

ABSTRACT
The type I signal peptidase of Staphylococcus aureus, SpsB, is an attractive antibacterial target because it is essential for viability and extracellularly accessible. We synthesized compound 103, a novel arylomycin-derived inhibitor of SpsB with significant potency against various clinical S. aureus strains (MIC of ~1 µg/ml). The predominant clinical strain USA300 developed spontaneous resistance to compound 103 with high frequency, resulting from single point mutations inside or immediately upstream of cro/cI, a homolog of the lambda phage transcriptional repressor cro. These cro/cI mutations led to marked (>50-fold) overexpression of three genes encoding a putative ABC transporter. Overexpression of this ABC transporter was both necessary and sufficient for resistance and, notably, circumvented the essentiality of SpsB during in vitro culture. Mutation of its predicted ATPase gene abolished resistance, suggesting a possible role for active transport; in these bacteria, resistance to compound 103 occurred with low frequency and through mutations in spsB. Bacteria overexpressing the ABC transporter and lacking SpsB were capable of secreting a subset of proteins that are normally cleaved by SpsB and instead were cleaved at a site distinct from the canonical signal peptide. These bacteria secreted reduced levels of virulence-associated proteins and were unable to establish infection in mice. This study reveals the mechanism of resistance to a novel arylomycin derivative and demonstrates that the nominal essentiality of the S. aureus signal peptidase can be circumvented by the upregulation of a putative ABC transporter in vitro but not in vivo.
IMPORTANCE
The type I signal peptidase of Staphylococcus aureus (SpsB) enables the secretion of numerous proteins by cleavage of the signal peptide. We synthesized an SpsB inhibitor with potent activity against various clinical S. aureus strains. The predominant S. aureus strain USA300 develops resistance to this inhibitor by mutations in a novel transcriptional repressor (cro/cI), causing overexpression of a putative ABC transporter. This mechanism promotes the cleavage and secretion of various proteins independently of SpsB and compensates for the requirement of SpsB for viability in vitro. However, bacteria overexpressing the ABC transporter and lacking SpsB secrete reduced levels of virulence-associated proteins and are unable to infect mice. This study describes a bacterial resistance mechanism that provides novel insights into the biology of bacterial secretion.
INTRODUCTION
Discovery of novel antibiotics has become an important goal for biomedical research in both academia and industry, mainly driven by the growing problem of widespread antibiotic resistance (1). Staphylococcus aureus is a major threat to human health and can cause life-threatening invasive infections, such as bacteremia, endocarditis, pneumonia, and osteomyelitis (2). Infections with S. aureus have become increasingly difficult to treat because of the emergence of methicillin resistance and high failure rates of standard-of-care antibiotics like vancomycin (3).
The number of suitable antibiotic targets in S. aureus is rather limited compared to the number of genes found to be essential in genetic screens (4), largely because of the difficulty of finding cell-active inhibitors for essential enzymes that can penetrate the bacterial cell wall and avoid efflux mechanisms. The type I signal peptidase of S. aureus, SpsB, is an attractive antibiotic target because it is essential for viability and accessible on the extracellular side of the bacterial membrane, while its active site is different from that of human signal peptidases (5). SpsB substrates are membrane-bound preproteins secreted by the Sec secretion pathway that contain a canonical Ala-X-Ala motif in the signal sequence (6, 7). Inhibition of SpsB activity leads to a loss of bacterial viability (8, 9).
A recently discovered natural compound, arylomycin (10, 11), has been reported to inhibit type I signal peptidases of various bacterial species by occupying the enzymatic groove that accommodates natural substrates during the process of secretion (12). However, the potency of arylomycin against S. aureus is too low to be clinically useful for treatment of S. aureus infections (5, 13).
This study describes a novel SpsB inhibitor, the arylomycin analog compound 103, which has enhanced potency against S. aureus. We found that resistance to this inhibitor occurs with high frequency in vitro and is mediated by robust overexpression of a putative ABC transporter that results from loss-of-function mutations in cro/cI, a homolog of the lambda phage transcriptional repressor cro gene. The overexpression of this ABC transporter prevented bacterial lethality caused by disruption of the spsB gene. This was associated with secretion of a subset of proteins that are normally cleaved by SpsB and were cleaved at a site distinct from the canonical SpsB cleavage site. Bacteria reliant on this secretion mechanism secreted reduced levels of functional virulence-associated proteins and were unable to infect mice, demonstrating an absolute requirement for SpsB activity during infection. This study reveals a novel bacterial resistance mechanism that led to the discovery of an alternative system for cleavage and secretion of signal peptide-containing proteins that counteracts the essentiality of SpsB in vitro but not in vivo.
RESULTS AND DISCUSSION
Anti-S. aureus activity of SpsB inhibitor compound 103.
Arylomycins are a naturally occurring family of structurally related antibiotics that inhibit SpsB of S. aureus, albeit with low antibacterial potency (5, 13). In an effort to create novel antibiotics to treat S. aureus infections, we synthesized compound 103 (see Fig. S1A in the supplemental material), a new analog of arylomycin (see Fig. S1B) with improved activity against S. aureus. The MIC of compound 103 for the predominant clinical methicillin-resistant S. aureus (MRSA) strain USA300 was 1.0 µg/ml, whereas the MIC for arylomycin A16 was 32 µg/ml (this study). Compound 103 exhibited MICs ranging from 0.5 to 1.4 µg/ml for a panel of eight clinical S. aureus strains (Table 1). Compound 103 dose dependently inhibited the enzymatic activity of recombinant SpsB (see Fig. S1C). The antibacterial activity of compound 103 occurred specifically through SpsB, as confirmed by a reduced MIC for a USA300 strain that underexpresses SpsB and an increased MIC for an SpsB-overexpressing strain (Table 1).
TABLE 1 .
MICs of compound 103 for S. aureus strains
| S. aureus strain | Resistance typea | MIC of compound 103 (μg/ml) |
|---|---|---|
| USA300 wild type | MRSA | 1.0 |
| USA300 SpsB-high | MRSA | 64 |
| USA300 SpsB-low | MRSA | 0.125 |
| COL | MRSA | 0.7 |
| MRSA252 | MRSA | 1.0 |
| Mu50 | VISA | 1.0 |
| USA100 | MRSA | 0.5 |
| USA1000 | MRSA | 1.0 |
| USA400 | MRSA | 1.4 |
| Newman | MSSA | 1.0 |
MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus; VISA, vancomycin-intermediate S. aureus.
