

Abstract
Reconsolidation updating is a form of memory modification in which an existing memory can become destabilized upon retrieval and subsequently be modified via protein-synthesis-dependent reconsolidation. However, not all memories appear to destabilize upon retrieval and thus are not modifiable via reconsolidation updating approaches and the neurobiological basis for this remains poorly understood. Here, we report that auditory fear memories created with 10 tone–shock pairings are resistant to retrieval-dependent memory destabilization and are associated with an increase in the synaptic GluN2A/GluN2B ratio in neurons of the basal and lateral amygdala (BLA) compared with weaker fear memories created via one or three tone–shock pairings. To increase the GluN2A/GluN2B ratio after learning, we generated a line of mice that expresses an inducible and doxycycline-dependent GFP-GluN2A transgene specifically in α-CaMKII-positive neurons. Our findings indicate that increasing the GluN2A/GluN2B ratio in BLA α-CaMKII-positive neurons after a weak fear memory has consolidated inhibits retrieval-dependent memory destabilization and modification of the fear memory trace. This was associated with a reduction in retrieval-dependent AMPA receptor trafficking, as evidenced by a reduction in retrieval-dependent phosphorylation of GluR1 at serine-845. In addition, we determined that increasing the GluN2A/GluN2B ratio before fear learning significantly impaired long term memory consolidation, whereas short-term memory remained unaltered. An increase in the GluN2A/GluN2B ratio after fear learning had no influence on fear extinction or expression. Our results underscore the importance of NMDAR subunit composition for memory destabilization and suggest a mechanism for why some memories are resistant to modification.
SIGNIFICANCE STATEMENT Memory modification using reconsolidation updating is being examined as one of the potential treatment approaches for attenuating maladaptive memories associated with emotional disorders. However, studies have shown that, whereas weak memories can be modified using reconsolidation updating, strong memories can be resistant to this approach. Therefore, treatments targeting the reconsolidation process are unlikely to be clinically effective unless methods are devised to enhance retrieval-dependent memory destabilization. Currently, little is known about the cellular and molecular events that influence the induction of reconsolidation updating. Here, we determined that an increase in the GluN2A/GluN2B ratio interferes with retrieval-dependent memory destabilization and inhibits the initiation of reconsolidation updating.
Keywords: amygdala, Glun2A, memory, NMDAR, Pavlovian fear conditioning, reconsolidation
Introduction
Reconsolidation updating is a process by which consolidated memories can be modified upon their retrieval (Nader et al., 2000). Retrieval of an existing memory can induce the memory to enter a destabilized state, necessitating its restabilization through reconsolidation, which is a protein-synthesis-dependent process (Tronson and Taylor, 2007, Lee, 2009, Alberini, 2011, Finnie and Nader, 2012). Targeting the reconsolidation process may provide a potential treatment for attenuating memories associated with pathologies such as posttraumatic stress disorder and drug addiction (Taylor et al., 2009, Milton and Everitt, 2010, Pitman, 2011, Surís et al., 2013). However, recent clinical evidence indicates that the effectiveness of this approach may be limited (Wood et al., 2015), possibly due to the fact that strong memories can be resistant to retrieval-dependent memory destabilization (i.e., the induction of reconsolidation updating; Wang et al., 2009, Winters et al., 2009). The molecular basis of reconsolidation induction mechanisms is still poorly understood and it remains unknown why strong memories are resistant to retrieval-dependent memory destabilization.
Previous studies have demonstrated a critical role for NMDARs in the initiation and restabilization phases of reconsolidation updating (Pedreira et al., 2002, Ben Mamou et al., 2006, Sadler et al., 2007, Brown et al., 2008, Itzhak, 2008, Lee and Everitt, 2008, Milton et al., 2008). NMDARs are composed of two obligatory GluN1 subunits and two GluN2 subunits (Sheng et al., 1994, Cull-Candy et al., 2001, Prybylowski and Wenthold, 2004). In the hippocampus and amygdala, the GluN2A and GluN2B subtypes are the predominant GluN2 subtypes (Monyer et al., 1992, Lopez de Armentia and Sah, 2003). Complete blockade of NMDARs using DL-2-amino-5-phosphonovaleric acid (APV) or GluN2B-specific blockade using infenprodil in the basal and lateral amygdala (BLA) are effective at inhibiting retrieval-dependent memory destabilization (Ben Mamou et al., 2006, Milton et al., 2013). In contrast, inhibition of the GluN2A subunit appears to influence selectively the restabilization phase of reconsolidation (Milton et al., 2013). These differences likely in part reflect inherent differences between the GluN2A and GluN2B subunits. GluN2A-containing receptors have a higher open probability, faster decay times, and lower sensitivity to glutamate and glycine than GluN2B-containing receptors (Lau and Zukin, 2007, Paoletti et al., 2013). GluN2A and GluN2B differ considerably in their cytoplasmic tails and these differences confer subunit-specific abilities to bind to postsynaptic scaffold proteins that regulate the induction of plasticity (Gardoni et al., 1998, Strack and Colbran, 1998). Therefore, an increase in the GluN2A/GluN2B ratio leads to an increase in the threshold for the induction of plasticity (Yashiro and Philpot, 2008). During learning, this ratio has been observed to be changed dynamically, leading to an increase in the GluN2A/GluN2B ratio (Quinlan et al., 2004). Notably, strong fear memories that do not undergo retrieval-dependent destabilization are associated with a reduction in GluN2B subunits in the BLA (Wang et al., 2009). It has been speculated that learning-induced changes in NMDAR subunit composition serve as a mechanism for maintaining the integrity of the memory and prevent memory destabilization upon retrieval (Zinebi et al., 2003, Quinlan et al., 2004, Wang et al., 2009); however, this has never been tested experimentally.
We hypothesized that an increase in intensity of auditory fear conditioning training would lead to an increase in the GluN2A/GluN2B ratio at BLA synapses and that this increase would be sufficient to prevent retrieval-dependent memory destabilization and therefore inhibit the induction of reconsolidation. To test this directly, we developed an inducible transgenic mouse line designed to express a GFP-GluN2A transgene in BLA pyramidal neurons, allowing us to examine how increasing the GluN2A/GluN2B ratio influences mnemonic processing. Our findings indicate that increasing the GluN2A/GluN2B ratio inhibits retrieval-dependent memory destabilization of a weak memory trace and therefore prevents its modification, similar to reconsolidation-resistant fear memories. These findings have importance for our understanding of the reconsolidation of fearful memories and may open new avenues for how to prevent or even reverse the transition of weak fear memories into strong, long-lasting memories.
Materials and Methods
Generation of inducible transgenic mice.
The standard Tet-off system was used (i.e., transgene expression is repressed upon administration of tetracycline or its analog doxycycline, Dox) to achieve inducible expression of GluN2A. The open reading frame for GFP-GluN2A was PCR amplified from pCI-GFP-GluN2A [a generous gift from Andres Barria (Barria and Malinow, 2002) using the following PCR primers (GFP-GluN2A attB FP: 5′-GGGACAAGTTTGTACAAAAAAGCAGGCTGCCGCCACCATGGGCAGATTGGGCTACTGG-3′ GFP-GluN2A attB RP: 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCTCCTAGTAGGGTGCTATC-3′). The PCR product was subsequently inserted into pDONR221 (Invitrogen) via a BP clonase reaction to create pDONR221-GFP-GluN2A. This vector was DNA sequenced to ensure the integrity of the GFP-GluN2A ORF. The pTet-Splice vector was generated by obtaining Addgene plasmid #17696 (Zheng et al., 2000) and digesting it with HindIII to release the pNTPTP α ORF and religating the vector backbone to reconstitute the original pTet-Splice vector. This vector was subsequently modified by inserting a MluI site by ligating into the XhoI site the following annealed DNA primers: 5′-TCGAGACGCGTctaggagagatac-3′ and 5′-TCGAgtatctctcctagACGCGTC-3′. Subsequently, we converted this vector to a Gateway-compatible destination vector (Invitrogen) by transferring the recombination cassette containing the ccdB gene from pLenti7.3 (Invitrogen) via the EcoRV sites to create pTet-Splice (+MluI)-ccdB Dest. The GFP-GluN2A ORF from pDONR221-GFP-GluN2A was transferred to pTet-Splice (+MluI)-ccdB Dest, via an LR clonase reaction (Invitrogen) to create pTet-Splice (+MluI)-Dest-GFP-GluN2A. After this, the pTet promoter was removed from this vector and exchanged with the TRE3G promoter. The TRE3G promoter was amplified from Addgene vector #27569 using the following DNA primers: FP: 5′-ATCCACGCGTTTTACTCCCTATCAGTGATAGAGAACG-3′ RP: 5′-TAAAATCGATTTTACGAGGGTAGGA AGTGGTAC-3′ and cloned into pTet-Splice (+MluI)-Dest-GFP-GluN2A via Mlu1 and Cla1 sites to generate pTRE3G-Splice (+MluI)-Dest-GFP-GluN2A. Dox-dependent transgene regulation using this vector was validated in vitro (data not shown) before generation of the GFP-GluN2A+/− transgenic mice. DNA harboring the TRE3G-GFP-GluN2A-pA transgene (∼7.3 kb) was excised via the XhoI-NotI sites from pTRE3G-Splice (+MluI)-Dest-GFP-GluN2A and purified and microinjected into fertilized oocytes from C57BL/6 mice at the Gene Targeting and Transgenic Facility at the University of Connecticut. The offspring obtained from this procedure were then genotyped to identify the GFP-GluN2A+/− founding line. The founding line was subsequently crossed with C57BL/6 mice to expand the line and this produced litters with healthy pups with expected Mendelian ratios. Upon receipt into our animal facility at the University of Texas at Dallas, GFP-GluN2A+/− mice were bred with α-CaMKII-tTA+/− mice that expressed the transcriptional activator tTA regulated by the α-CaMKII promoter (Jackson Laboratories; Mayford et al., 1996) to generate double-transgenic mice (GFP-GluN2A+/tTA+); this produced litters with healthy pups at the expected Mendelian ratios. The GluN2A expression was regulated by administering Dox (200 mg, LabDiet) through the animals' diet. All GFP-GluN2A+/tTA+ mice were placed on a diet containing Dox at weaning.
Subjects.
Behavior, electrophysiology, and molecular experiments were conducted on adult mice aged 8–12 weeks. Male and female mice were used in all experiments and were equally distributed across the experimental groups. All mice were individually housed and maintained on a 12 h light/dark cycle with ad libitum access to food and water. All procedures were in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the University of Texas at Dallas.
Genotyping.
Before weaning, ear punches were obtained from mice under anesthesia and processed for DNA extraction. The tissue samples were heated in 75 μl of 25 mm NaOH/0.2 mm EDTA for 1 h at 98°C and were then neutralized using 75 μl of 40 mm Tris-HCl, pH 5.5. The resulting solution was centrifuged for 3 min at 4000 rpm and 1 μl of the supernatant was used per 12 μl of Platinum-Taq (Invitrogen) PCR assay for genotyping. GFP-GluN2A+/− mice were identified using the following PCR primers: FP: 5′-GCCTCGAGAGTTGGCTTTAC-3′; RP: 5′-CCAGTAGCCCAATCTGCCCATGG-3′, which amplified a 464 bp DNA fragment that specifically identified the presence of the TRE3G-GFP-GluN2A transgene. The presence of the α-CaMKII-tTA transgene was identified using the following PCR primers: FP: 5′-CGCTGTGGGGCATTTTACTTTAG-3′; RP: 5′-CATGTCCAGATCGAAATCGTC-3′, which amplified a 450 bp DNA fragment specific to the α-CaMKII-tTA transgene described previously (Mayford et al., 1996). After PCR amplification, the samples were electrophoresed on a 1.5% agarose 1XTAE gel for identification of the GFP-GluN2A+/− and α-CaMKIItTA+/− positive mice.
