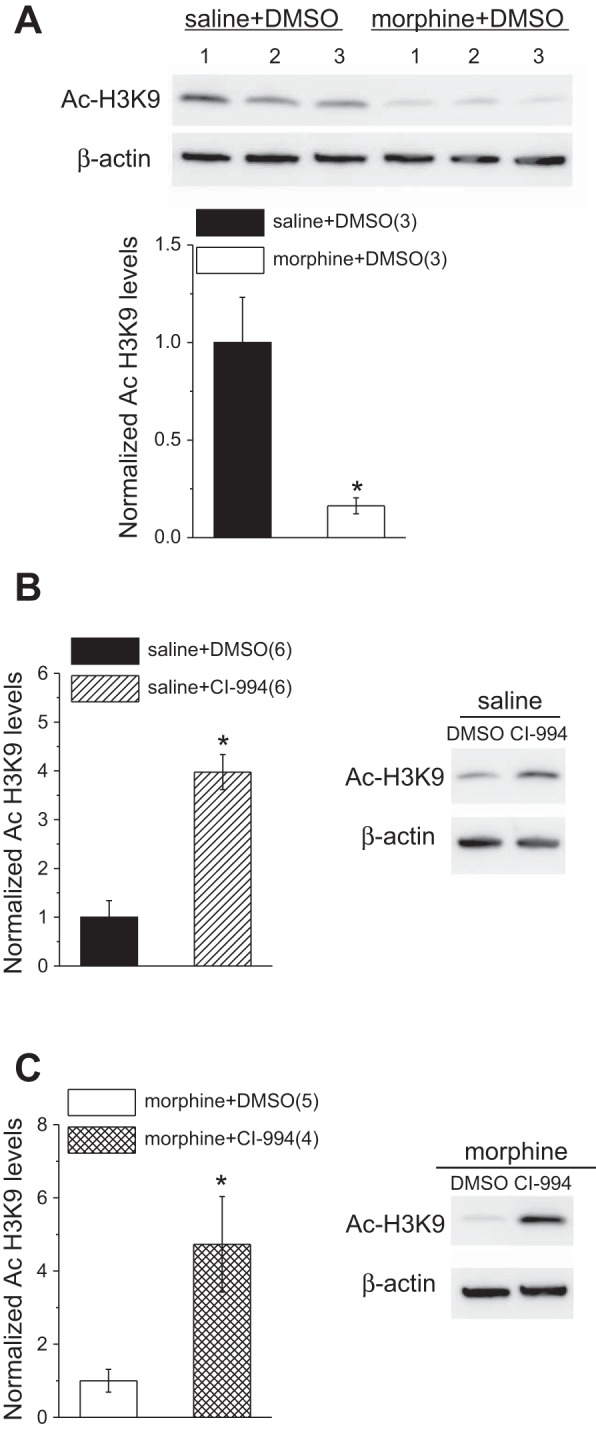
Histone acetylation at H3K9 is decreased after a single exposure to morphine and is recovered by in vitro HDAC inhibition with CI-994. A: representative Western blots (top) and quantitative data (bottom) of Ac-H3K9 and β-actin (control) in VTA homogenates from saline- and morphine-treated rats. B and C: representative Western blots and quantitative data of Ac-H3K9 and β-actin in VTA homogenates from saline (B)- and morphine (C)-treated rats incubated either in DMSO or CI-994. *Statistical significance.
Alteration of glutamatergic transmission by morphine was reversed after HDAC inhibition.