Resistance to compound 103 is caused by mutations in SAUSA300_0350 (cro/cI).
To determine the mechanism of spontaneous resistance against compound 103, we selected 40 independently generated resistant mutants of S. aureus USA300, which arose at a frequency of 3 × 10−7 from cultures on agar containing compound 103 at fourfold its MIC. The MICs of compound 103 for all of these mutants were increased by at least 16-fold compared to the MIC of wild-type (WT) USA300.
Whole-genome sequencing of all 40 mutants revealed that resistance was associated with a single mutation inside or just upstream of gene SAUSA300_0350 in all of these clones. Based on homology to the lambda phage Cro protein, SAUSA300_0350 is annotated as “Cro/CI transcriptional regulator-like protein” and will be referred to as cro/cI throughout this paper. We identified mutations in 16 of the 67 amino acids of the predicted Cro/CI protein (Fig. 1A; see also Table S1 in the supplemental material), including multiple substitutions or stop codons and one insertion. We also identified two single-nucleotide substitutions immediately upstream of the cro/cI translational start associated with resistance (Fig. 1B), i.e., a change of G to T 14 bp upstream of cro/cI, located in a predicted ribosomal binding site (RBS) (AGGAGT) in the cro/cI promoter and likely leading to defective Cro/CI protein translation, and a change of G to A 62 bp upstream of cro/cI.
FIG 1 .

cro/cI mutations that are associated with resistance to SpsB inhibitor compound 103. (A) Predicted full-length amino acid sequence of Cro/CI protein. Amino acids that were found to be mutated in resistant clones are indicated in red, with their respective substitutions underneath. sc, stop codon; in, insertion of CGTTTCAAGCGGG resulting in multiple stop codons. (B) Single-base-pair substitutions that were found in the promoter region of the cro/cI open reading frame (ORF) and that were associated with resistance to compound 103 were located 62 bp and 14 bp upstream of the cro/cI ORF. All mutations inside (A) or upstream of (B) the cro/cI ORF are associated with a >16-fold increase in the MIC of compound 103.
Plasmid-encoded expression of exogenous WT cro/cI reversed the resistant phenotype of clones with mutations inside cro/cI or at 14 bp upstream of cro/cI to a sensitive phenotype (Table 2, strains GNE0109 and GNE0097). This confirms that loss-of-function mutations in cro/cI are the cause of resistance to compound 103. WT cro/cI could not complement the phenotype of the G-to-A mutation 62 bp upstream of cro/cI (Table 2, strain GNE0100), indicating that this mutation is presumably located in the putative Cro/CI binding site, by analogy to the lambda phage cro gene (14, 15). Mutation of cro/cI did not reduce sensitivity to other tested antibiotics, including vancomycin, linezolid, oxacillin, daptomycin, rifampin, gentamicin, clindamycin, erythromycin, gatifloxacin, and novobiocin (see Table S2 in the supplemental material). The loss-of-function cro/cI mutation did not result in a significant alteration in virulence in a mouse infection model (see Fig. S2).
TABLE 2 .
Roles of cro/cI and putative ABC transporter genes in resistance of USA300-derived strains to compound 103
| Strain | Relevant genotypea | MIC of compound 103 (fold increase versus MIC for USA300 WT) |
Expression of SAUSA300_0352 (fold increase versus level in USA300 WT) |
|---|---|---|---|
| GNE0163 | USA300 WT(pMK4) | 1.0 | 1.0 |
| GNE0162 | psarA-SAUSA300_0351_0352_0353 | >32 | 56.9 |
| GNE0107 | cro/cI(M1V)(pMK4) | >32 | 66.2 |
| GNE0109 | cro/cI(M1V)(pcro/cI-cro/cI) | 1.7 | 1.0 |
| GNE0096 | cro/cI(−14G→T)(pMK4) | >32 | 10.9 |
| GNE0097 | cro/cI(−14G→T)(pcro/cI-cro/cI) | 3.0 | 0.1 |
| GNE0099 | cro/cI(−62G→A)(pMK4) | >32 | 5.5 |
| GNE0100 | cro/cI(−62G→A)(pcro/cI-cro/cI) | >32 | 8.0 |
| GNE0173 | Δmcr cro/cI(Q16stop) | >32 | 105 |
| GNE0220 | Δmcr cro/cI(Q16stop) Δ(SAUSA300_0351-SAUSA300_0352-SAUSA300_0353) | 2.0 | NDb |
| GNE0209 | Δmcr cro/cI(Q16stop) ΔSAUSA300_0351 | >32 | 93 |
| GNE0175 | Δmcr cro/cI(Q16stop) SAUSA300_0352(K44A) | 1.0 | 106 |
| GNE0210 | Δmcr cro/cI(Q16stop) ΔSAUSA300_0353 | 1.0 | ND |
All strains are USA300 derived, and all plasmids are pMK4 (empty plasmid) or constructed in pMK4. A more detailed description of each strain is given in Table S5 in the supplemental material. SAUSA300_0351 encodes a putative membrane protein, SAUSA300_0352 encodes a putative ABC transporter ATPase, and SAUSA300_0353 encodes a putative permease.
ND, not determined.
A recent study suggested a Gln at position 22 (Gln22) in a cro/cI homolog in S. aureus strain N315 to be associated with resistance to M131, another arylomycin derivative (16). According to the public NCBI database, Gln22 is present in the sequences of several S. aureus strains that are sensitive to compound 103, such as COL, Mu50, and MRSA252, as well as the USA300 strain we used in the present study (Table 1). Therefore, a Gln at position 22 in cro/cI does not confer resistance to compound 103. Instead, we did observe that the replacement of Gln22 with a stop codon (Q22stop) was associated with resistance to compound 103 (Fig. 1A).
We next investigated whether S. aureus cro/cI mutations preexist in nature. Among a panel of 102 globally diverse clinical S. aureus isolates, 7 isolates showed resistance to compound 103, each exhibiting a MIC of >32 µg/ml. Compared with the corresponding sequences from 5 compound 103-sensitive strains (strains USA300, COL, MRSA252, USA400, Mu50) (Table 1), all 7 resistant isolates showed an AG-to-TA nucleotide substitution at 67/68 bp upstream of cro/cI, while their spsB sequences were identical to the spsB sequences of these 5 sensitive strains. Analogous to the mutation we detected 62 bp upstream of cro/cI in USA300 (Fig. 1B), we speculate that the resistance of these 7 clinical isolates to compound 103 may be caused by mutation of the Cro/CI binding site in the operator region.