Electrophysiology.
Electrophysiological recordings were obtained from adult (60–84 d postnatal) mice of both sexes. Mice were anesthetized with an overdose of urethane (3 g/kg) and quickly decapitated. Brains were placed in ice-cold artificial CSF (aCSF) consisting of the following (in mm): 110 choline, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 25 dextrose, 11.6 Na+-ascorbate, and 3.1 Na+-pyruvate bubbled with 95% O2/5% CO2. Coronal slices (350 μm) containing the BLA were cut on a vibratome (VT1000S; Leica) and then transferred for a minimum of 45 min into incubating aCSF consisting of the following (in mm): 126 NaCl, 2.5 KCl, 1.2 Na2HPO4, 25 Na2HCO3, 10 glucose, 2 CaCl2, and 1 MgCl2 bubbled with 95% O2/5% CO2, and maintained at 35°C. Whole-cell voltage-clamp recordings were obtained from pyramidal cells in the BLA at room temperature using recording aCSF consisting of the following (in mm): 126 NaCl, 2.5 KCl, 1.2 Na2HPO4, 25 Na2HCO3, 10 glucose, 2 CaCl2, and 1 MgCl2 bubbled with 95% O2/5% CO2. Electrodes (4–6 MΩ open tip resistance) were filled with (in mm) 130 CsCl, 20 TEA, 10 HEPES, 2 MgCl2, 0.5 EGTA, 4 Na-ATP, 0.3 Na-GTP, 14 phosphocreatine, and 2 QX-314. Recordings were performed using an Axon Multiclamp 700B amplifier (Molecular Devices) and data were acquired and analyzed using Axograph X (Axograph Scientific). Synaptic potentials were evoked by focal stimulation (0.2 ms pulse duration every 15 s) of the external capsule using theta-glass pipettes (Warner Instruments) and a stimulus isolator (WPI). To investigate the contributions of the GluN2A and GluN2B subunits individually, recordings were performed at +40 mV with 75 μm picrotoxin and CNQX in the bath and the subunit-specific current were isolated pharmacologically using 0.5 μm of the GluN2A antagonist PEAQX (NVP-AAM077 tetrasodium hydrate, [[[(1S)-1-(4-bromophenyl)ethyl]amino](1,2,3,4-tetrahydro-2,3-dioxo-5-quinoxalinyl) methyl] phosphonic acid tetrasodium hydrate) and 0.5 μm of the GluN2B antagonist Ro25-6981 ([R-(R*,S*)]-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol- hydrochloride hydrate), respectively. All drugs were obtained from Sigma-Aldrich. The relative contribution of GluN2A and GluN2B currents was estimated by first recording at least 15 sweeps of the total NMDAR current before bath applying either PEAQX or Ro25-6981, respectively. Ten minutes after wash-in of the drug, another 15–20 sweeps were recorded. Series resistance was monitored throughout the recordings as the response to small hyperpolarizing current injections and recordings that showed a >20% change were excluded. Traces were averaged and the relative contribution of GluN2A or GluN2B currents was calculated by subtracting the averaged maximum amplitude after drug application from the averaged amplitude before. Decay time was calculated in Axograph X using a two-exponential simplex fit.
Purification of PSDs from BLA tissue.
At the appropriate time, mice were deeply and briefly anesthetized using CO2 and the brains were immediately extracted and frozen at −80°C. Two hundred micrometers of frozen coronal brain sections containing the amygdala were taken on a cryostat and the lateral and basal nuclei were dissected using a 0.5 mm punch tool (Fine Science Tools) and frozen at −80°C until homogenization. Tissue punches from left and right amygdala nuclei were pooled and processed for isolation of PSDs as described previously (Migues et al., 2010). All solutions were supplemented with cOmplete Protease Inhibitors (Roche) and all steps were performed on ice or at 4°C. In addition, solutions were also supplemented with Phosphatase Inhibitor Cocktail (Sigma-Aldrich) while blotting for phosphorylated proteins. Briefly, tissue punches were lysed in 70 μl of ice-cold homogenizing buffer (30 mm Tris-HCl, 4 mm EDTA, 1 mm EGTA) using a dounce homogenizer and centrifuged for 10 min at 800 × g at 4°C. The resulting supernatant was ultracentrifuged in polycarbonate centrifuge tubes (8 × 34 mm) Beckman catalog #343776, in a TLA 120.1 rotor at 100,000 × g for 1 h at 4°C. The pellet obtained was resuspended in 30 μl of homogenizing buffer (30 mm Tris-HCl, 4 mm EDTA, 1 mm EGTA, 0.05% Triton-X) and incubated on ice for 20 min. The resuspended solution was overlaid over 100 μls of 1 m sucrose, centrifuged at 100,000 × g for 1 h at 4°C, and the resulting pellet resuspended in 20 μl of homogenizing buffer. Protein concentrations were measured using a BCA protein estimation kit (Fisher Scientific).
Western blotting.
Equal amounts of protein samples (6 μg) were loaded onto SDS-PAGE and subsequently transferred onto a PVDF membrane overnight at 4°C as described previously (Ploski et al., 2008). The membranes were rinsed for 20 min at room temperature with TTBS (20 mm Tris HCl, 50 mm NaCl, 0.1% Tween) and then blocked at room temperature for 1 h with BSA or nonfat dry milk (NFDM) in TTBS or PBS (1× PBS, pH 7.4) and probed with primary antibodies overnight at 4°C. The blots were then rinsed three times with TTBS and probed with secondary antibody tagged with HRP for 1 h at room temperature. After secondary antibody incubation, the blots were rinsed three times with TTBS and developed using the ECL Chemiluminescence assay (Fisher Scientific). For multiple antibodies, the blots were first stripped with 0.2 m NaOH for 20 min at room temperature and rinsed 3 times with TTBS before incubation with another antibody. The conditions for each antibody were as follows: GluR1-Ser845 (Millipore, 04-1073; blocking: 3% BSA in TTBS; primary: 1:1000 in 1% BSA in TTBS; secondary: anti-rabbit 1:10,000 in 1% BSA in TTBS); GluR1 (Cell Signaling Technology, D4N9 V; blocking: 5% NFDM in TTBS; primary: 1:1000 in 1% NFDM in TTBS; secondary: anti-rabbit 1:10,000 in 1% NFDM in TTBS); GluN2A (Millipore, 07-632; blocking: 5% NFDM in TTBS; primary: 1:1000 in 3% NFDM in TTBS; secondary: anti-rabbit 1:10,000 in 3% NFDM in TTBS); GluN2B (Millipore, 06-600; blocking: 3% NFDM in PBS; primary: 1:1000 in 3% NFDM in PBS; secondary: anti-rabbit 1:20,000 in 3% NFDM in PBS); GluN1 (Cell Signaling Technology, D65B7; blocking: 3% BSA in TTBS; primary: 1:1000 in 1% BSA in TTBS; secondary: anti-rabbit: 1:10,000 1% BSA in TTBS); PSD-95 (Cell Signaling Technology, D27E11; blocking: 3% BSA in TTBS; primary: 1:1000 1% BSA in TTBS; secondary: anti-rabbit 1:10,000 1% BSA in TTBS).
Quantification.
Western blots were scanned using DCP 7030 scanner (Brother) and the protein bands were quantified by calculating average integrated density using ImageJ. To control for variability in protein loading, the average integrated density for each sample was divided by its respective average integrated density for PSD-95 as described previously (Reissner et al., 2012, Delaney et al., 2013). This value was averaged across different samples per group.
Surgery.
Mice under ketamine (100 mg/kg) and xylazine (10.0 mg/kg) anesthesia were stereotaxically implanted with 22 gauge guide cannula with 4 mm projection (Plastics One) targeting the BLA (AP − 1.6 ML ± 3.3 DV − 4.95). The guide cannula was secured in place using Ketacfil-Applicap Glass ionomer restorative (3M, 3784269) and inserted with a dummy cannula (Plastics One) to prevent clogging. The mice were then returned to their home cages and allowed to heal for 5–6 d before behavioral procedures.
Histology.
Mice were killed by perfusion as described previously (Holehonnur et al., 2014). Briefly, the mice were anesthetized with an overdose of chloral hydrate (250 mg/kg) and then perfused with ∼100 ml of PBS (1× phosphate buffer, 150 mm NaCl) and then with 10% buffered formalin (Fisher Scientific). After brain extraction, the brains were then fixed in 10% formalin for 4–5 h, followed by cryoprotection in 1×PBS, pH 7.4, 30% sucrose for 2–4 d. Forty-micrometer coronal sections were obtained using a cryostat and collected on Superfrost slides (Fisher Scientific).
Transgene expression characterization.
Double transgenic GFP-GluN2A+/tTA+ and GFP-GluN2A+ (control) mice were killed and sectioned as described above. Coronal sections from different brain regions (bregma −6.12 to +1.98) were imaged for GFP expression at 50× using an Olympus BX51 upright fluorescence microscope and processed using an Olympus DP71 digital camera and DP manager software.
Cannulation placement.
Cannulated mice were killed and their brains were sectioned as described above. Coronal sections containing amygdala (bregma −2.3 to −0.58) were collected and processed for cresyl violet staining. Briefly, the slices were dehydrated using a 50% chloroform/ethanol mixture and stained with 0.5% cresyl violet solution, followed by dehydration steps with ethanol and xylene. The slides were then coverslipped with DPX mount solution. The brains were analyzed for cannulation tip placement using an Olympus BX51 upright microscope and images were processed with an Olympus DP71 Digital Camera and DP manager software. Only mice with correct bilateral amygdala cannula placements were included in the analysis of the behavior experiments.
Drugs and infusion.
Anisomycin (Sigma-Aldrich) was solubilized fresh before every behavior experiment at a concentration of 125 μg/μl in 1 m HCl and adjusted to pH 7.4 using 1 m NaOH (Nader et al., 2000). On the day of reactivation, mice were bilaterally infused with 0.3 μl of anisomycin at a rate of 0.2 μl/min using a 28 gauge infuser that projected 1 mm below the guide cannula, as described previously (Holehonnur et al., 2014). After infusion, the infusers were left in for an additional minute to allow the drug to diffuse away from the cannula, after which time they were withdrawn, the dummy cannula were inserted into the guide cannula, and the mice were returned to their home cages.
Fear conditioning.
Mice were fear conditioned and tested using a standard auditory fear conditioning system equipped with video monitoring (Coulbourn Instruments). All mice were habituated by handling for 2 d before fear conditioning.
Training.