Potentiation of glutamatergic transmission onto VTA DA neurons in rodents is a well-characterized adaptation in both acute and long-term exposure to drugs of abuse, including cocaine and morphine (Saal et al. 2003). Here we tested whether a single injection of morphine was sufficient to trigger glutamatergic LTP in VTA DA neurons of rats. To accomplish this, we measured AMPA-to-NMDA ratios, a commonly used indicator of glutamatergic potentiation (Saal et al. 2003) and AMPAR-mediated mEPSCs. AMPA-to-NMDA ratios from morphine-treated animals were significantly increased compared with those from saline-treated control animals (Fig. 3, C and D). Our cumulative probability analyses showed that both amplitude and frequency of mEPSCs were significantly increased in morphine-treated rats relative to saline-treated control rats, although we detected no significant difference between the average amplitude and frequency of mEPSCs in morphine-treated rats vs. saline control rats (Fig. 3, A and B; saline, n = 7: 12.43 ± 1.25 pA, 1.18 ± 0.22 Hz; morphine, n = 7: 12.91 ± 1.09 pA, 3.03 ± 0.87 Hz; unpaired Student's t-tests: P = 0.77 for amplitude, P = 0.06 for frequency; KS tests for cumulative distribution curves: P < 0.05 for amplitude, P < 0.01 for frequency). We also calculated the baseline PPRs from paired evoked EPSCs in saline- and morphine-treated rats (Fig. 3C; PPR for saline: n = 6, 1.33 ± 0.17; PPR for morphine: n = 6, 1.2 ± 0.08; unpaired t-test, P = 0.49). There was no significant difference in PPRs between the two groups, suggesting that the change in mEPSC frequency is consistent with a postsynaptic effect (unsilencing of postsynaptic receptors).
Fig. 3.

Morphine-induced glutamatergic plasticity is reversed with HDAC inhibition. A: sample AMPAR-mediated mEPSC traces and average bar graphs of mEPSC amplitude and frequency from saline-treated and morphine-treated rats. Calibration bars, 10 pA, 5 s. B: cumulative probability plots of amplitude and frequency (interevent interval) for all mEPSCs in saline-treated and morphine-treated rats. Morphine induced an increase in amplitude and frequency. C: baseline PPRs from paired evoked EPSCs in saline- and morphine-treated rats. There was no significant difference in PPRs between the 2 groups. D: sample traces of AMPA and NMDA EPSCs recorded at +40 mV from VTA DA neurons of saline-treated and morphine-treated rats. Calibration bars, 50 pA, 25 ms. E: summary of AMPAR-to-NMDAR ratios obtained from saline-treated and morphine-treated rats with DMSO (vehicle) or CI-994 (20 μM). The morphine-induced increase in AMPAR-to-NMDAR ratio was reversed by incubation with CI-994. F: summary of AMPAR EPSC rectification indexes obtained from saline-treated and morphine-treated rats with DMSO (vehicle) and CI-994. CI-994 did not reverse the morphine-induced increase in rectification index but increased this index in slices from saline-treated rats. All recordings (D–F) were performed with intrapipette inclusion of spermine. *Statistical significance.
We next tested whether HDAC inhibition was sufficient to reverse this morphine-induced plasticity. To accomplish this, we incubated midbrain slices prepared from both morphine-treated and saline-treated rats killed 24 h after in vivo injection in either vehicle (DMSO) or CI-994 for 2–4 h and measured AMPA-to-NMDA ratios. When incubated in CI-994, AMPA-to-NMDA ratios in slices from morphine-treated rats were significantly reduced compared with morphine-treated controls incubated in carrier solution. In fact, CI-994 treatment fully reversed the elevated AMPA-to-NMDA ratios after morphine treatment back to normal levels as seen in control rats. Furthermore, AMPA-to-NMDA ratios recorded in slices from saline-treated rats incubated in CI-994 showed no significant difference from saline-treated controls incubated in carrier solution [Fig. 3, D and E; saline + DMSO, n = 6: 0.52 ± 0.108; morphine + DMSO, n = 6: 2.02 ± 0.34; saline + CI-994, n = 7: 0.63 ± 0.14; morphine + CI-994, n = 7: 0.79 ± 0.09; 2-way ANOVA, treatment: morphine vs. saline: F(1,22) = 19.12, P < 0.0001; drug: DMSO vs. CI-994: F(1,22) = 8.7, P = 0.007; interaction: F(1,22) = 12.56, P = 0.0018; Bonferroni post hoc analyses after 1-way ANOVA showed significant differences between saline + DMSO- and morphine + DMSO-treated slices: P < 0.0001 and between morphine + DMSO- and morphine + CI-994 treated slices: P < 0.0001; 1-way ANOVA, F(3,22) = 12.72, P < 0.0001].