In summary, our comprehensive analysis identified a number of recessive mutations in cro/cI or in its predicted RBS and a mutation in a putative operator region of cro/cI that was quite similar to a naturally occurring mutation in about 7% of clinical S. aureus isolates tested, all of which could lead to loss of Cro/CI function. Thus, we hypothesized that loss of Cro/CI function is the most common mechanism of resistance to SpsB inhibitor compound 103.
Cro/CI is a previously uncharacterized transcriptional regulator of S. aureus and suppresses the expression of a putative ABC transporter.
Lambda phage Cro regulates the expression of target genes that are located in its operon, through interaction at an upstream operator sequence (14, 15). We tested the hypothesis that the cro/cI gene product in S. aureus functions as a transcriptional regulator by analyzing the transcription profiles of cro/cI mutants.
RNA sequencing of four cro/cI mutants (carrying Cro/CI mutations of M to V at position 1 [M1V] [strain GNE0117], R11stop [strain GNE0215], E34D [strain GNE0214], and N53K/54stop [strain GNE0217]) revealed that the expression levels of four genes were 42- to 91-fold greater than in the WT, while the expression levels of all other genes ranged within a 1- to 6-fold difference from their levels in wild-type bacteria (Fig. 2A; see also Table S3 in the supplemental material). These four most highly overexpressed genes are located in a predicted operon that includes the following genes: cro/cI itself; a gene encoding a putative ABC transporter protein, annotated as “putative membrane protein,” i.e., SAUSA300_0351; SAUSA300_0352, whose product is annotated as “multidrug ABC transporter ATP-binding domain”; and SAUSA300_0353, whose product is annotated as “putative multidrug ABC transporter permease.” Reverse transcription-quantitative PCR (qRT-PCR) analysis confirmed overexpression of the cro/cI operon genes by more than 50-fold in five different cro/cI mutants (Fig. 2B).
FIG 2 .

cro/cI mutations enhance the expression of all 4 genes in the cro/cI-ABC transporter operon. (A) RNA sequencing showing differential expression of 4 genes in S. aureus USA300 cro/cI(M1V) mutant (strain GNE0117) versus WT USA300. This revealed a significant enhancement in transcript expression of four genes sharing an operon, including SAUSA300_0350 (cro/cI) itself (89-fold enhanced) and three genes encoding a putative ABC transporter, i.e., SAUSA300_0351 (0351, product annotated as a putative membrane protein; 91-fold upregulated), SAUSA300_0352 (0352, encoding a putative ATPase; 78-fold), and SAUSA300_0353 (0353, encoding a putative permease; 42-fold). Similar results were obtained for three other cro/cI mutants, i.e., cro/cI(R11stop) (strain GNE0215), cro/cI(E34D) (strain GNE0214), and cro/cI(N53K/54stop) (strain GNE0217) mutants. FC, fold change compared to expression in WT USA300; AU, arbitrary units. Table S3 in the supplemental material shows all other genes that were upregulated by at least twofold. (B) Transcript expression of genes upstream of or inside the cro/cI operon was quantified by qRT-PCR in five different USA300 cro/cI mutants (with E34D, M1V, R11stop, Q22stop, and N53K/54stop mutations). This confirmed the robust (>50-fold) overexpression of genes located inside the cro/cI operon, i.e., SAUSA300_0350 (cro/cI), SAUSA300_0352, and SAUSA300_0353. The expression of two genes located immediately upstream of cro/cI, i.e., SAUSA300_0347 (annotated as tatC) and SAUSA300_0348 (tatA), was not affected. Shown is the fold change for each mutant versus the expression in WT USA300, representing the average ± standard deviation (SD) of three independent values. (C) Hypothetical model for the role of cro/cI-mediated regulation of the putative ABC transporter in S. aureus resistance to compound 103. In WT S. aureus (top), Cro/CI protein binds to the operator sequence, suppressing transcription of the ABC transporter by blocking RNA polymerase activity, analogous to the effects of Cro of lambda phage (15). In a cro/cI mutant (bottom), either no Cro/CI protein or a nonfunctional Cro/CI protein is produced, enabling polymerase activity that leads to overexpression of the ABC transporter, which causes resistance to compound 103.
Exogenous expression of plasmid-encoded WT cro/cI in a cro/cI(M1V) mutant reversed the transcription of SAUSA300_0352 to WT levels (Table 2, strain GNE0109), demonstrating that intact cro/cI actively suppresses the transcription of the putative ABC transporter. The transcription of two genes located immediately upstream of cro/cI, i.e., SAUSA300_0347 (whose product is annotated as “TatC secretion protein”) and SAUSA300_0348 (whose product is annotated as “TatA secretion protein”), was not affected by cro/cI mutations (Fig. 2B). These data confirm that S. aureus Cro/CI is a transcriptional repressor with the putative ABC transporter as its critical target.
A proposed model for the regulation of the ABC transporter by Cro/CI is shown in Fig. 2C. In WT bacteria, the expression of the putative ABC transporter genes (SAUSA300_0351, _0352, and _0353) is suppressed by binding of Cro/CI to the operator, located upstream of cro/cI (analogous to lambda phage Cro [14, 15]). In cro/cI mutants, either no Cro/CI protein or a nonfunctional Cro/CI protein is produced, or the affinity of the Cro/CI-binding site is reduced, in each case enabling RNA polymerase to initiate transcription of the operon genes, leading to overexpression of the ABC transporter.
The putative ABC transporter is necessary and sufficient for high-level resistance to compound 103.
A series of genetic experiments established the role of the putative ABC transporter in resistance to compound 103. Deletion of all 3 ABC transporter genes (SAUSA300_0351, _0352, and _0353) reversed the resistant phenotype of a USA300 cro/cI mutant to a sensitive phenotype (Table 2, strain GNE0220). Conversely, overexpression of these 3 genes in WT USA300 induced resistance to compound 103 (Table 2, strain GNE0162). Thus, overexpression of the genes for the predicted ABC transporter is necessary and sufficient to mediate full resistance to compound 103.