Mice were fear conditioned with one, three, or 10 tone–shock pairings (TSPs). The training consisted of a 150 s acclimation period (preshock period), followed by the presentation of a 30 s, 5 kHz, 75 dB tone that coterminated with a 2 s, 0.75 mA foot shock in a dimly lit training chamber. Three TSP training used an intertrial interval of 120 s and 10 TSP training used a random ITI of 60–450 s, similar to a previous study (Wang et al., 2009). Mice remained in the training chamber for an additional minute after the delivery of the last foot shock (postshock period). A shock intensity of 0.75 mA was used for all fear conditioning experiments following previously established protocols for cued fear conditioning studies in C57BL/6J mice (Stiedl et al., 1999, Anagnostaras et al., 2000).
Memory retrieval.
Mice were subjected to retrieval by exposing them to a novel chamber with distinct tactile, visual, and olfactory cues. They were allowed a 150 s acclimation period, were presented with one tone presentation for 30 s, and then were removed from the chamber 30 s after the tone presentation. Percentage freezing was measured during the tone presentation.
Postretrieval long-term memory (PR-LTM) testing.
PR-LTM testing occurred 24 h after memory retrieval in a novel chamber with distinct tactile, visual, and olfactory cues. The mice were presented with five tone presentations: a 120 s acclimation period and five 30 s tones with 120 s ITIs. Percentage freezing was measured during each tone presentation.
Short-term memory (STM) testing.
STM testing occurred 3 h after training in a novel chamber with distinct tactile, visual, and olfactory cues and consisted of a 150 s acclimation period, followed by the presentation of three 30 s tone presentations with 120 s ITIs. Percentage freezing was measured during each tone presentation.
LTM testing.
LTM testing occurred 24 or 48 h after training in a novel chamber with distinct tactile, visual, and olfactory cues and consisted of a 150 s acclimation period, followed by the presentation of three or five 30 s tone presentations with 120 s ITIs. Percentage freezing was measured during each tone presentation.
Extinction.
Extinction training consisted of a 150 s acclimation period, followed by the presentation of 10 30 s tone presentations with 120 s ITIs per day for 4 d. Percentage freezing was measured during each tone presentation and computed from the recorded videos using Freezeframe software (Coulbourn Instruments). Data extraction and analysis were conducted by multiple experimenters blinded to the experimental conditions.
Experiment 1: Does the intensity of auditory fear conditioning affect memory strength?
Wild-type mice were auditory fear conditioned with either three TSPs (n = 12) or 10 TSPs (n = 12) (Fig. 1A). Two days after fear conditioning, mice were exposed to three tone presentations in a novel testing chamber and the degree of freezing was measured to determine the strength of LTM. Data points in Figure 1A indicate average percentage freezing across three tones.
Figure 1.

Auditory fear memories that are resistant to retrieval-dependent destabilization are associated with an increase in the GluN2A/GluN2B subunit ratio in the BLA. A, Mice trained with 10 TSPs froze significantly more than mice trained with TSPs during an LTM test, indicating that 10 TSP training results in stronger fear memory compared with three TSP training. B, Behavior timeline for a reconsolidation experiment using mice trained with three TSPs. Mice were auditory fear conditioned with three TSPs and, 2 d later, retrieval of the fear memory was induced by exposing the mice to a single tone presentation. After this, the mice were immediately infused with either anisomycin or vehicle into the BLA. One day later, the mice were reexposed to tones in a novel context during a PR-LTM test and freezing was measured. C, There were no differences in percentage freezing during retrieval, but the anisomycin group froze significantly less compared with the vehicle group during the PR-LTM test, indicating that these mice did exhibit retrieval-dependent memory destabilization. D, Behavior timeline for a reconsolidation experiment using 10 TSPs. Mice were auditory fear conditioned with 10 TSPs and, 2 d later, retrieval of the fear memory was induced by exposing the mice to a single tone presentation. After this, the mice were immediately infused with either anisomycin or vehicle into the BLA. One day later, the mice were reexposed to five tones in a novel context during a PR-LTM test and freezing was measured. E, During retrieval, mice trained with 10 TSPs did not exhibit differences in freezing between the vehicle and anisomycin groups. During PR-LTM, mice trained with 10 TSPs did not exhibit differences in freezing between the vehicle and anisomycin groups, indicating that these mice did not exhibit retrieval-dependent memory destabilization (*p < 0.05). F, Timeline for a Western blotting experiment performed with BLA PSD fractions to determine the levels of GluN1, GluN2A, and GluN2B levels in mice fear conditioned with one TSP, three TSPs, and 10 TSPs. GluN1, GluN2A, and GluN2B protein levels were normalized to PSD-95 levels to account for variability in protein loading and group means were reported. G–J, Western blot and quantification of protein levels in BLA PSDs extracted from mice 48 h after training. One-way ANOVA between groups that were trained with one TSP, three TSPs, or 10 TSPs revealed that there were no differences in GluN1 levels (G), GluN2A levels (H), or GluN2B levels. However, there was a trend for an increase in GluN2A levels and a trend for a decrease in GluN2B as TSP increased, respectively. J, The GluN2A/GluN2B ratio was significantly increased in mice trained with 10 TSPs compared with one TSP and three TSPs and no differences were observed between one TSP and three TSPs (*p < 0.05). Error bars indicate SEM.
Experiments 2 and 3: Does the intensity of auditory fear conditioning affect retrieval-dependent memory destabilization?
Wild-type mice were bilaterally cannulated into the BLA. After recovery, the mice were auditory fear conditioned with either three TSPs (Experiment 2) or 10 TSPs (Experiment 3) (Fig. 1C,E). Two days later, mice were subjected to retrieval by exposing them to a single tone presentation. The mice were then immediately infused bilaterally with 0.3 μl of vehicle (0.9% saline) or anisomycin into the BLA as described previously (Nader et al., 2000). Twenty-four hours after retrieval, mice were subjected to PR-LTM by exposing them to five tone presentations. Data points in Figure 1, C and E, indicate the average percentage freezing across one tone for retrieval and five tones for PR-LTM. For Experiment 2 (three TSPs): vehicle, n = 10; anisomycin, n = 8. For Experiment 3 (10 TSPs): vehicle, n = 10; anisomycin, n = 9.
Experiment 4: Does the intensity of auditory fear conditioning affect synaptic GluN levels?
Wild-type mice were auditory fear conditioned with one TSP (n = 8), three TSPs (n = 10), or 10 TSPs (n = 13) as described above (Fig. 1G–J). Two days after fear conditioning, mice were killed to examine the differences in GluN1, GluN2A, and GluN2B subunit levels as a result of training. The mice were rapidly decapitated after brief sedation with CO2. The brains were rapidly dissected and frozen using powdered dry ice and stored at −80°C until further processing.
Experiment 5: What is the spatial distribution of GFP-GluN2A transgene in the brain of GFP-GluN2A+/tTA+ mice?
GFP-GluN2A+ (control) and GFP-GluN2A+/tTA+ mice maintained on a normal diet of laboratory chow were killed at ∼2 months of age by perfusion (Fig. 2B–F) and their brains were sectioned and analyzed for GFP expression as described above. Figure 2, B–F, illustrates the representative patterns of transgene expression.
Figure 2.
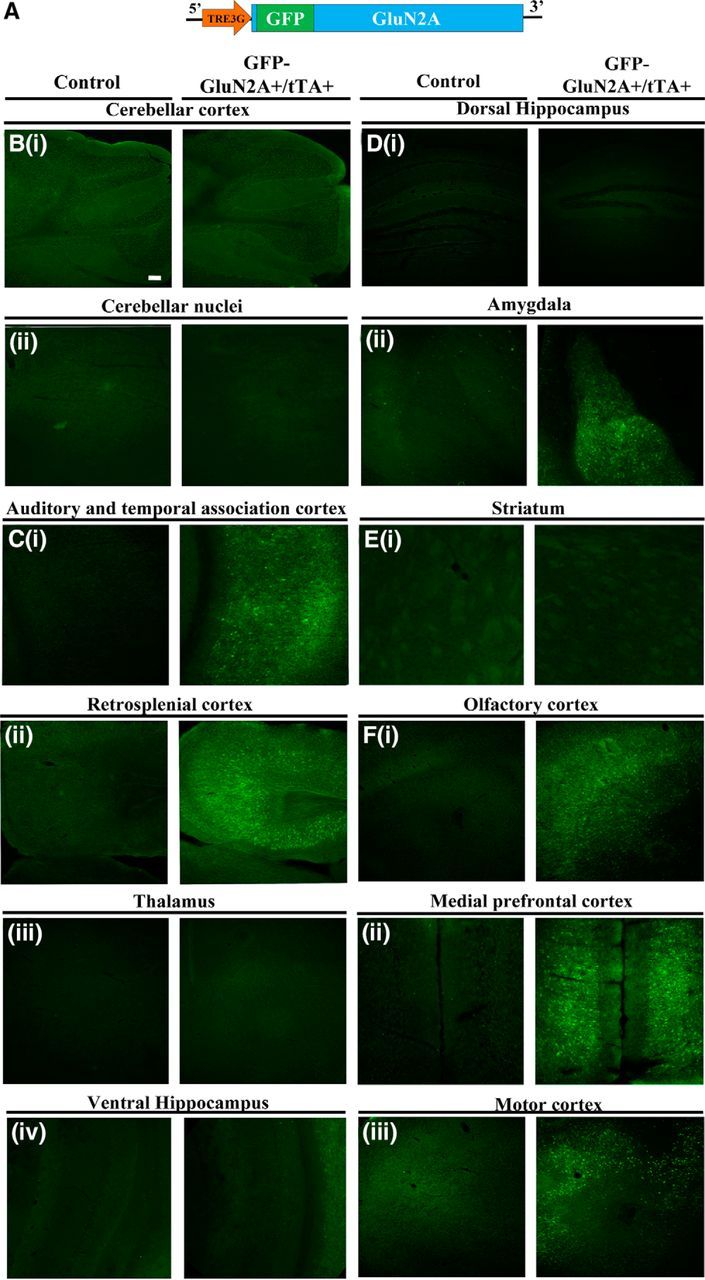
Spatiotemporal characterization of the GFP-GluN2A transgene expression in GFP-GluN2A+/tTA+ mice and its regulation using Dox. A, GFP-GluN2A transgene used to generate an inducible transgenic line expressing GFP-GluN2A+ from a TRE3G promoter. B–F, Representative coronal images depicting GFP-GluN2A transgene expression (GFP fluorescence) throughout the brain in double-transgenic mice (GFP-GluN2A+/tTA+) mice compared with controls (GFP-GluN2A+). Coronal images from bregma (−6.12; B) demonstrates no transgene expression in cerebellar cortex (Bi) and cerebellar nuclei (Bii). C, Bregma (−3.52) demonstrates positive transgene expression in auditory and temporal association cortex (Ci) and retrosplenial cortex (Cii), but not in thalamus (Ciii) or ventral hippocampus (Civ). D, Bregma (−1.82) demonstrates no transgene expression in dorsal hippocampus and central nucleus (Di), but positive transgene expression in basal and lateral amygdala (Dii). Ei, Bregma (−0.10) demonstrates no transgene expression in striatum. F, Bregma (+1.98) demonstrates positive transgene expression in olfactory cortex (Fi), medial prefrontal cortex (Fii), and motor cortex (Fiii). Scale bar, 100 μm (50×).
Experiment 6: Can the expression of the GFP-GluN2A transgene be regulated using Dox?