To further investigate the effects of HDAC inhibition after acute in vivo morphine exposure on AMPAR-mediated synaptic function, we recorded evoked AMPAR-mediated EPSCs at different holding potentials (while intracellularly applying spermine) from DA neurons in midbrain slices incubated in either vehicle or CI-994 prepared 24 h after saline or morphine treatment. This allowed us to measure the relative contribution of Ca2+-permeable/GLuA2-lacking AMPARs to synaptic transmission. AMPA EPSC rectification index was significantly higher in morphine-treated animals compared with saline control animals (Fig. 3F). An increase in rectification suggests that morphine induced a loss of GluR2-containing AMPARs that were replaced by GluR2-lacking AMPARs. Despite the demonstrated recovery of AMPA-to-NMDA ratios by HDAC inhibition in slices from morphine-treated rats, the morphine-induced increase in the rectification index could not be reversed to normal levels by the HDACi, suggesting that the mechanisms underlying the increases in AMPA-to-NMDA ratio and rectification index may differ. However, there was also a significant increase in rectification indexes measured from DA neurons in CI-994-incubated slices prepared from saline-treated rats compared with those from saline-treated control rats incubated in carrier solution. The level of increase in rectification after CI-994 in control rat slices was comparable with those seen in slices from morphine-treated rats, potentially masking the anticipated reversal effects of HDAC inhibition on rectification indexes in morphine-treated rats [Fig. 3F; saline + DMSO, n = 6: 1.53 ± 0.34; morphine + DMSO, n = 6: 2.45 ± 0.12; saline + CI-994, n = 6: 2.54 ± 0.16; morphine + CI-994, n = 7: 2.57 ± 0.07; 2-way ANOVA, treatment: morphine vs. saline: F(1,21) = 5.79, P = 0.025; drug: DMSO vs. CI-994: F(1,21) = 8.25, P = 0.009; interaction: F(1,21) = 5.17, P = 0.033; Bonferroni post hoc analyses after 1-way ANOVA showed significant differences between saline + DMSO- and morphine + DMSO-treated slices (P = 0.022) and between saline + DMSO- and saline + CI-994-treated slices (P = 0.01), whereas morphine + DMSO and morphine + CI-994-treated slices did not show a significant difference (P = 1.01); 1-way ANOVA, F(3,21) = 6.29, P = 0.003].
Morphine-induced pre- and postsynaptic changes of GABAergic synaptic transmission were reversed by HDAC inhibition.
So far, our data suggest the possible involvement of HDAC2 and H3K9 acetylation in maintenance of morphine-induced glutamatergic plasticity that could be reversed by HDAC inhibition. We further examined the effects of CI-994 treatment on morphine-induced GABAergic plasticity. Midbrain slices containing the VTA from both saline- and morphine-treated animals killed 24 h after saline/morphine exposure were incubated in either CI-994 or vehicle (DMSO) for 2–4 h as described previously (Authement et al. 2015). The carrier solution, DMSO, did not have any significant effect on either mIPSC amplitude or frequency; therefore the data from DMSO-free control experiments were pooled together with DMSO controls. Both amplitude and frequency of mIPSCs were significantly reduced in morphine-treated rats relative to saline-treated control rats (Fig. 4, A, B, and D; saline, n = 12: 38.13 ± 2.67 pA, 6.46 ± 0.68 Hz; morphine, n = 13: 30.07 ± 2.94 pA, 2.76 ± 0.40 Hz; unpaired Student's t-tests: P = 0.02 for amplitude, P = 0.0012 for frequency; KS tests for cumulative distribution curves: P < 0.0001 for amplitude, P < 0.0001 for frequency). Consistent with a presynaptic GABAergic LTD, baseline PPRs from paired evoked IPSCs calculated in morphine-treated rats were significantly higher than those in saline-treated rats (PPR for saline: n = 10, 1.02 ± 0.05; PPR for morphine: n = 11, 1.27 ± 0.09; unpaired t-test, P = 0.023).