Next, we determined which of these three individual genes were required for resistance. Deletion of SAUSA300_0351 (encoding the putative membrane protein) in a resistant cro/cI mutant had no effect on the resistance phenotype (Table 2, strain GNE0209). qRT-PCR analysis confirmed the absence of transcripts of SAUSA300_0351 in this deletion mutant (not shown), while the overexpression of SAUSA300_0352 (Table 2) and SAUSA300_0353 (not shown) transcripts was similar to their transcription in the parental cro/cI mutant (strain GNE0193). Substitution of Lys to Ala at position 44 (K44A) within the Walker A motif of the gene encoding the predicted ATPase, SAUSA300_0352, known to prevent nucleotide binding in other ABC transporters (17, 18), reversed the resistant phenotype of a cro/cI mutant to a sensitive phenotype (Table 2, strain GNE0175). This SAUSA300_0352(K44A) point mutant showed unaltered transcript overexpression of SAUSA300_0352 itself (Table 2) and of SAUSA300_0353 (not shown). Thus, high-frequency resistance to compound 103 requires ATP binding by the SAUSA300_0352 gene product and, presumably, involves active transport by the putative ABC transporter. Deletion of SAUSA300_0353 also reversed the resistant phenotype of the cro/cI mutant, consistent with the predicted role of its product as an ABC transporter permease (Table 2, strain GNE0210).
Inactivation of the putative ABC transporter by a K44A substitution in SAUSA300_0352 in a WT USA300 background (generating strain GNE0174) reduced the frequency of spontaneous resistance to compound 103 by approximately 2 orders of magnitude, to 2 × 10−9. In the resistant mutants that arose from this strain, we identified mutations in spsB leading to substitutions of 4 amino acids in SpsB (Y30, V49, H68, and V167), which were each associated with an ~4- to 8-fold increased MIC for compound 103 (Table 3), presumably due to reduced affinity of SpsB for compound 103. Thus, inactivation of the putative ABC transporter eliminates the effects of cro/cI mutations, resulting in low-frequency resistance of USA300 to compound 103 that is associated with on-target mutations in spsB.
TABLE 3 .
SpsB point mutations found in the USA300 Δmcr cro/cI(Q16stop) SAUSA300_0352(K44A) mutanta
| Amino acid substitution | MIC of compound 103 (fold increase versus MIC for USA300 WT) |
|---|---|
| Y30C | 4 |
| Y30N | 4 |
| Y30D | 8 |
| Y30S | 4 |
| V49L | 4 |
| H68Y | 8 |
| H69L | 4 |
| V167D | 8 |
| V167F | 8 |
Resistant clones were selected from USA300 Δmcr cro/cI(Q16stop) SAUSA300_0352(K44A) (strain GNE0175) bacteria cultured on agar containing compound 103 at fourfold its MIC. The SpsB mutations were identified by sequencing PCR products.
Together, these data indicate that overexpression of the putative ABC transporter, induced by derepression of its transcription as the result of mutations in the transcriptional repressor cro/cI, is the main mechanism of S. aureus resistance to compound 103.
We then tested the effects of inactivation of the ABC transporter in the absence of cro/cI mutations. In a neutropenic thigh mouse model of infection, S. aureus USA300 with a K44A point mutation in SAUSA300_0352 in a WT cro/cI background (strain GNE0174) was more sensitive than its parental USA300 WT strain (strain GNE0023) to treatment with a compound 103 analog (see Fig. S3A in the supplemental material). In vitro, the K44A mutation had no effect on sensitivity to the compound 103 analog (the MIC was 0.25 µg/ml for both strains). A general phenotypic characterization of the K44A mutant showed no difference from the WT strain with regard to growth rate in Mueller-Hinton broth (MHB) in the absence or presence of serum (see Fig. S3B), sensitivity to antimicrobial peptide protamine (the MIC was 8 µg/ml for both strains) or nisin (the MIC was 200 µg/ml for both strains), or the ability to form biofilm in vitro (see Fig. S3C). A recent study showed that transcription of the putative ABC transporter can be transiently induced in WT S. aureus (16). Based on those and our observations, we speculate that the putative ABC transporter may become overexpressed in WT S. aureus during infection, leading to reduced sensitivity to SpsB inhibition.
Overexpression of the ABC transporter in the absence of SpsB partially restores viability and protein secretion but not infectivity.
The above-described data showed that overexpression of the putative ABC transporter mediates resistance to the SpsB inhibitor. Therefore, we hypothesized that overexpression of the putative ABC transporter would overcome the essentiality of SpsB for S. aureus viability. To test this, we generated a mutant containing an insertion in chromosomal spsB and coexpressing a plasmid-encoded LtrA that removes the insertion from transcripts by RNA splicing; this mutant is normally viable at permissive temperature (Fig. 3A, strain GNE0190). Loss of the LtrA-encoding plasmid by culture at a nonpermissive temperature resulted in nonproductive spsB transcripts and complete cessation of growth (Fig. 3A), confirming the requirement of SpsB. In the LtrA-containing strain with the spsB insertion (strain GNE0190), we then generated a cro/cI(M1V) mutation by selection with compound 103. After loss of the LtrA-encoding plasmid at the nonpermissive temperature, which was confirmed by culture on antibiotic-selective medium and by plasmid-specific PCR, this USA300 spsB cro/cI double mutant (GNE0191) showed normal growth compared to that of the WT strain (Fig. 3A). We confirmed both the absence of SpsB protein in this strain (by Western blotting) (Fig. 3B) and overexpression of the ABC transporter (by qRT-PCR) (SAUSA300_0352 transcript expression was 162- ± 13-fold higher than in the WT USA300 strain). These data demonstrate that S. aureus is able to survive without SpsB in vitro when the ABC transporter is overexpressed. This is consistent with data from another study describing a strain of S. aureus N315 that carries a deletion of spsB and a point mutation in a gene homologous to cro/cI (19).
FIG 3 .