GFP-GluN2A+/tTA+ mice were maintained on a diet containing Dox for 6 or 8 weeks or were maintained on the same diet for 6 weeks and then taken off Dox for 2 weeks before they were perfused and their brains sectioned and analyzed for GFP expression as described above (Fig. 3B). Figure 3B shows representative images of transgene regulation using Dox.
Figure 3.
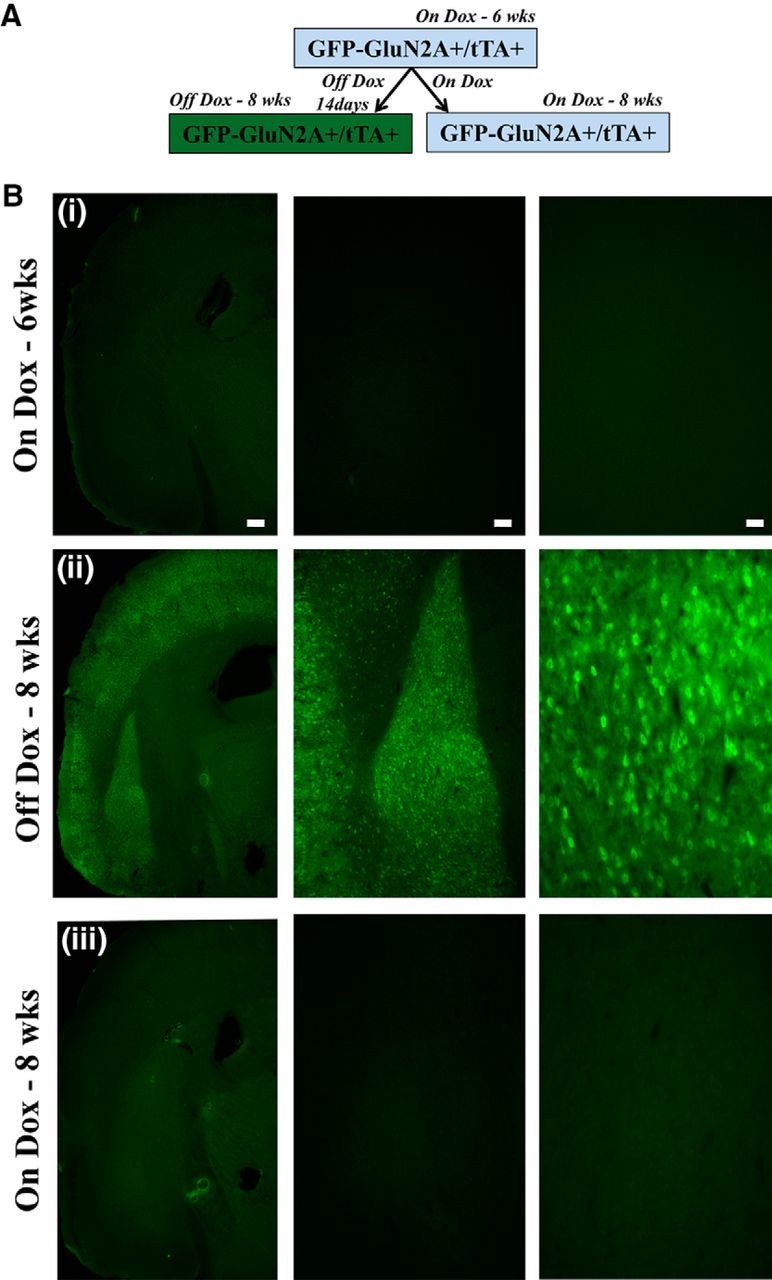
Regulation of transgene expression in GFP-GluN2a+/tTA+ using Dox. A, Timeline for temporal regulation of transgene expression using Dox. B, Transgene expression in GFP-GluN2A+/tTA+ mice is tightly regulated using Dox. Representative coronal images containing BLA show no GFP-GluN2A transgene expression in GFP-GluN2A+/tTA+ mice that were On-Dox at 6 weeks (Bi) and 8 weeks (Biii), but Bii depicts robust transgene expression when Dox is removed from the mice diet for 2 weeks. Scale bars (left to right), 250 μm (20×), 100 μm (50×), 50 μm (100×).
Experiments 7 and 8: Does overexpression of GFP-GluN2A transgene lead to an increase in synaptic GluN2A subunits?
GFP-GluN2A+ (control) and GFP-GluN2A+/tTA+ mice maintained on a normal diet of laboratory chow were used for electrophysiology and Western blotting experiments as described above. For Experiment 7 (electrophysiology): GFP-GluN2A+ (control) 4B, n = 15; 4D, n = 8; GluN2A+/tTA+, 4B, n = 16; 4D, n = 7. For Experiment 8 (Western blotting): GFP-GluN2A+ (control), n = 7; GFP-GluN2A+/tTA+, n = 5 (Fig. 4).
Figure 4.

Overexpression of GFP-GluN2A alters NMDA currents in pyramidal neurons and leads to an increase in the GluN2A/GluN2B ratio within the BLA. A, Example traces of evoked EPSCs in control (left column) and GFP-GluN2A expressing neurons. Pharmacologically isolated NMDA currents (black traces) were recorded at +40 mV and then either the GluN2AR antagonist PEAQX (top row) or the GluN2BR antagonist Ro25-6981 (bottom row) was bath applied. The residual currents are shown in gray. B, Summary graph of the data shown in A. Cells from mice overexpressing GFP-GluN2ARs (gray bars) show a much larger reduction in current after PEAQX application than those from control littermates (black bars). GluN2BR-mediated currents, measured as a reduction after Ro25-6981 application, were not differentially affected between control and GFP-GluN2A+/tTA+ mice. C, Normalized sample traces of total NMDA (left) and isolated GluN2A currents (Ro25, top right) and GluN2B currents (PEAQX, bottom right). D, Expression of GluN2AR decreases decay time of the total NMDA current, but does not significantly affect currents in the presence of either Ro25-6981 or PEAQX. E–H, Western blot and quantification of GluN1, GluN2A, and GluN2B levels in BLA PSDs extracted from transgenic mice (GFP-GluN2A+/tTA+) expressing GFP-GluN2A compared with controls (GFP-GluN2A+). E, No differences in GluN1 levels were detected. F, G, A significant increase in GluN2A levels and a significant reduction in GluN2B levels were detected in GFP-GluN2A+/tTA+ mice. H, A significant increase in the GluN2A/GluN2B ratio was detected in GFP-GluN2A+/tTA+ mice. Error bars indicate SEM (*p < 0.05).
Experiments 9 and 10: Does an increase in the GluN2A/GluN2B ratio influence modification of an existing memory trace via reconsolidation updating?
GFP-GluN2A+ (control mice) in Experiment 9 and GFP-GluN2A+/tTA+ mice in Experiment 10 were maintained on a diet containing Dox and then were cannulated bilaterally into the BLA (Fig. 5B,D). After recovery, the mice were fear conditioned using three TSPs as described above. Two days after training, the Dox-containing diet was replaced with regular laboratory chow for half of the GFP-GluN2A+/tTA+ mice (Off-Dox) and half of the control mice (Off-Dox). The remaining half of GFP-GluN2A+/tTA+ mice (On-Dox) and control mice (On-Dox) remained on the Dox-containing diet. Fourteen days later, mice were subjected to memory retrieval by exposing them to a single tone presentation. The mice were then immediately infused bilaterally with 0.3 μl of vehicle (0.9% saline) or anisomycin into the BLA. Twenty-four hours after retrieval, mice were subjected to PR-LTM by exposing them to five tone presentations. Data points in Figure 5, B and D, represent average percentage freezing across one tone for retrieval and five tones for PR-LTM. For Experiment 9: GFP-GluN2A+ (control), On-Dox/vehicle, n = 8; Off-Dox/vehicle, n = 7; On-Dox/anisomycin, n = 6; Off-Dox/anisomycin, n = 6. For Experiment 10: GFP-GluN2A+/tTA+, On-Dox/vehicle, n = 13; Off-Dox/vehicle, n = 12; On-Dox/anisomycin, n = 12; Off-Dox/anisomycin, n = 12).
Figure 5.

An increase in the GluN2A/GluN2B ratio within BLA neurons inhibits the modification of an existing memory trace and reduces retrieval-dependent AMPAR trafficking. A, Behavior timeline for a reconsolidation experiment using control mice (GFP-GluN2A+ only) trained with three TSPs. Two days after training, half of the mice were removed from the Dox-containing diet. Fourteen days later, the mice were induced to retrieve the fear memory by exposing them to a single tone presentation and then the mice were immediately infused into the BLA with either anisomycin or vehicle. One day later, the mice were reexposed to five tones in a novel context during a PR-LTM test and freezing was measured. B, During retrieval, control mice did not exhibit a significant difference in freezing levels. During PR-LTM, there was a significant main effect of percentage freezing. The On-A group froze significantly less than the On-V group and the Off-V group. Further, the Off-A group froze significantly less than the Off-V group and the On-V group. However, there were no differences in percentage freezing due to Dox administration alone in the A or V groups (V = vehicle, A = anisomycin; *p < 0.05). C, Behavior timeline for a reconsolidation experiment using GFP-GluN2A+/tTA+ mice trained with three TSPs using a similar methodology as described in A. D, During retrieval, GluN2A+/tTA+ mice did not exhibit a significant difference in freezing levels. During PR-LTM, there was a significant main effect of percentage freezing. The On-A group froze significantly less than the On-V group, the Off-V group, and the Off-A group, but there were no differences between the Off-A and Off-V and On-V groups. There were also no differences in percentage freezing due to Dox administration alone in the vehicle group. E, Timeline for Western blotting experiment on BLA PSDs to determine the levels of pGluR1(Ser-845) and tGluR1 in control mice 1 h after retrieval (V = vehicle, A = anisomycin; *p < 0.05). F, In control mice, there was a significant effect of group on the pGluR1/tGluR1 ratio. The pGluR1/tGluR1 ratio was significantly higher in the On-Ret group compared with the On-No Ret and Off-No Ret groups. Further, the pGluR1/tGluR1 ratio was significantly higher in the Off-Ret group compared with the Off-No Ret and On-No Ret groups. However, there were no differences in the ratio due to Dox administration alone in the retrieval or no retrieval groups. G, Timeline for a Western blotting experiment on BLA PSDs to determine the levels of pGluR1(Ser-845) and tGluR1 in GFP-GluN2A+/tTA+ mice 1 h after retrieval (*p < 0.05). H, There was a significant effect of group on the pGluR1/tGluR1 ratio. The On-Ret group had significantly higher pGluR1/tGluR1 levels compared with the On-No Ret, Off-No Ret, and Off-ret groups. There were no significant differences in the pGluR1/tGluR1 ratio in the Off-Ret compared with Off-No Ret and the On-No Ret groups. In addition, there was no effect of Dox administration alone in the no retrieval groups (*p < 0.05). Error bars indicate SEM.
Experiments 11 and 12: Does an increase in the GluN2A/GluN2B ratio influence retrieval-dependent AMPAR trafficking?