Given the ability of endocannabinoid (eCB) signaling to suppress neurotransmitter release at GABAergic synapses (Morena et al. 2016), we sought to investigate the role of eCB signaling in the morphine-induced inhibitory plasticity. We found that the GABAergic depression induced in response to the CB1 agonist WIN 55212-2 (2 μM) was absent in slices from morphine-treated rats while WIN 55212-2 successfully induced depression of evoked IPSCs of DA neurons in slices from saline-treated animals. Given that mGluRs have been shown to trigger LTD at GABAergic synapses through production of eCBs (Chevaleyre et al. 2006), we also tested the effects of mGluR agonist DHPG on GABAergic transmission in VTA DA neurons. Interestingly, DHPG (50 μM) was still able to induce LTD at GABAergic synapses in both saline- and morphine-treated animals, suggesting that morphine affects CB1 receptor-mediated GABAergic plasticity independently from mGluR-mediated LTD. In fact, it has been shown that mGluR-mediated LTD induced in response to DHPG is postsynaptic and independent of eCB signaling, while eCBs only mediate the presynaptic short-term depression induced by DHPG (Yu et al. 2013) [Fig. 4C; saline + WIN55212-2, n = 4, 71.5 ± 1.4% of baseline values, F(8.18,16.35) = 4.55, P = 0.004; morphine + WIN55212-2, n = 5, 99.5 ± 1.4% of baseline values, F(10.08,30.25) = 0.67, P = 0.74; saline + DHPG, n = 6, 58.2 ± 1.08% of baseline values, F(6.64,19.94) = 3.93, P = 0.015; morphine + DHPG, n = 6, 74 ± 2.2% of baseline values, F(3.8,19.05) = 3.11, P = 0.04].
Midbrain slices from morphine-treated rats incubated in CI-994 displayed larger amplitudes and higher frequencies of mIPSCs compared with slices from morphine-treated rats incubated either in DMSO or DMSO-free solutions. This suggested that CI-994 was able to recover normal GABAergic synaptic transmission onto VTA DA neurons after acute morphine exposure (Fig. 4, A, B, and F; morphine, n = 13: 30.07 ± 2.94 pA, 2.76 ± 0.4 Hz; morphine + CI-994, n = 11: 42.46 ± 3.41 pA, 4.40 ± 0.49 Hz; unpaired Student's t-tests: P = 0.011 for amplitude, P = 0.017 for frequency; KS tests for cumulative distribution curves: P < 0.0001 for amplitude, P < 0.0001 for frequency). Interestingly, saline control rats treated with CI-994 also showed significant decreases in both mIPSC amplitude and frequency, although no significant difference between the average amplitude of mIPSCs was detected after HDACi treatment (Fig. 4, A, B, and E; saline, n = 12: 38.13 ± 2.67 pA, 6.46 ± 0.68 Hz; saline + CI-994, n = 5: 31.91 ± 1.88 pA, 2.46 ± 0.46 Hz; unpaired Student's t-tests: P = 0.092 for amplitude, P < 0.0001 for frequency; KS tests for cumulative distribution curves: P = 0.012 for amplitude, P < 0.0001 for frequency).