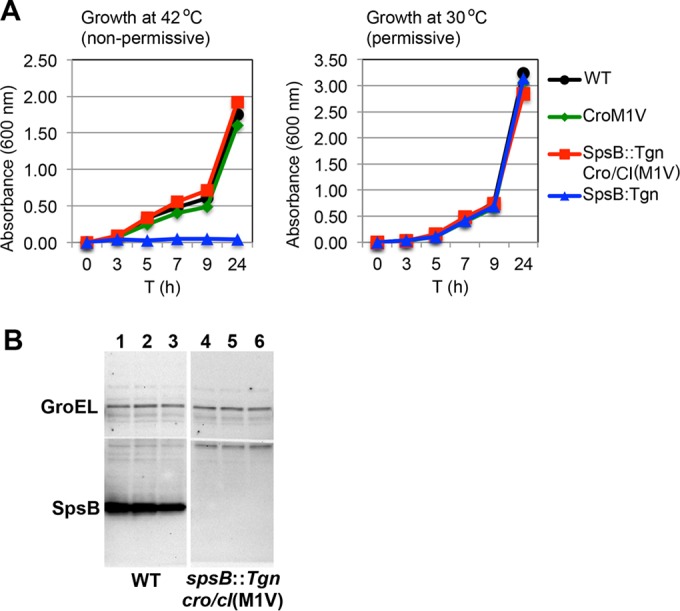
Viability of S. aureus USA300 lacking SpsB is restored by cro/cI mutation in vitro. (A) cro/cI mutation restores in vitro growth of a mutant lacking SpsB. At nonpermissive temperature (42°C), plasmid pNL9164-ltrA is cured from strain USA300 Δmcr spsB::Tgn(pNL9164-ltrA) (strain GNE0190) and the TargeTron insertion in spsB prevents spsB expression, resulting in complete cessation of growth and confirming the essentiality of spsB. Remarkably, the growth of this strain is restored by a mutation in cro/cI [USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191)]. At permissive temperature (30°C), plasmid pNL9164-ltrA mediates splicing of the TargeTron insertion that removes it from the spsB transcript, allowing SpsB expression and growth of USA300 Δmcr spsB::Tgn pNL9164-ltrA (strain GNE0190). Circles, USA300 Δmcr (WT; strain GNE0023); triangles, USA300 Δmcr spsB::Tgn pNL9164-ltrA (strain GNE0190); squares, USA300 Δmcr spsB::Tgn cro/cI(M1V) (after curing plasmid pNL9164-ltrA) (strain GNE0191); diamonds, USA300 Δmcr cro/cI(M1V) (strain GNE0117). (B) Western blotting using a polyclonal rabbit anti-SpsB antibody showed a complete lack of SpsB protein in 3 independent whole-cell lysates from USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191) (lanes 4 to 6), compared to normal SpsB expression in USA300 Δmcr (WT, strain GNE0023) (lanes 1 to 3). Rabbit IgG anti-GroEL served as the loading control.
We next determined the secretion profile of the USA300 spsB cro/cI double mutant by mass spectrometry. This identified 25 proteins that were released into the culture supernatant without signal peptide cleavage. For 12 of these secreted proteins, an alternative, SpsB-independent cleavage site was identified (see Table S4 in the supplemental material), for which we generated a consensus cleavage motif (see Fig. S4A). Thus, the ABC transporter compensates at least in part for the absence of SpsB by promoting the secretion of a subset of proteins, presumably through an alternative cleavage mechanism.
We then performed a quantitative assessment of the secretion of individual functional proteins in the absence of SpsB. First, the levels of surface expression of several sortase A-anchored proteins, i.e., FnbpA, ClfA, IsdA, and protein A, in the USA300 spsB cro/cI double mutant were significantly reduced, as shown by flow cytometry (Fig. 4A). Second, the USA300 spsB cro/cI double mutant demonstrated a severe defect in the release of active proteases (Fig. 4B). All four sortase A-anchored proteins tested contribute to the virulence of S. aureus through adherence, iron uptake, and evasion of immune responses, while the secreted proteases facilitate tissue penetration and impair immune responses (20–22). Since the expression of both groups of proteins was quantitatively diminished, we assessed the virulence of this strain in a mouse model of systemic infection. The USA300 spsB cro/cI double mutant was completely unable to establish infection (Fig. 4C). Thus, in the absence of SpsB, the overexpression of the ABC transporter restores the secretion of a subset of proteins, likely through an alternative cleavage mechanism. While this allows growth in vitro, it does not overcome the requirement of SpsB during infection.
FIG 4 .
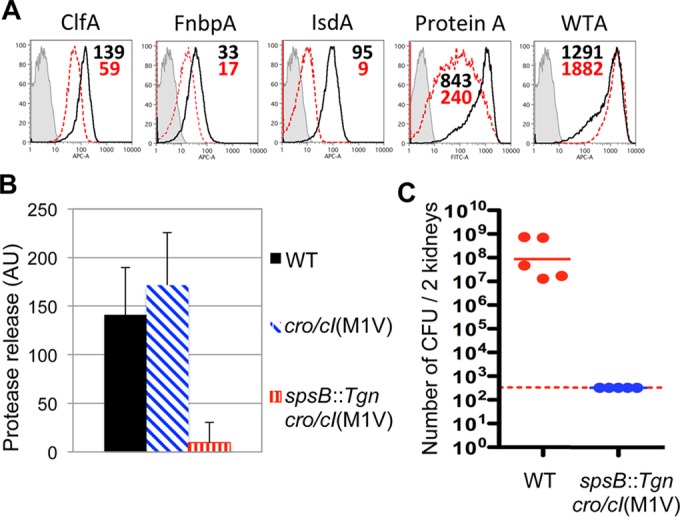
spsB cro/cI double mutant exhibits reduced expression of virulence factors and impaired infectivity. (A) Reduced surface expression of cell wall proteins on spsB cro/cI double mutant. Whole S. aureus USA300 WT (black lines, with mean fluorescence intensity [MFI] in black numbers; strain GNE0023) or USA300 Δmcr spsB::Tgn cro/cI(M1V) double mutant (red dotted lines, with MFI in red numbers; strain GNE0191) bacteria were incubated with MAbs against surface antigens, or WT was incubated with medium alone (gray shading and lines), followed by flow cytometry analysis. The data show significant reductions in surface expression of clumping factor A (ClfA), fibronectin-binding protein A (FnbpA), iron-regulated surface determinant A (IsdA), and protein A, but not of the nonproteinaceous antigen wall teichoic acid (WTA). APC-A, allophycocyanin channel; FITC-A, fluorescein isothiocyanate channel. (B) Defective release of active proteases in culture supernatant of USA300 spsB::Tgn cro/cI double mutant. Filled black, WT USA300 Δmcr (strain GNE0023); hatched blue, USA300 Δmcr cro/cI(M1V) (strain GNE0117); vertically striped red, USA300 spsB::Tgn cro/cI(M1V) (strain GNE0191). (C) spsB::Tgn cro/cI double mutant is unable to establish infection. Mice were injected intravenously with S. aureus USA300 Δmcr (WT; strain GNE0023) or USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191) double mutant, and numbers of CFU per kidney were determined at day 3 after infection. Dashed line, detection limit.