GFP-GluN2A+ (control mice) in Experiment 11 and GFP-GluN2A+/tTA+ mice in Experiment 12 were maintained on a diet containing Dox (Fig. 5F,H). These mice were then fear conditioned with three TSPs. Two days after training, the Dox-containing diet was replaced with regular laboratory chow for half of the GFP-GluN2A+/tTA+ mice (Off-Dox) and half of the control mice (Off-Dox). The remaining half of GFP-GluN2A+/tTA+ mice (On-Dox) and control mice (On-Dox) remained on the Dox-containing diet. Fourteen days later, mice in the retrieval group (On-Ret and Off-Ret) were subjected to memory retrieval by exposing them to a single tone presentation. Mice in the no retrieval group (On-No-Ret and Off-No-Ret) received no tone presentation. One hour later, mice in both groups were rapidly decapitated after brief sedation with CO2. Brains were rapidly dissected and frozen using powdered dry ice and immediately stored at −80°C until further processing. For Experiment 11: GFP-GluN2A+ (control), On-Dox/no retrieval, n = 11; On-Dox/retrieval, n = 11; Off-Dox/no retrieval, n = 10; Off-Dox/retrieval, n = 11. For Experiment 12: GFP-GluN2A+/tTA+, On-Dox/no retrieval, n = 13; On-Dox/retrieval, n = 12; Off-Dox/no retrieval, n = 12; Off-Dox/retrieval, n = 13.
Experiment 13: Does an increase in the GluN2A/GluN2B ratio influence consolidation of fear memory?
Wild-type, α-CaMKII-tTA+, GFP-GluN2A+, and GFP-GluN2A+/tTA+ mice were maintained on Dox (Fig. 6B,C). Dox was then removed from their diet for 14 d before mice were fear conditioned with three TSPs as described above. Three hours after training, mice were subjected to the STM test by exposing them to three tone presentations. Twenty-four hours after training, mice were subjected to an LTM test by exposing them to five tone presentations. Data points in Figure 6, B and C, represent average percentage freezing across three tones for STM and five tones for LTM. Wild-type, n = 11; α-CaMKII-tTA+, n = 14; GFP-GluN2A+, n = 11; GFP-GluN2A+/tTA+, n = 12.
Figure 6.

An increase in the GluN2A/GluN2B ratio before fear learning impairs LTM consolidation. A, Behavior timeline for fear learning experiment. Wild-type, α-CaMKII-tTA+, GFP-GluN2A+, and GFP-GluN2A+/tTA+ mice were maintained on Dox, Dox was removed from their diet for 14 d, and then the mice were fear conditioned with three TSPs. Three hours after training, the mice conditioned fear responses to the auditory cue were examined in an STM test in which the mice were exposed to three tones. Twenty-one hours later, LTM was examined where the mice were exposed to five tones. B, For STM, there was no significant main effect of genotype on percentage freezing (wild-type, α-CaMKII-tTA+, GFP-GluN2A+, or GFP-GluN2A+/tTA+). C, For LTM, there was a significant main effect of genotype on percentage freezing. The GFP-GluN2A+/tTA+ group froze significantly less compared with the wild-type, α-CaMKII-tTA+, and GFP-GluN2A+ groups. However, there were no differences in percentage freezing among the wild-type, α-CaMKII-tTA, and GFP-GluN2A+ groups (wild-type vs α-CaMKII-tTA, α-CaMKII-tTA vs GFP-GluN2A+, GFP-GluN2A+ vs wild-type (*p < 0.05). Error bars indicate SEM.
Experiment 14: Does an increase in the GluN2A/GluN2B ratio influence fear expression and extinction?
GFP-GluN2A+/tTA+ were maintained on a diet containing Dox (Fig. 7B,C). At ∼2 months of age, the mice were fear conditioned with three TSPs as described above. Two days after training, the Dox-containing diet was replaced with regular laboratory chow for half of the GFP-GluN2A+/tTA+ mice (Off-Dox) and the remaining half of the GFP-GluN2A+/tTA+ mice (On-Dox) remained on the Dox-containing diet. Fourteen days later, the mice were subjected to extinction training. Figure 7B shows the percentage freezing averaged across the first three tones during LTM1. Figure 7C shows percentage freezing averaged across 10 tones for each LTM session during extinction (days 1–4). On-Dox, n = 9; Off-Dox, n = 9.
Figure 7.

An increase in the GluN2A/GluN2B ratio after learning has no influence on fear memory extinction. A, Behavior timeline for fear expression and fear extinction experiment. GFP-GluN2A+/tTA+ mice were maintained on a diet containing Dox and then fear conditioned with three TSPs. Two days after training, half of the mice had Dox removed from their diet. Fourteen days later, the mice were exposed to 10 tones once per day over a period of 4 d. For expression of fear, freezing behavior was reported for the first three tones presented during LTM1. For fear extinction behavior, freezing behavior during LTMs 1, 2, 3, and 4 is presented. B, For fear expression, there was no significant effect of group on percentage freezing. C, For fear extinction, there was no significant effect of group on percentage freezing during LTM1, LTM2, LTM3, and LTM4. Error bars indicate SEM.
Statistics.
For fear conditioning experiments, columns/data points in the graph indicate average percentage freezing across tones presented during the session and error bars indicate SEM. One-way-ANOVA with repeated measures and Fisher's PLSD post hoc test were used to determine the statistical significance for all behavior experiments. For Western blotting experiments, columns represent average of average integrated density for each group and error bars indicate SEM. One-way ANOVA and Fisher's PLSD post hoc test were used to determine statistical significance for Western blotting. p-values <0.05 were considered significant. Electrophysiological data were presented as group means, with error bars representing the SEM. Statistical differences were determined using Student's t tests (two-tailed). p-values <0.05 were considered significant.
Results
Auditory fear memories that are resistant to retrieval-dependent destabilization are associated with an increase in the GluN2A/GluN2B subunit ratio in the BLA
In our first set of experiments, we aimed to determine how the intensity of fear conditioning training influences the strength of auditory fear memory and the ability of these memories to destabilize upon retrieval. To this end, we auditory fear conditioned mice with either 3 TSPs (3 pairings of a 30 s 5 kHz, 75 dB tone that coterminated with a 2 s, 0.75 mA foot shock), or 10 TSPs (10 pairings of a 30 s 5 kHz, 75 dB tone that coterminated with a 2 s, 0.75 mA foot shock), respectively. Two days after training, the mice were exposed to 3 presentations of the conditioned stimulus (CS; 30 s, 5 kHz tone) in a novel context and the strength of the fear memory was measured as the amount of tone-induced freezing the mice exhibited (Fig. 1A). The 10 TSP group (n = 12) exhibited significantly higher tone-induced freezing than the 3 TSP group (n = 12), indicating that the 10 TSP training resulted in stronger memories (F(1,22) = 7.086; p = 0.0142). Next, mice were implanted bilaterally with cannula into the BLA and auditory fear conditioned with either three or 10 TSPs. Two days after training, the mice were exposed to a single presentation of the CS (30 s, 5 kHz tone) in a novel context to induce retrieval of the fear memory and tone-induced freezing was measured. Immediately after retrieval, the mice were infused bilaterally with 0.3 μl of vehicle or anisomycin into the BLA. Twenty-four hours after retrieval, the mice were exposed to five tones and the strength of the fear memory was assessed by measuring the degree of tone-induced freezing that the mice exhibited during a PR-LTM test. For mice trained with 3 TSPs, a repeated-measures ANOVA indicated no significant differences in freezing levels between the vehicle-treated group (n = 10) and the anisomycin-treated group (n = 8) during retrieval (F(1,16) = 0.031; p = 0.8635). However, there was a significant decrease in freezing in the anisomycin group compared with the vehicle group during the PR-LTM test (F(1,16) = 18.505; p = 0.0005; Fig. 1C). These results are consistent with previous studies showing that postretrieval anisomycin administration produces amnesia (Nader et al., 2000). This is believed to be due to the fact that blocking protein synthesis impairs reconsolidation of the retrieved destabilized memory. For mice trained with 10 TSPs, there were no significant differences in freezing levels between the vehicle-treated (n = 10) or anisomycin-treated (n = 9) groups during retrieval (F(1,17) = 0.11; p = 0.7435) or PR-LTM (F(1,17) = 0.125; p = 0.7284) (Fig. 1E), indicating that administration of anisomycin did not cause amnesia in the mice that underwent 10 TSP training. This may be due to the fact that memories formed via 10 TSP training do not undergo retrieval-dependent memory destabilization, thus rendering these auditory fear memories immune to anisomycin. These results are consistent with similar experiments reported previously in rats (Wang et al., 2009).
Previous studies have indicated that NMDAR activity at the very moment of memory retrieval is crucial to induce memory destabilization (Ben Mamou et al., 2006) and that the NMDAR may undergo training-dependent alterations in its subunit composition that endure past the consolidation window (Wang et al., 2009). Because NMDAR subunit composition has been shown to alter NMDAR function (Barria and Malinow, 2005, Foster et al., 2010, Paoletti et al., 2013), we hypothesized that there might be training-dependent changes in the synaptic GluN2A/GluN2B subunit ratio that endure past the consolidation window that serve to regulate retrieval-dependent fear memory destabilization and essentially serve to regulate the ability of a memory to be modified via reconsolidation updating. To begin to test this hypothesis, we auditory fear-conditioned mice with differing training intensities consisting of 1 TSP (n = 8), 3 TSPs (n = 10), or 10 TSPs (n = 13) and 2 d after training, the mice were killed and postsynaptic density (PSD) fractions were purified from BLA tissue. We then examined the synaptic levels of GluN1, GluN2A, and GluN2B via Western blotting. We found that GluN1 levels did not differ across training intensities (F(2,28) = 0.271; p = 0.7848; Fig. 1G). Furthermore, a one-way ANOVA also revealed that training intensity did not influence the levels of either GluN2A (F(2,28) = 2.362; p = 0.1128; Fig. 1H) or GluN2B (F(2,28) = 2.010; p = 0.1528; Fig. 1I) significantly. However, pairwise comparisons between the groups indicated a trend toward an increase in GluN2A levels as training intensity increased (1 TSP vs 10 TSPs: p = 0.114; 3 TSPs vs 10 TSPs: p = 0.031) and a decrease in GluN2B levels as training intensity increased (1 TSP vs 10 TSPs: p = 0.018; 3 TSPs vs 10 TSPs: p = 0.266). Considering this trend, we speculated that the increase in the ratio of GluN2A/GluN2B subunits might result from the increase in training intensity. A one-way ANOVA revealed significant main effects for the intensity of training and the GluN2A/GluN2B ratio (F(2,28) = 5.989; p = 0.0068; Fig. 1J). Post hoc comparisons using Fisher's PLSD furthermore showed a significant increase in the GluN2A/GluN2B ratio in the 10 TSP group compared with the 1 TSP (p = 0.0027) and the 3 TSP (p = 0.0279) groups. However, the 1 TSP and 3 TSP groups did not differ in their GluN2A/GluN2B ratios (p = 0.298). Together, these data indicate that an increase in auditory fear conditioning training intensity is correlated with an increase in the GluN2A/GluN2B subunit ratio.