DISCUSSION
The acute effects of morphine on VTA synaptic plasticity are well documented (Brown et al. 2010; Jalabert et al. 2011; Nugent et al. 2007; Saal et al. 2003). Here we provide the first demonstration of the involvement of HDAC2-mediated changes in histone acetylation profile upon a single in vivo morphine exposure in this phenomenon. We show that morphine increased HDAC2 activity in VTA DA neurons and reduced histone H3K9 acetylation in the VTA 24 h after injection. We further demonstrated that 2- to 4-h in vitro incubation of midbrain slices with a class I-specific HDACi significantly increased acetylation of histone H3K9 in morphine-treated rats (Fig. 1, Fig. 2). Consistent with these reversible epigenetic modifications, HDAC inhibition reversed morphine-induced glutamatergic potentiation while also reversing depression of GABAergic synaptic transmission onto VTA DA neurons. HDACi incubation also increased levels of Ac-H3K9 and triggered plasticity at GABAergic and glutamatergic synapses onto VTA DA neurons of saline-treated rats. Furthermore, we also provide the first evidence that morphine-induced depression of GABAergic transmission is subsequent to morphine-induced potentiation of glutamatergic transmission and mediated by eCB signaling. Our results suggest that acute morphine-induced changes in VTA DA activity and synaptic transmission engage HDAC2 activity locally in the VTA to maintain synaptic modifications through histone hypoacetylation. This work highlights the fact that reversible epigenetic dysregulation through HDACs may serve as a possible mechanism by which initial VTA synaptic plasticities in response to opioids could be maintained.
Consistent with findings in mice and rats demonstrating NMDAR-dependent LTP of glutamatergic transmission onto VTA DA neurons and increased postsynaptic insertion of inwardly rectifying Ca2+-permeable GluA2-lacking AMPARs following acute exposure to morphine (Baimel and Borgland 2015; Brown et al. 2010; Saal et al. 2003), we found that a single exposure to morphine increased AMPA-to-NMDA ratios, the amplitude of mEPSCs, and rectification indexes in DA neurons (Fig. 3). Previous studies have also shown that a single in vivo injection of morphine is able to trigger long-lasting changes in synaptic strength of GABAergic synapses (Dacher and Nugent 2011; Nugent et al. 2007). Here we demonstrated that morphine per se induced a reduction in GABAergic synaptic strength by suppressing GABA release and decreasing the number or function of postsynaptic GABAARs (Fig. 4). Consistent with this, a recent study established that a single dose of morphine was able to elicit a decrease in presynaptic GABA release facilitated by orexin signaling (Baimel and Borgland 2015). However, their results did not support an induction of a postsynaptic change in GABAAR number or function by morphine, which is likely due to the use of higher amplitude cutoff for detecting mEPSCs. Interestingly, we provide new evidence that the presynaptic component of morphine-induced suppression of GABA release is in fact eCB mediated (Fig. 4). This raises the possibility that, similar to an acute stress, acute morphine may downregulate CB1 receptors (Crosby et al. 2011; Morena et al. 2016). On the other hand, eCB signaling arising from morphine-induced glutamatergic plasticity could retrogradely depress GABAergic signaling onto VTA DA neurons. Consistent with this, cocaine-induced LTD of GABAergic synapses onto VTA DA neurons also required eCB signaling (Pan et al. 2008).