Concluding remarks.
The present study demonstrates that resistance of the clinical S. aureus strain USA300 to compound 103, a potent inhibitor of the type I signal peptidase SpsB, is caused by robust overexpression of a putative ABC transporter, occurring as a result of mutations in cro/cI. We showed that the cro/cI gene product functions as a novel transcriptional regulator in S. aureus, by repressing the three genes of the putative ABC transporter that are carried in the same operon as cro/cI. Overexpression of this ABC transporter is both necessary and sufficient to induce resistance to compound 103. A K44A substitution in the predicted ATPase gene of the transporter prevents the development of resistance, indicating that the resistance mechanism involves active transport.
The precise molecular mechanism by which the ABC transporter mediates resistance to SpsB inhibitor compound 103 remains to be determined. The striking observation that, in bacteria lacking SpsB, overexpression of the ABC transporter restores in vitro viability and promotes alternative cleavage of secreted proteins suggests that an extracellular protease is linked to transporter activity and resistance. Since the alternative cleavage sites were located N terminal to the SpsB cleavage site, this putative protease may be embedded in the membrane.
Although overexpression of the ABC transporter restored in vitro viability of SpsB-deficient S. aureus bacteria, it did not rescue the secretion of virulence-associated proteins and in vivo virulence in a mouse infection model. This in vivo requirement of SpsB may explain its strong conservation as the sole signal peptidase through the evolution of staphylococci as human pathogens.
MATERIALS AND METHODS
Bacterial strains and media.
Wild-type clinical S. aureus strains were obtained from the Network on Antibiotic Resistance in Staphylococcus aureus (NARSA/BEI) program. MRSA strain USA300 refers throughout this paper to strain NRS384 from NARSA/BEI. A global panel of 102 clinical S. aureus isolates was selected from the collection of International Health Management Associates, Inc. (IHMA; Schaumburg, IL), based on diversity of geographic locations, antibiotic resistance profiles, and sites of infection. Bacteria were cultured on cation-adjusted Mueller-Hinton agar (MHA) plates or in cation-adjusted Mueller-Hinton broth (MHB). Bacterial strains used in this study are listed in Table S5 in the supplemental material.
MICs and frequency of resistance.
MICs were determined according to the standard protocol from the Clinical and Laboratory Standards Institute (23). Briefly, S. aureus bacteria were harvested from regular MHA plates that had been cultured for ~20 h at 37°C. Serial dilutions of compound 103 were incubated with bacteria at approximately 106 CFU (CFU)/ml in MHB in round-bottom 96 well plates (Corning, Corning, NY) for 22 h at 37°C. MICs were determined by the lowest concentration of compound 103 showing absence of growth by visual inspection.
To determine the frequency of spontaneous resistance, suspensions containing approximately 5 × 109 CFU of S. aureus were cultured on MHA plates containing compound 103 at fourfold the MIC; serial dilutions were cultured in parallel on regular MHA to determine the actual number of CFU (input). The frequency of spontaneous resistance was defined as the number of viable colonies able to grow in the presence of compound 103 after culture at 37°C for 2 days (output) divided by input.
Mutations and genetic engineering of S. aureus.
Compound 103-resistant mutants derived from S. aureus USA300 were selected after culture on MHA containing 4 µg/ml of compound 103 for 2 days. Single colonies were restreaked onto MHA with 4 µg/ml of compound 103 to confirm resistance and establish clonality.
For transformations, we used the methylation-deficient Δmcr strain of USA300 (strain GNE0023), which exhibits improved transformability while retaining unaltered in vivo virulence (24). To generate mutations in SAUSA300_0351, _0352, and _0353 genes by homologous recombination, we transformed USA300 Δmcr using targeting constructs created in the vector pIMAY (24). To generate mutations in SAUSA300_0351, _0352, and _0353 genes in a cro/cI mutant background, we selected colonies from USA300 Δmcr cultured on plates containing fourfold the MIC of compound 103. Sequencing of PCR products of the cro/cI gene from resistant colonies yielded a clone with a Q16stop codon mutation in cro/cI (strain GNE0173). We further mutated SAUSA300_0351, _0352, and _0353 in this USA300 Δmcr cro/cI (Q16stop) clone to study the contributions of these three genes to cro/cI-mediated resistance.
A conditional SpsB-deficient strain was generated by inserting a TargeTron (Tgn; a Lactococcus lactis LI.LtrB group II intron) into the spsB gene and coexpressing a temperature-sensitive plasmid, pNL9164-ltrA, that encodes LtrA (Sigma, St. Louis, MO) (25) in USA300 Δmcr (clone GNE0190). In the presence of LtrA, the LtrB group II intron is spliced out of spsB mRNA, enabling the synthesis of functional SpsB protein. In this clone, we generated a mutation in cro/cI (i.e., M1V) by selection on agar containing compound 103 at a concentration of fourfold the MIC. Plasmid pNL9164-ltrA was cured out by culture at nonpermissive temperature (42°C), which was confirmed by absence of growth on antibiotic-selective medium and by plasmid-specific PCR, and which generated USA300 Δmcr spsB::Tgn cro/cI(M1V) (clone GNE0191).
A strain of USA300 Δmcr overexpressing SpsB (USA300 SpsB-high; GNE0028) was engineered by cloning SpsB under the constitutive sarA promoter into vector pMK4, resulting in SpsB expression that was ~20-fold higher than the wild-type level as determined by Western blotting. A USA300 Δmcr strain that underexpresses SpsB (USA300 SpsB-low; GNE0028) was generated by genomic incorporation of spsB under the isopropyl-β-d-thiogalactopyranoside (IPTG)-sensitive promoter spac; without IPTG, this strain expresses SpsB at ~5% of the wild-type level as determined by Western blotting.
RNA isolation and transcript analysis by qRT-PCR.
To isolate S. aureus RNA, ~108 CFU from a 22-h culture in MHB were treated with RNAprotect bacterial reagent (Qiagen, Valencia, CA) for 10 min at room temperature and lysed for 30 min at 37°C in 30 mM Tris (pH 8.0) containing 1 mM EDTA, 1 mg/ml of lysostaphin (Sigma), and protease K (Qiagen), followed by mechanical disruption using a bead beater (BioSpec Products, Bartlesville, OK). RNA was purified using an RNeasy RNA purification kit (Qiagen); RNA concentrations were determined using a NanoDrop 8000 instrument (Thermo Scientific, Waltham, MA).