Spatiotemporal, electrophysiological, and molecular characterization of the GFP-GluN2A transgene expression in GFP-GluN2A+/tTA+ mice
Based on the experiments outlined above, we wondered whether an increase in the GluN2A/GluN2B ratio is sufficient to prevent retrieval-dependent memory destabilization. To test this hypothesis more directly, we genetically engineered mice in which the GluN2A/GluN2B ratio could be increased selectively after learning. Therefore, we generated an inducible transgenic mouse line designed to express a GFP-GluN2A transgene from a third-generation tetracycline-responsive element containing promoter, TRE3G (Fig. 2A). When these mice are crossed with the transgenic line designed to express the tTA transcriptional activator under the regulation of α-CaMKII promoter (Mayford et al., 1996), the GFP-GluN2A transgene expression is restricted to α-CaMKII positive neurons in the mouse forebrain. Administration of Dox to the GFP-GluN2A+/tTA+ mice results in repression of the GFP-GluN2A transgene, allowing for temporal control over when this transgene is expressed.
To identify the spatial distribution of the GFP-GluN2A transgene expression in the GFP-GluN2A+/tTA+ mice, we examined GFP fluorescence in coronal slices from double-transgenic mice (GFP-GluN2A+/tTA+) and GFP-GluN2A+ only transgenic mice (control). The images were acquired at the same exposure times to allow for direct comparison of region-specific transgene expression in GFP-GluN2A+/tTA+ and control mice (Fig. 2C). Across the brain regions examined (bregma −6.12 to +1.98), there was robust GFP-GluN2A transgene expression within pyramidal neurons in the temporal association and auditory cortex (2Ci), retrosplenial cortex (2Cii), BLA (2Dii), olfactory cortex (2Fi), medial prefrontal cortex (2Fii), and motor cortex (2Fiii) in GFP-GluN2A+/tTA+ mice compared with controls. However, no GFP-GluN2A expression was observed in the cerebellar cortex (2Bi), cerebellar nuclei (2Bii), thalamus (2ciii), CeA (2Dii), ventral hippocampus (2Civ), dorsal hippocampus (2Di), and the striatum (2Ei). The specific pattern of GFP-GluN2A expression across the brain may in part be due to a positional effect of where the GFP-GluN2A+ transgene was inserted into the genome of the founding line of mice. Collectively, these results demonstrate the absence of leaky expression in single-transgenic mice (controls) and robust GFP-GluN2A transgene expression within neurons in the BLA and cortex of GFP-GluN2A+/tTA+ mice. In addition, there was no difference in baseline expression between GFP-GluN2A+ only and wild-type mice (data not shown), indicating the absence of leaky expression in GFP-GluN2A+ mice. Next, we examined the temporal regulation of the GFP-GluN2A transgene expression using Dox. To this end, we obtained coronal sections from GFP-GluN2A+/tTA+ mice that received Dox for 6 and 8 weeks or received Dox for 6 weeks and then were taken off Dox for 2 weeks. Analysis of GFP expression from these slices indicated successful repression of the GFP-GluN2A transgene when Dox was administered to the mice for 6 (3Bi) and 8 (3Biii) weeks. However, if the mice were on Dox for 6 weeks and then removed from Dox for 2 weeks, then the GFP-GluN2A transgene showed robust expression (3Bii). These data indicate that Dox administration can repress GFP-GluN2A transgene expression successfully and that 2 weeks off Dox is sufficient to induce robust GFP-GluN2A transgene expression (Fig. 3B).
Next, we investigated whether expression of the GFP-GluN2A transgene led to an increase in synaptic GluN2A subunits that can contribute to NMDAR signaling. To this end, we used whole-cell patch-clamp recordings in slices from adult GFP-GluN2A+/tTA+ or GFP-GluN2A+ (control) mice. We first recorded total NMDAR currents and then bath applied selective antagonists of GluN2A or GluN2B, respectively, to measure the relative change in the evoked response (Fig. 4A). To measure the contribution of GluN2A subunits, we applied 0.5 μm PEAQX and then digitally subtracted the remaining current, which reflects the current through synaptic GluN2B and potentially other subunits. Similarly, to isolate the GluN2B contribution to the total current, we bath applied the GluN2B antagonist Ro25-6981 (0.5 μm) and then subtracted the remaining current. All recordings used low stimulation intensities (between 100 and 200 μV) in an effort to isolate monosynaptic connections. Within this narrow range of stimulation intensities, there was no difference in the average amplitude of total NMDA currents before application of subunit-specific antagonists (control = 47.07 ± 5.2 pA, n = 22; GFP-GluN2A+/tTA+ = 47.04 ± 6.1 pA, n = 27). However, overexpression of GFP-GluN2A in GFP-GluN2A+/tTA+ mice led to a significant increase in GluN2A-mediated currents compared with controls (t(24) = 4.916; p = 0.00005; Fig. 4B). In addition, a small, nonsignificant decrease in GluN2B-mediated currents was also observed (t(21) = 0.722; p = 0.478; Fig. 4B). Overexpression of GluN2A also decreased the decay time of the total NMDAR current (Fig. 4C), reflecting the faster decay times of GluN2A subunits (t(26) = 2.468; p = 0.021; Fig. 4D). However, the decay times of isolated GluN2A-mediated currents (t(13) = 0.542; p = 0.597) or GluN2B-mediated currents (t(11) = 0.346; p = 0.736; Fig. 4D) did not differ significantly between GFP-GluN2A+/tTA+ and control groups. Together, these data indicate that overexpression of GFP-GluN2A leads to an increase in functional synaptically localized GluN2A receptors and a change in the kinetics of the total NMDAR current. In addition, these data also suggest that overexpression of GFP-GluN2A in GluN2A+/tTA+ mice might result in a small compensatory reduction of GluN2B receptors in BLA pyramidal neurons.
We further corroborated this electrophysiological characterization of NMDARs in GFP-GluN2A+/tTA+ mice using Western blotting. We purified PSDs from BLA of GFP-GluN2A+/tTA+ mice (n = 5) and GFP-GluN2A+ only mice (control, n = 7). Western blotting for GluN1, GluN2A, and GluN2B protein levels showed comparable GluN1 levels between the two groups (t(10) = 0.20; p = 0.41; Fig. 4E). In GFP-GluN2A+/tTA+ mice, synaptic GluN2A levels were increased significantly (t(10) = 2.52;p = 0.01; Fig. 4F) and synaptic GluN2B levels were decreased significantly (t(10) = 2.04; p = 0.03; Fig. 4G) compared with controls. These results indicated that the expression of GFP-GluN2A in GFP-GluN2A+/tTA+ mice leads to an increase in the number of synaptically localized GluN2A-containing NMDARs and a decrease in the synaptic GluN2B-containing NMDARs, as evidenced by a significant upregulation in the GluN2A/GluN2B subunit ratio (t(10) = 3.467; p = 0.003; Fig. 4H) in GFP-GluN2A+/tTA+ mice. These findings are consistent with previous findings in organotypic neurons showing that ectopically expressed GluN2A leads to an increase in synaptically localized GluN2A and a displacement of GluN2B from the synapse (Barria and Malinow, 2002).
Upregulation of the GluN2A/GluN2B ratio in BLA neurons inhibits the modification of an existing fear memory trace and reduces retrieval-dependent AMPAR trafficking
Next, we investigated whether increasing the GluN2A/GluN2B ratio in BLA neurons after a fear memory has formed can render this memory resistant to retrieval-dependent memory destabilization. To accomplish this, we inserted cannulas bilaterally into the BLA of GFP-GluN2A+ (controls) and GFP-GluN2A+/tTA+ mice, which had been maintained on a diet of Dox food. After recovery, we auditory fear conditioned the mice with three TSPs, a protocol that produces fear memories that are susceptible to retrieval-dependent memory destabilization in unaltered mice. Two days after training, we removed Dox from the diet of half of the control mice and half of the GFP-GluN2A+/tTA+ mice to induce transgene expression. Two weeks later, the mice were subjected to memory retrieval using a single presentation of the CS, followed by infusions of vehicle or anisomycin into the BLA. Twenty-four hours after retrieval, the mice were exposed to multiple CS's and freezing was assessed during a PR-LTM test. Control mice did not exhibit significantly different freezing levels during memory retrieval (F(3,23) = 0.230; p = 0.874; On-Dox/vehicle, n = 8; Off-Dox/vehicle, n = 7; On-Dox/anisomycin, n = 6; Off-Dox/anisomycin, n = 6). However, there was a significant main effect of treatment on percentage freezing during the PR-LTM test (F(3,23) = 5.018; p = 0.0080; Fig. 5B). Fisher's PLSD post hoc tests revealed that both anisomycin groups froze significantly less than vehicle control groups (p < 0.05), but Dox administration did not influence freezing levels for either the anisomycin groups (p = 0.5795) or the vehicle groups (p = 0.5650). Collectively, these data indicate that control mice exhibited retrieval-dependent memory destabilization that was independent of Dox administration.
GFP-GluN2A+/tTA+ mice did not exhibit significantly different freezing levels during memory retrieval (F(3,45) = 0.090; p = 0.965; On-Dox/vehicle, n = 13; Off-Dox/vehicle, n = 12; On-Dox/anisomycin, n = 12; Off-Dox/anisomycin, n = 12). A one-way ANOVA indicated a significant main effect of treatment on percentage freezing during PR-LTM (F(3,45) = 3.384; p = 0.026). Fisher's PLSD post hoc test revealed that the On-Dox/anisomycin group froze significantly less than all other groups during PR-LTM (p < 0.0168), consistent with an anisomycin-induced weakening of a retrieved memory. However, the Off-Dox/anisomycin group did not exhibit significantly different freezing levels compared with the vehicle control groups (p > 0.7850) and the vehicle control groups did not significantly differ in their levels of freezing (p = 0.8015; Fig. 5D). Collectively, these results indicate that increasing the GluN2A/GluN2B ratio in BLA neurons (Off-Dox group) interfered with retrieval-dependent memory destabilization, as indicated by a lack of anisomycin-induced amnesia in the Off-Dox GFP-Glu2A+/tTA+ mice. These data also indicate that the expression of GFP-GluN2A does not influence the expression of fear because the On-Dox vehicle group and the Off-Dox vehicle group did not differ in their levels of freezing (p = 0.8015).