Increasing evidence suggests that HDACs play an important role in memory and cognition (Penney and Tsai 2014). Histone acetylation is tightly regulated by the opposing actions of histone acetyltransferases, which promote histone acetylation, and HDACs, which promote histone deacetylation. Typically, histone acetylation promotes expression of genes that positively regulate synaptic plasticity, while HDAC activity (deacetylation) negatively regulates plasticity. Furthermore, other studies have shown that inhibition of HDAC activity through the use of HDACis such as sodium butyrate, suberoylanilide hydroxamic acid (SAHA), and CI-994, is able to facilitate LTP induction and increase LTP magnitude in the hippocampus and amygdala (Guan et al. 2009; Levenson et al. 2004; Monsey et al. 2011; Vecsey et al. 2007). We recently demonstrated that in vitro HDAC inhibition with CI-994 was able to reverse VTA GABAergic synaptic abnormalities in a rodent model of severe early-life stress; i.e., a 24-h maternal deprivation procedure (Authement et al. 2015). Here we confirmed that this short-term in vitro HDACi treatment of the VTA is indeed sufficient to increase histone H3K9 acetylation. Morphine increased HDAC activity selectively in VTA DA neurons, which subsequently resulted in decreased levels of Ac-H3K9 (Fig. 1, Fig. 2). CI-994 treatment reversed this histone modification as well as glutamatergic and GABAergic plasticity induced by a single injection of morphine (Figs. 2–4). In closer relation to our work, early repeated maternal deprivation increases opiate consumption in adult rats through epigenetic modifications that enhance HDAC expression and activity in the NAc and treatment with the HDACi sodium valproate normalized opioid consumption (Tesone-Coelho et al. 2015). HDACis have also been shown to facilitate extinction of conditioned place preference and reduce development of morphine behavioral sensitization in rodents exposed to morphine (Jing et al. 2011; Wang et al. 2010). Furthermore, specific knockdown of class I HDACs suppresses cocaine-induced behavioral plasticity through a chromatin-mediated decrease in GABAergic inhibition in the NAc (Kennedy et al. 2013). In contrast, HDAC inhibition and HDAC3 deletions enhance morphine-induced locomotor sensitization and drug-induced conditioned place preference (Rogge et al. 2013; Sanchis-Segura et al. 2009). It must be noted that the timing and regimen of HDACi administration differs in these studies, which may account for the conflicting results. While in some studies animals were pretreated with HDACis, other studies used HDACis in conjunction with addictive drugs and during addiction behavioral assays. Our work here supports the hypothesis that postmorphine HDAC inhibition in the VTA per se is sufficient to reverse acute morphine-induced epigenetic dysregulation and synaptic adaptations.
The exact mechanisms linking morphine-induced synaptic changes and HDAC activity remain unknown. However, it is likely that morphine-induced changes in VTA DA neuron activity and synaptic transmission precede upregulation of HDAC2. The increased levels of HDAC2 and hypoacetylation of histones may then be maintaining the morphine-induced changes in synaptic transmission. Therefore, when HDAC activity is inhibited this maintenance is no longer present, allowing synaptic transmission to return to levels comparable to saline-treated animals. Transcriptional repression of mechanisms governing AMPAR trafficking could maintain morphine-induced postsynaptic glutamatergic LTP. The opposing actions of kinases such as protein kinase A (PKA) and phosphatases such as calcineurin (CaN) regulate synaptic insertion and maintenance of AMPARs and GABAARs during synaptic plasticity through phosphorylation and dephosphorylation of synaptic receptors (Luscher and Keller 2004; Sanderson and Dell'acqua 2011). In fact, CaN seems to be a negative regulator of memory, whereas PKA activity enhances learning and memory (Giese and Mizuno 2013; Malleret et al. 2001). CaN overexpression also prevents behavioral sensitization to amphetamine and morphine, suggesting that decreased CaN expression and activity at glutamatergic synapses may allow PKA activity to favor glutamatergic LTP underlying sensitization (Biala et al. 2005). Therefore, we assume that HDAC2-mediated transcriptional repression of PKA regulatory subunits (which promotes the formation of a persistently active PKA) or of CaN at glutamatergic synapses could favor phosphorylation of AMPARs and maintain morphine-triggered LTP (Biala et al. 2005; Chain et al. 1999). Our previous work in the VTA demonstrated that HDAC inhibition was able to reverse early-life stress-induced GABAergic synaptic abnormalities through restoration of A-kinase anchoring protein (AKAP)150 signaling. AKAP150 is a scaffolding protein that functions as an integrator of signaling molecules to glutamatergic and GABAergic synapses, where it can modulate synaptic trafficking and plasticity (Jurado et al. 2011; Lu et al. 2007, 2008; Sanderson et al. 2012). However, AKAP150 in the VTA has been shown to only modulate synaptic trafficking at GABAergic synapses (Dacher et al. 2013). Therefore, it is reasonable that treatment with the HDACi CI-994 recovers a morphine-induced disruption in AKAP-mediated trafficking of GABAARs, thereby reversing morphine-induced reduction in the number of postsynaptic GABAARs.