Transcript levels were analyzed by qRT-PCR as described previously (26). Briefly, purified RNA was treated with DNase (DNA-free kit; Ambion) and reverse transcribed using gene-specific reverse primers and the Superscript III reverse transcription kit (Invitrogen, Carlsbad, CA), followed by PCRs with gene-specific primers, 6-carboxyfluorescein (FAM)-labeled probes (see Table S6 in the supplemental material), and TaqMan universal PCR master mix (Applied Biosystems). PCRs were recorded using the 7500 real-time PCR system (Applied Biosystems). Delta cycle threshold (ΔCT) values were determined by subtracting the CT values of the rRNA gene of S. aureus (rrsA), used for normalization, from the CT value of each test gene. Data were expressed as the fold change (FC) of transcript expression in mutants relative to the expression of the transcript in wild-type bacteria and were calculated using the following formula: FC = 2^[ΔCT (wild-type) − ΔCT (mutant)].
Quantitative analysis of cell wall protein expression and secretion of active proteases.
Human IgG1 monoclonal antibodies (MAbs) against S. aureus fibronectin-binding protein A (FnbpA; clone 6931), clumping factor A (ClfA; clone 4675), iron-regulated surface determinant A (IsdA; clone 4402), and wall teichoic acid (WTA; clone 7574) were cloned from peripheral B cells of patients after S. aureus infection using the Symplex technology (27–29). Murine IgG2a against protein A (MAb 8F9) was generated by immunizing BALB/c mice with USA300 bacteria. To quantify surface protein expression, approximately 107 bacteria were incubated with 200 µg/ml of rabbit IgG (Sigma, St. Louis, MO), 2 µg/ml of MAbs against surface antigens, and fluorochrome-labeled anti-human or anti-mouse IgG F(ab′)2 (Jackson ImmunoResearch, West Grove, PA). Fluorescence was analyzed using a fluorescence-activated cell sorter (FACSAria; Becton, Dickinson) and FlowJo software (version 8.4.5), and the fluorescence intensity served as a measure for surface expression of cell wall proteins.
To quantify the secretion of active proteases, culture supernatants from bacteria cultured in MHB at 37°C for 6 h were centrifuged at 2,000 × g for 10 min and passaged through a 0.2-µm filter. The amounts of active proteases released into the culture supernatants were quantified using the EnzCheck assay kit (Invitrogen), which detects the activity of multiple classes of proteases based on fluorescent casein cleavage.
Mouse infection.
For systemic infection, 7-week-old female A/J mice (Jackson, Bar Harbor, ME) were injected intravenously through the tail vein with 100 µl phosphate-buffered saline (PBS) containing approximately 2 × 106 CFU of log-phase S. aureus. At 2, 4, or 7 days after infection, kidneys were harvested and homogenized using a gentleMACS dissociator (Miltenyi Biotec, Inc., San Diego, CA), and serial dilutions were plated on agar to determine the number of CFU per organ. Mouse experiments were approved by the Institutional Animal Care and Use Committee and conducted in an AAALACi-accredited facility.
SUPPLEMENTAL MATERIAL
Supplemental materials and methods. Download
Compound 103: structure and inhibition of SpsB enzyme activity. Chemical structure of (A) compound 103 and (B) arylomycin A16. (C) Compound 103 inhibits enzymatic activity of purified SpsB with a calculated 50% inhibitory concentration (IC50) of 6.86 ± 0.27 nM. All values are the average ± SD from three independent experiments. Download
USA300 cro/cI mutant bacteria are normally virulent in mice. A/J mice were infected with S. aureus USA300 Δmcr (WT; strain GNE0023) (left two panels) or USA300 Δmcr cro/cI(T29N) (strain GNE0219) (right two panels). At 2 (D2) and 7 (D7) days after infection, the numbers of CFU in the kidneys were determined. The cro/cI mutation was not associated with a significant in vivo fitness cost as determined by the Mann-Whitney t test (P > 0.05). Dotted line, detection limit. Download
Inactivation of the putative ABC transporter sensitizes S. aureus to SpsB inhibition in vivo. (A) Neutropenic mice were given an intramuscular injection with 2 × 105 CFU of S. aureus and treated by continuous infusion with PBS (0) or with two different doses of compound 103 analog, which inhibits SpsB, reaching a plasma concentration at steady state (Css) of either 2 or 8 µg/ml. One day after infection, the numbers of viable bacteria in the thighs were determined. The SAUSA300_0352(K44A) point mutant with inactive ABC transporter in a WT cro/cI background showed a lower bacterial burden in the thigh muscle than did its parental WT strain. The MICs of the compound 103 analog were equal for both strains (0.25 µg/ml). The dotted line represents the number of bacteria in the inoculum. (B) The SAUSA300_0352(K44A) mutant shows no difference from the WT strain (GNE0023) in growth rate when cultured at 37°C in MHB either in the absence (left) or presence (right) of 25% normal human serum (NHS). (C) The ability to form biofilm in vitro was similar for both strains. WT, S. aureus USA300 WT (strain GNE0023); K44A, S. aureus USA300 SAUSA300_0352(K44A) mutant (strain GNE0174). Download
SpsB-independent protein secretion. Supernatants of S. aureus USA300 spsB::Tgn cro/cI(M1V) double mutant (strain GNE0191) and of WT USA300 Δmcr (strain GNE0023) were harvested from 18-h cultures grown at 37°C in MHB and analyzed by mass spectrometry and gel electrophoresis. (A) Logogram showing an alternative cleavage site identified by mass spectrometry in proteins secreted independently of SpsB in the spsB cro/cI double mutant (strain GNE0191). The alternative cleavage site appeared at various positions between 13 and 22 (N terminal to the signal peptide) in 12 of 25 secreted proteins (listed in Table S4 in the supplemental material). (B) Gel image of secreted proteins. Supernatants were separated by ultrafiltration using 10,000 nominal molecular weight limit (NMWL) cellulose filters into flowthrough and 20-fold-concentrated retentate fractions. Retentates (estimated to be 25 µg/lane) were fractionated by SDS-PAGE, and the gel was stained with Coomassie blue G-250. Lane 1, MW marker; lane 2, WT USA300 Δmcr (strain GNE0023); lane 3, USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191). This image shows that the secretion profile of the spsB cro/cI double mutant strain is generally different from that of the WT. Download
cro/cI nucleotide and amino acid changes leading to reduced sensitivity to compound 103. Shown are all mutations found inside or upstream of the SAUSA300_0350 (cro/cI) gene. Each of these mutations resulted in an increase in the MIC for compound 103 by at least 16-fold compared to the MIC for USA300 WT.
cro/cI(M1V) mutant of S. aureus USA300 exhibits unaltered sensitivity to antibiotics of various classes.