Next, we examined how increasing the GluN2A/GluN2B ratio might influence retrieval-dependent molecular changes that occur at BLA synapses during memory destabilization. It has been demonstrated previously that retrieval-dependent memory destabilization induces phosphorylation of GluR1 at the Ser-845 site (pGluR1), which serves a marker for increased membrane trafficking of GluR1 (Jarome et al., 2012). This increase in GluR1 is believed to facilitate synaptic plasticity by converting calcium-impermeable AMPARs to calcium-permeable AMPARs (Hong et al., 2013). In addition, NMDAR activity is believed to be crucial for this GluR1 membrane trafficking (Hong et al., 2013). Therefore, we tested the hypothesis that increasing the GluN2A/GluN2B ratio would prevent retrieval-dependent AMPAR trafficking, as measured by reducing retrieval-dependent pGluR1845 levels. To this end, we auditory fear-conditioned GFP-GluN2A+/tTA+ and control (GFP-GluN2A+) mice and had them retrieve the fear memory as described above. One hour after retrieval, we killed the mice and subsequently purified PSDs from the lateral amygdala. We then used Western blotting to determine the synaptic levels of pGluR1Ser845 and total GluR1 (tGluR1). Consistent with previous findings (Monfils et al., 2009, Jarome et al., 2012), we observed a significant main effect on the pGluR1/tGluR1 ratio in control mice (F(3,39) = 3.603; p = 0.0217; On-Dox/no retrieval, n = 11; On-Dox/retrieval, n = 11; Off-Dox/no retrieval, n = 10; Off-Dox/retrieval, n = 11; Fig. 5F). Further, Fisher's PLSD post hoc tests revealed that both retrieval groups had significantly higher pGluR1/tGluR1 ratios compared with the no-retrieval groups (p < 0.05) and Dox administration did not influence the pGluR1/tGluR1 ratios for the no-retrieval groups (p = 0.9141) or the retrieval groups (p = 0.5811). In the GFP-GluN2A+/tTA+ group, there was a significant main effect of treatment on the pGluR1/tGluR1 ratio (F(3,46) = 3.036; p = 0.0385; On-Dox/no retrieval, n = 13; On-Dox/retrieval, n = 12; Off-Dox/no retrieval, n = 12; Off-Dox/retrieval, n = 13; Fig. 5H). Fisher's post hoc tests revealed that the On-Dox/retrieval group had significantly higher pGluR1/tGluR1 ratios compared with the no retrieval group (p = 0.0108); however, there was no difference in the pGluR1/tGluR1 ratio between the retrieval and no retrieval groups in Off-Dox group (p = 0.5775). Collectively, these results indicate that an increase in the GluN2A/GluN2B ratio decreased retrieval-dependent phosphorylation of GluR1 Ser 845, which is a marker for retrieval-dependent memory destabilization.
Increase in the GluN2A/GluN2B ratio before fear learning impairs LTM consolidation
It has been demonstrated previously that genetic overexpression of GluN2A in neurons of the mouse forebrain impairs long-term fear memory consolidation (Cui et al., 2013). Therefore, we investigated whether we could similarly affect fear memory consolidation using our inducible transgenic system designed to express GFP-GluN2A. To this end, we auditory fear conditioned mice Off-Dox using three TSPs. Three hours after training, mice were exposed to the CS (tone) in a novel context and freezing was assessed to measure STM retention. Twenty-one hours later, LTM was assessed by exposing the mice to the CS in a novel context, in a similar manner as STM was conducted. There was no main effect of genotype on percentage freezing during the STM test (F(3,44) = 1.036; p = 0.385; wild-type, n = 11; α-CaMKII-tTA+, n = 14; GFP-GluN2A+, n = 11; GFP-GluN2A+/tTA+, n = 12; Fig. 6B). However, there was a significant effect of genotype on percentage freezing during the LTM test (F(3,44) = 3.614; p = 0.0204; Fig. 6C). Fisher's PLSD post hoc tests indicated that mice with an increased GluN2A/GluN2B ratio (GFP-GluN2A+/tTA+) froze significantly less than the other groups (p < 0.045). There was no difference in freezing levels among the wild-type, α-CaMKII-tTA+, and GFP-GluN2A+ mice (p > 0.2918).
Increase in the GluN2A/GluN2B ratio after learning has no influence on fear memory extinction
Next, we sought to determine whether increasing the GluN2A/GluN2B ratio once a fear memory had formed would inhibit its extinction. Therefore, we auditory fear conditioned GFP-GluN2A+/tTA+ mice on Dox using three TSPs. Two days after training, we removed Dox from the diet of half of the mice to induce GFP-GluN2A transgene expression. Two weeks later, we subjected the mice to extinction training, which involved exposing the mice to CS's (10 tones) in a novel context over 4 subsequent days. On day 1, we measured fear expression by averaging the mice freezing to the first 3 tones. Subsequently, we measured and averaged freezing over 10 tones across 4 d (LTM1–4) to determine the rate of extinction. Our data indicate there was not a significant effect of group on fear expression on day 1 (F(1,16) = 0.694; p = 0.4170; On-Dox, n = 9; Off-Dox, n = 9; Fig. 7B). There was also no significant effect of treatment on extinction during LTM1–4 (p > 0.4286); LTM1 (F(1,16) = 0.660, p = 0.4286), LTM2 (F(1,16) = 0.006, p = 0.9374), LTM3 (F(1,16) = 0.018, p = 0.8956), and LTM4 (F(1,16) = 0.698, p = 0.4157; On-Dox, n = 9; Off-Dox, n = 9; Fig. 7C). Collectively, these results indicate that an increase in the GluN2A/GluN2B ratio does not influence fear memory expression or fear memory extinction.
Discussion
We show that increasing the intensity of auditory fear conditioning results in an increase in the GluN2A/GluN2B ratio in BLA neurons and an impairment in retrieval-dependent memory destabilization. We developed a novel transgenic line of mice that show robust expression of a GFP-GluN2A transgene in cortical regions and the BLA of GFP-GluN2A+/tTA+ mice and this expression can be regulated by Dox. Electrophysiological and biochemical analyses indicate that overexpression of GFP-GluN2A leads to an increase in the GluN2A/GluN2B ratio. Behavioral analysis furthermore indicated that increasing GluN2A levels after fear learning led to an inhibition in retrieval-dependent memory destabilization and that this was associated with a reduction in retrieval-dependent phosphorylation of GluR1 at serine-845. Overexpression of GluN2A impaired LTM, but did not influence fear memory expression or the extinction of fear memory.
NMDAR subunit composition influences plasticity and mnemonic processing
GluN2 subunits play an important role in normal and pathological functions of the CNS. Work on the role of NMDARs in synaptic plasticity shows that signaling through these receptors is crucial for learning and memory formation, including auditory fear conditioning and memory destabilization, during reconsolidation updating (Kim et al., 1991, LeDoux, 1993, Tang et al., 1999, Rodrigues et al., 2001, Pedreira et al., 2002, Ben Mamou et al., 2006, Lee et al., 2006, Milton et al., 2013). GluN2A and GluN2B subunits differ considerably in their properties and thus subunit composition can dramatically alter the properties of the NMDAR (Sheng et al., 1994, Cull-Candy et al., 2001, Prybylowski and Wenthold, 2004). The subunits show large differences in their cytoplasmic tails and these differences determine their ability to bind to postsynaptic proteins that regulate the induction of plasticity (Gardoni et al., 1998, Strack and Colbran, 1998). For example, α-CaMKII binds to GluN2B with much higher affinity than to GluN2A and this interaction is a major determinant of the magnitude of hippocampal LTP (Barria and Malinow, 2005).
Recent studies using genetic and pharmacological approaches indicate that the physical presence of GluN2 receptors at the PSD may serve critical functions that are separate from the ion channel function of the receptor (Foster et al., 2010). This fact underscores the potential limitation of using purely pharmacological approaches that block channel function to decipher the contribution of these receptors to mnemonic processing. For example, it was found recently that GluN2B ion channel function is dispensable for LTP; however, the physical presence of GluN2B at the PSD is required for LTP induction and this is likely due to the ability of GluN2B to recruit critical factors such as α-CaMKII to the neuronal membrane via its cytoplasmic tail (Foster et al., 2010). In addition, GluN2A's cytoplasmic tail was found to inhibit LTP induction (Foster et al., 2010). Consistent with the view that the physical presence of these receptors can serve important functions, the GluN2A/GluN2B ratio has been found to be important for regulating plasticity and metaplasticity (Kirkwood et al., 1996, Quinlan et al., 1999a, Quinlan et al., 1999b, Philpot et al., 2001).
During development, an important change in the GluN2A/GluN2B ratio occurs (Monyer et al., 1994, Sheng et al., 1994, Sans et al., 2000). Early in development, GluN2B is the primary subunit present and then the levels of synaptic GluN2A gradually increase. However, the ratio of these subunits can be regulated dynamically (Philpot et al., 2001, Paoletti et al., 2013) and sensory experience and synaptic activity also increase the GluN2A/GluN2B ratio (Quinlan et al., 1999a, Philpot et al., 2001, Ehlers, 2003, Matta et al., 2011). We hypothesized that synapses in the amygdala that are relevant for fear conditioning undergo a change during learning, so GluN2B subunits become replaced by GluN2A subunits, and this shift in the GluN2A/GluN2B ratio provides a constraint on the induction of future plasticity. As the strength of the memory increases, so might the ratio of GluN2A/GluN2B. This process could serve as a mechanism to preserve the integrity of the memory trace and thus serve to inhibit destabilization and modification of the memory. A previous study indicated that fear conditioning that results in fear memories that are resistant to destabilization after retrieval are associated with a downregulation of the GluN2B receptor, whereas GluN1 levels remain unchanged (Wang et al., 2009). However, this previous study did not examine synaptic levels of the receptors and did not examine the levels of GluN2A. Therefore, we examined GluN2B, GluN2A, and GluN1 levels on BLA PSD fractions via Western blotting from mice that were fear conditioned at various intensities. We observed an increase in the GluN2A/GluN2B ratio in mice trained with 10 TSPs compared with three TSPs and one TSP. This increase in the ratio correlated with the ability of these memories to undergo destabilization after retrieval because mice trained with 10 TSPs were insensitive to the amnestic effects of anisomycin, whereas mice trained with three TSPs demonstrated the predicted anisomycin-induced memory impairments. To test directly whether altering the GluN2A/GluN2B ratio in BLA neurons could serve as a mechanism to inhibit the modification of an existing memory, we generated an inducible transgenic mouse line in which we could overexpress the GluN2A subunit in α-CaMKII-positive neurons selectively after fear conditioning. This design allowed us to increase specifically the GluN2A/GluN2B ratio after learning while maintaining normal conditions for initial learning. Our data agree with our hypothesis that increasing the GluN2A/GluN2B ratio prevents memory destabilization and the subsequent updating of the memory.
Increasing the GluN2A/GluN2B ratio before fear learning impaired LTM consolidation significantly. We observed that an increase in the GluN2A/GluN2B ratio had no effect on STM; however, GFP-GluN2A+/tTA+ mice froze significantly less during the LTM test compared with their control littermates. Such a selective impairment in consolidation was also observed in another study that looked at chronic forebrain GluN2A overexpression (Cui et al., 2013). That study found that overexpression of GluN2A led to abnormally compressed LTD, specifically in the 3–5 Hz range, and the investigators speculated that an LTD-like process at this frequency is required for postlearning sculpting of synapses to consolidate the memory trace.
Our results indicate that an increase in the GluN2A/GluN2B ratio due to expression of GFP-GluN2A does not influence fear memory extinction or the expression of fear. Previous studies showed that pharmacological blockade of GluN2A subunits in the amygdala does not affect fear memory extinction (Dalton et al., 2012), whereas GluN2B antagonism does (Sotres-Bayon et al., 2007, Dalton et al., 2012). However, in our model, we increased the GluN2A/GluN2B ratio genetically by overexpressing GluN2A subunits. This makes it difficult to draw direct comparisons with other studies that used pharmacological approaches to study fear memory extinction. Therefore, one of the reasons that we did not observe an impairment in fear extinction might be the fact that overexpression of GluN2A does not influence negatively the induction of 1 Hz-induced LTD (Cui et al., 2013, Jacobs et al., 2014), which is required for extinction learning (Dalton et al., 2012).