We also found that HDACi incubation of midbrain slices from saline-treated animals increased histone H3K9 acetylation and reduced the strength of GABAergic inhibition in DA neurons pre- and postsynaptically (Fig. 2 and Fig. 4). This is consistent with our previous findings that HDACi incubation of midbrain slices from non-maternally deprived animals induced a postsynaptic LTD at GABAergic synapses and modulated GABAergic spike timing-dependent plasticity (Authement et al. 2015). CI-994 treatment may promote the insertion of GluA2-lacking AMPARs into the postsynaptic membrane, allowing an increased influx of Ca2+ to the postsynaptic neuron facilitating plasticity at the synapse. HDACi-induced histone modifications and plasticity at GABAergic synapses and glutamatergic plasticity in the VTA could both increase levels of DA, which is shown to be necessary for hippocampal LTP and memory formation (Li et al. 2003). These findings reinforce the idea that HDACis can act as cognitive enhancers (Guan et al. 2009; Penney and Tsai 2014; Vecsey et al. 2007).
Given that the pattern and nature of epigenetic modifications induced by drugs of abuse vary from one brain region to another, it is critical to identify cell- and brain region-specific epigenetic mechanisms underlying drug-triggered DA dysfunction, thereby advancing our knowledge of epigenetics of addiction.
GRANTS
This work was supported by National Institute on Drug Abuse Grant R01 DA-039533 (F. S. Nugent). The funding agency did not contribute to writing this article or deciding to submit it.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.E.A., B.M.C., and F.S.N. conception and design of research; M.E.A., L.D.L., H.K., S.G., M.D., and R.D.S. performed experiments; M.E.A., L.D.L., H.K., S.G., and F.S.N. analyzed data; M.E.A., L.D.L., H.K., B.M.C., and F.S.N. interpreted results of experiments; M.E.A., L.D.L., H.K., and F.S.N. prepared figures; M.E.A. and F.S.N. drafted manuscript; M.E.A., L.D.L., H.K., B.M.C., and F.S.N. edited and revised manuscript; M.E.A., L.D.L., H.K., S.G., M.D., R.D.S., B.M.C., and F.S.N. approved final version of manuscript.
ACKNOWLEDGMENTS
The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as official or reflecting the views of the Uniformed Services University of the Health Sciences or the Department of Defense or the Government of the United States.
Present address of M. Dacher: Université Pierre et Marie Curie, Physiologie de l'Insecte, Signalisation et Communication-UMR 1272, Versailles, France.
REFERENCES
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 8: 57–64, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Authement ME, Kodangattil JN, Gouty S, Rusnak M, Symes AJ, Cox BM, Nugent FS. Histone deacetylase inhibition rescues maternal deprivation-induced GABAergic metaplasticity through restoration of AKAP signaling. Neuron 86: 1240–1252, 2015. [DOI] [PubMed] [Google Scholar]
- Baimel C, Borgland SL. Orexin signaling in the VTA gates morphine-induced synaptic plasticity. J Neurosci 35: 7295–7303, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic D-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci 21: 3089–3096, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Bellone C, Mameli M, Labouebe G, Bocklisch C, Balland B, Dahan L, Lujan R, Deisseroth K, Luscher C. Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS One 5: e15870, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron 22: 147–156, 1999. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29: 37–76, 2006. [DOI] [PubMed] [Google Scholar]
- Crosby KM, Inoue W, Pittman QJ, Bains JS. Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron 71: 529–541, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, Gouty S, Dash S, Cox BM, Nugent FS. A-kinase anchoring protein-calcineurin signaling in long-term depression of GABAergic synapses. J Neurosci 33: 2650–2660, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M, Nugent FS. Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology 61: 1166–1171, 2011. [DOI] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD. DNA methylation regulates associative reward learning. Nat Neurosci 16: 1445–1452, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Mizuno K. The roles of protein kinases in learning and memory. Learn Mem 20: 540–552, 2013. [DOI] [PubMed] [Google Scholar]
- Graff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, Rei D, Bero AW, Phan TX, Wagner F, Holson E, Xu J, Sun J, Neve RL, Mach RH, Haggarty SJ, Tsai LH. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156: 261–276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459: 55–60, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Tsai LH. Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol Learn Mem 96: 41–52, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598, 2006. [DOI] [PubMed] [Google Scholar]
- Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci USA 108: 16446–16450, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Luo J, Zhang M, Qin WJ, Li YL, Liu Q, Wang YT, Lawrence AJ, Liang JH. Effect of the histone deacetylase inhibitors on behavioural sensitization to a single morphine exposure in mice. Neurosci Lett 494: 169–173, 2011. [DOI] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci 13: 1053–1055, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Heidmets LT, Shen HY, Zharkovsky A, Chen JF. Histone deacetylase inhibitors modulates the induction and expression of amphetamine-induced behavioral sensitization partially through an associated learning of the environment in mice. Behav Brain Res 181: 76–84, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci 8: 844–858, 2007. [DOI] [PubMed] [Google Scholar]
- Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, Bassel-Duby R, Olson EN, Nestler EJ. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci 16: 434–440, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48: 303–314, 2005. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279: 40545–40559, 2004. [DOI] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci USA 102: 19186–19191, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci 6: 526–531, 2003. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J 26: 4879–4890, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, Hell JW. AKAP150-anchored PKA activity is important for LTD during its induction phase. J Physiol 586: 4155–4164, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther 102: 195–221, 2004. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 104: 675–686, 2001. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ, Zachariou V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci 25: 6005–6015, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One 6: e19958, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morena M, Patel S, Bains JS, Hill MN. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 41: 80–102, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci 32: 108–117, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature 446: 1086–1090, 2007. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28: 1385–1397, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Elsevier Academic, 2007. [Google Scholar]
- Penney J, Tsai LH. Histone deacetylases in memory and cognition. Sci Signal 7: re12, 2014. [DOI] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE 3rd, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62: 335–348, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56: 517–529, 2007. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12: 623–637, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge GA, Singh H, Dang R, Wood MA. HDAC3 is a negative regulator of cocaine-context-associated memory formation. J Neurosci 33: 6623–6632, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge GA, Wood MA. The role of histone acetylation in cocaine-induced neural plasticity and behavior. Neuropsychopharmacology 38: 94–110, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582, 2003. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Lopez-Atalaya JP, Barco A. Selective boosting of transcriptional and behavioral responses to drugs of abuse by histone deacetylase inhibition. Neuropsychopharmacology 34: 2642–2654, 2009. [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Dell'acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist 17: 321–336, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML. AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci 32: 15036–15052, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D, Neve RL, Zachariou V, Shen L, Nestler EJ. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci 32: 17454–17464, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesone-Coelho C, Morel LJ, Bhatt J, Estevez L, Naudon L, Giros B, Zwiller J, Dauge V. Vulnerability to opiate intake in maternally deprived rats: implication of MeCP2 and of histone acetylation. Addict Biol 20: 120–131, 2015. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587, 2001. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci 27: 6128–6140, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang Y, Qing H, Liu M, Yang P. The extinction of morphine-induced conditioned place preference by histone deacetylase inhibition. Neurosci Lett 483: 137–142, 2010. [DOI] [PubMed] [Google Scholar]
- Yu F, Zhong P, Liu X, Sun D, Gao HQ, Liu QS. Metabotropic glutamate receptor I (mGluR1) antagonism impairs cocaine-induced conditioned place preference via inhibition of protein synthesis. Neuropsychopharmacology 38: 1308–1321, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]