Increasing the GluN2A/GluN2B ratio inhibits retrieval-induced phosphorylation of GluR1
Fear memory retrieval induces phosphorylation of AMPA receptor subunit GluR1 at serine-845, which is a marker for the induction of enhanced GluR1 membrane trafficking to convert calcium-impermeable AMPA receptors (CI-AMPARs) to calcium-permeable AMPA receptors (CP-AMPARs; Monfils et al., 2009, Clem and Huganir, 2010). The GluR2 subunit, which blocks calcium entry through this receptor complex, is endocytosed and exchanged for GluR1 subunits, thus creating CP-AMPARs at the membrane. Infusion of TAT-GluR23Y peptides that inhibit the endocytosis of GluR2 into the BLA prevents the conversion of CI-AMPARs to CP-AMPARs and inhibits memory destabilization, indicating that the endocytosis of GluR2 and the conversion of CI-AMPARs to CP-AMPARs are critical steps leading to memory destabilization (Rao-Ruiz et al., 2011, Hong et al., 2013). Notably, retrieval-dependent conversion of CI-AMPARs to CP-AMPARs and endocytosis of GluR2 are dependent on NMDAR activity, thus linking NMDAR activity directly to AMPA receptor subunit trafficking (Hong et al., 2013). It has been demonstrated that retrieved memories that are resistant to modification via anisomycin treatment have reduced phosphorylated GluR1 subunits and an increase in the pGluR1/tGluR1 ratio after retrieval corresponds to a temporal window during which memory modification is possible (Jarome et al., 2012). As a result, a retrieval-induced increase in the pGluR1/tGluR1 ratio serves as a molecular indicator for the initiation of reconsolidation updating. We hypothesized that increasing the GluN2A/GluN2B ratio would lead to a decrease in retrieval-dependent AMPA receptor trafficking. Here, we observed that increasing the GluN2A/GluN2B ratio inhibits retrieval-induced phosphorylation of GluR1. In control mice, retrieval induced increases in the pGluR1/tGluR1 ratio in the On-Dox and Off-Dox groups, as expected, indicating an absence of nonspecific effects of Dox administration on AMPAR trafficking.
Together, these data are the first to demonstrate that increasing the GluN2A/GluN2B ratio inhibits retrieval-dependent memory destabilization and prevents the modification of an existing memory trace. Elucidating the role of the GluN2A/GluN2B ratio in fear memory modification improves our understanding of the molecular mechanisms that can regulate the induction of reconsolidation. This process will be further aided by identifying and targeting critical molecules that can enable the modification of reconsolidation-resistant memories.
Footnotes
This work was supported by the National Institutes of Health (Grant RMH096202A) and The University of Texas at Dallas. We thank Seth G.N. Grant, Marcelo P. Coba, Angela Mabb, and Paola Virginia Migues for constructive advice and technical expertise regarding the purification of PSDs; Mark Mayford for technical assistance with the Tet inducible system; Kelly Foster for sharing insight regarding the role of GluN2s in plasticity; and Andres Barria for supplying us with the pCI-GFP-GluN2A plasmid.
The authors declare no competing financial interests.
References
- Alberini CM. The role of reconsolidation and the dynamic process of long-term memory formation and storage. Front Behav Neurosci. 2011;5:12. doi: 10.3389/fnbeh.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostaras SG, Josselyn SA, Frankland PW, Silva AJ. Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice. Learn Mem. 2000;7:58–72. doi: 10.1101/lm.7.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/S0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9:1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Sorg BA. The NMDA antagonist MK-801 disrupts reconsolidation of a cocaine-associated memory for conditioned place preference but not for self-administration in rats. Learn Mem. 2008;15:857–865. doi: 10.1101/lm.1152808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Feng R, Jacobs S, Duan Y, Wang H, Cao X, Tsien JZ. Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci Rep. 2013;3:1036. doi: 10.1038/srep01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/S0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Dalton GL, Wu DC, Wang YT, Floresco SB, Phillips AG. NMDA GluN2A and GluN2B receptors play separate roles in the induction of LTP and LTD in the amygdala and in the acquisition and extinction of conditioned fear. Neuropharmacology. 2012;62:797–806. doi: 10.1016/j.neuropharm.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Sedlak PL, Autuori E, Power JM, Sah P. Synaptic NMDA receptors in basolateral amygdala principal neurons are triheteromeric proteins: physiological role of GluN2B subunits. J Neurophysiol. 2013;109:1391–1402. doi: 10.1152/jn.00176.2012. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Finnie PS, Nader K. The role of metaplasticity mechanisms in regulating memory destabilization and reconsolidation. Neurosci Biobehav Rev. 2012;36:1667–1707. doi: 10.1016/j.neubiorev.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, Sheng M. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci. 2010;30:2676–2685. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 1998;71:1733–1741. doi: 10.1046/j.1471-4159.1998.71041733.x. [DOI] [PubMed] [Google Scholar]
- Holehonnur R, Luong JA, Chaturvedi D, Ho A, Lella SK, Hosek MP, Ploski JE. Adeno-associated viral serotypes produce differing titers and differentially transduce neurons within the rat basal and lateral amygdala. BMC Neurosci. 2014;15:28. doi: 10.1186/1471-2202-15-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I, Kim J, Kim J, Lee S, Ko HG, Nader K, Kaang BK, Tsien RW, Choi S. AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc Natl Acad Sci U S A. 2013;110:8218–8223. doi: 10.1073/pnas.1305235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak Y. Role of the NMDA receptor and nitric oxide in memory reconsolidation of cocaine-induced conditioned place preference in mice. Ann N Y Acad Sci. 2008;1139:350–357. doi: 10.1196/annals.1432.051. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Cui Z, Feng R, Wang H, Wang D, Tsien JZ. Molecular and genetic determinants of the NMDA receptor for superior learning and memory functions. PLoS One. 2014;9:e111865. doi: 10.1371/journal.pone.0111865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Kwapis JL, Werner CT, Parsons RG, Gafford GM, Helmstetter FJ. The timing of multiple retrieval events can alter GluR1 phosphorylation and the requirement for protein synthesis in fear memory reconsolidation. Learn Mem. 2012;19:300–306. doi: 10.1101/lm.024901.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. N-methyl-D-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav Neurosci. 1991;105:126–133. doi: 10.1037/0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MC, Bear MF. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotional memory: in search of systems and synapses. Ann N Y Acad Sci. 1993;702:149–157. doi: 10.1111/j.1749-6632.1993.tb17246.x. [DOI] [PubMed] [Google Scholar]
- Lee JL. Reconsolidation: maintaining memory relevance. Trends Neurosci. 2009;32:413–420. doi: 10.1016/j.tins.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ. Appetitive memory reconsolidation depends upon NMDA receptor-mediated neurotransmission. Neurobiol Learn Mem. 2008;90:147–154. doi: 10.1016/j.nlm.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci. 2003;23:6876–6883. doi: 10.1523/JNEUROSCI.23-17-06876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Ashby MC, Sanz-Clemente A, Roche KW, Isaac JT. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron. 2011;70:339–351. doi: 10.1016/j.neuron.2011.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–2319. doi: 10.1111/j.1460-9568.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Butler VJ, Gardner R, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008;28:8230–8237. doi: 10.1523/JNEUROSCI.1723-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Merlo E, Ratano P, Gregory BL, Dumbreck JK, Everitt BJ. Double dissociation of the requirement for GluN2B- and GluN2A-containing NMDA receptors in the destabilization and restabilization of a reconsolidating memory. J Neurosci. 2013;33:1109–1115. doi: 10.1523/JNEUROSCI.3273-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Pedreira ME, Pérez-Cuesta LM, Maldonado H. Reactivation and reconsolidation of long-term memory in the crab Chasmagnathus: protein synthesis requirement and mediation by NMDA-type glutamatergic receptors. J Neurosci. 2002;22:8305–8311. doi: 10.1523/JNEUROSCI.22-18-08305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/S0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Pitman RK. Will reconsolidation blockade offer a novel treatment for posttraumatic stress disorder? Front Behav Neurosci. 2011;5:11. doi: 10.3389/fnbeh.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploski JE, Pierre VJ, Smucny J, Park K, Monsey MS, Overeem KA, Schafe GE. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of Pavlovian fear conditioning in the lateral amygdala. J Neurosci. 2008;28:12383–12395. doi: 10.1523/JNEUROSCI.1662-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ. N-Methyl-D-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem. 2004;279:9673–9676. doi: 10.1074/jbc.R300029200. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999a;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999b;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–192. doi: 10.1016/S0896-6273(03)00874-2. [DOI] [PubMed] [Google Scholar]
- Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB, Spijker S. Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci. 2011;14:1302–1308. doi: 10.1038/nn.2907. [DOI] [PubMed] [Google Scholar]
- Reissner KJ, Sartor GC, Vazey EM, Dunn TE, Aston-Jones G, Kalivas PW. Use of vivo-morpholinos for control of protein expression in the adult rat brain. J Neurosci Methods. 2012;203:354–360. doi: 10.1016/j.jneumeth.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler R, Herzig V, Schmidt WJ. Repeated treatment with the NMDA antagonist MK-801 disrupts reconsolidation of memory for amphetamine-conditioned place preference. Behav Pharmacol. 2007;18:699–703. doi: 10.1097/FBP.0b013e3282effb81. [DOI] [PubMed] [Google Scholar]
- Sans NA, Montcouquiol ME, Raymond J. Postnatal developmental changes in AMPA and NMDA receptors in the rat vestibular nuclei. Brain Res Dev Brain Res. 2000;123:41–52. doi: 10.1016/S0165-3806(00)00082-1. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- Stiedl O, Radulovic J, Lohmann R, Birkenfeld K, Palve M, Kammermeier J, Sananbenesi F, Spiess J. Strain and substrain differences in context- and tone-dependent fear conditioning of inbred mice. Behav Brain Res. 1999;104:1–12. doi: 10.1016/S0166-4328(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Surís A, Smith J, Powell C, North CS. Interfering with the reconsolidation of traumatic memory: sirolimus as a novel agent for treating veterans with posttraumatic stress disorder. Ann Clin Psychiatry. 2013;25:33–40. [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Olausson P, Quinn JJ, Torregrossa MM. Targeting extinction and reconsolidation mechanisms to combat the impact of drug cues on addiction. Neuropharmacology. 2009;56:186–195. doi: 10.1016/j.neuropharm.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- Wang SH, de Oliveira Alvares L, Nader K. Cellular and systems mechanisms of memory strength as a constraint on auditory fear reconsolidation. Nat Neurosci. 2009;12:905–912. doi: 10.1038/nn.2350. [DOI] [PubMed] [Google Scholar]
- Winters BD, Tucci MC, DaCosta-Furtado M. Older and stronger object memories are selectively destabilized by reactivation in the presence of new information. Learn Mem. 2009;16:545–553. doi: 10.1101/lm.1509909. [DOI] [PubMed] [Google Scholar]
- Wood NE, Rosasco ML, Surís AM, Spring JD, Marin MF, Lasko NB, Goetz JM, Fischer AM, Orr SP, Pitman RK. Pharmacological blockade of memory reconsolidation in posttraumatic stress disorder: three negative psychophysiological studies. Psychiatry Res. 2015;225:31–39. doi: 10.1016/j.psychres.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinebi F, Xie J, Liu J, Russell RT, Gallagher JP, McKernan MG, Shinnick-Gallagher P. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci. 2003;23:10283–10291. doi: 10.1523/JNEUROSCI.23-32-10283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]