Genes that are overexpressed by at least twofold in the cro/cI(M1V) mutant of USA300 (strain GNE0117) compared to their expression in WT USA300 by RNA sequencing. The essential role of the clearly outstanding top four genes, which are organized in an operon with cro/cI itself, in resistance to compound 103 was confirmed experimentally by mutagenesis analysis (this study). For more details, see the text and Fig. 2A
SpsB-independent processing of secreted proteins. Proteins secreted into the culture supernatants of S. aureus WT USA300 Δmcr (strain GNE0023) and USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191), cultured for 18 h at 37°C in MHB, were analyzed by mass spectrometry. Proteins detected in the supernatants of USA300 Δmcr spsB::Tgn cro/cI(M1V) were secreted independently of SpsB. SpsB cleavage sites in proteins secreted by WT USA300 Δmcr were confirmed either by the presence of a peptide whose N terminus matched the right flanking sequence in the trypsin digest of secreted proteins retained by a 10-kDa filter or by a peptide with the matching C-terminal sequence identified in the filter’s flowthrough. The observed N- or C-terminal flanking residues are indicated in boldface. For the first 12 proteins in this list, we predicted a consensus cleavage motif (see Fig. S4A in the supplemental material).
Bacterial strains and plasmids.
Primers and probes used for qRT-PCR analysis of gene expression. Shown are the sequences of the forward and reverse primers and probes used to determine the expression of the corresponding genes, indicated in the left column, by qRT-PCR (see Fig. 3B).
ACKNOWLEDGMENTS
We thank Robert Higuchi and Tucker Roberts (RQx Pharmaceuticals, Inc.) for synthesis of compound 103, Jeremy Stinson, Alex Abbas, and Mark McCreary for generation and analysis of whole-genome-sequencing data, Summer Park for in vivo experiments, Jiansheng Wu for protein purification, Magnus Strandh, Klaus Koefoed, and Peter Andersen (Symphogen A/S, Denmark) for generating MAbs 6931, 4675, 4402, and 7574, Anan Chuntharapai and Theresa Shek for generating MAb 8F9, Brad Friedman for RNA expression analysis, and Lee Swem, Mireille Nishiyama, Lydia Santell, Bob Lazarus, Chris Koth, and Jian Payandeh for helpful advice.
The Network on Antibiotic Resistance in Staphylococcus aureus (NARSA/BEI) program is supported under NIAID/NIH contract no. HHSN272200700055C.
Footnotes
Citation Morisaki JH, Smith PA, Date SV, Kajihara KK, Truong CL, Modrusan Z, Yan D, Kang J, Xu M, Shah IM, Mintzer R, Kofoed EM, Cheung TK, Arnott D, Koehler MFT, Heise CE, Brown EJ, Tan M-W, Hazenbos WLW. 2016. A putative bacterial ABC transporter circumvents the essentiality of signal peptidase. mBio 7(5):e00412-16. doi:10.1128/mBio.00412-16.
REFERENCES
- 1.Servick K. 2015. The drug push. Science 348:850–853. doi: 10.1126/science.348.6237.850. [DOI] [PubMed] [Google Scholar]
- 2.DeLeo FR, Chambers HF. 2009. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest 119:2464–2474. doi: 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Small PM, Chambers HF. 1990. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 34:1227–1231. doi: 10.1128/AAC.34.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roemer T, Boone C. 2013. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol 9:222–231. doi: 10.1038/nchembio.1205. [DOI] [PubMed] [Google Scholar]
- 5.Smitha Rao CV, Anné J. 2011. Bacterial type I signal peptidases as antibiotic targets. Future Microbiol 6:1279–1296. doi: 10.2217/fmb.11.109. [DOI] [PubMed] [Google Scholar]
- 6.Van Roosmalen ML, Geukens N, Jongbloed JD, Tjalsma H, Dubois JY, Bron S, van Dijl JM, Anné J. 2004. Type I signal peptidases of Gram-positive bacteria. Biochim Biophys Acta 1694:279–297. doi: 10.1016/j.bbamcr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Dalbey RE, Wickner W. 1985. Leader peptidase catalyzes the release of exported proteins from the outer surface of the Escherichia coli plasma membrane. J Biol Chem 260:15925–15931. [PubMed] [Google Scholar]
- 8.Smith PA, Romesberg FE. 2012. Mechanism of action of the arylomycin antibiotics and effects of signal peptidase I inhibition. Antimicrob Agents Chemother 56:5054–5060. doi: 10.1128/AAC.00785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Therien AG, Huber JL, Wilson KE, Beaulieu P, Caron A, Claveau D, Deschamps K, Donald RG, Galgoci AM, Gallant M, Gu X, Kevin NJ, Lafleur J, Leavitt PS, Lebeau-Jacob C, Lee SS, Lin MM, Michels AA, Ogawa AM, Painter RE, Parish CA, Park YW, Benton-Perdomo L, Petcu M, Phillips JW, Powles MA, Skorey KI, Tam J, Tan CM, Young K, Wong S, Waddell ST, Miesel L. 2012. Broadening the spectrum of beta-lactam antibiotics through inhibition of signal peptidase type I. Antimicrob Agents Chemother 56:4662–4670. doi: 10.1128/AAC.00726-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schimana J, Gebhardt K, Holtzel A, Schmid DG, Sussmuth R, Muller J, Pukall R, Fiedler HP. 2002. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomyces sp. Tu 6075. I. Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 55:565–570. doi: 10.7164/antibiotics.55.565. [DOI] [PubMed] [Google Scholar]
- 11.Höltzel A, Schmid DG, Nicholson GJ, Stevanovic S, Schimana J, Gebhardt K, Fiedler H, Jung G. 2002. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by streptomyces sp. Tu 6075. II. Structure elucidation. J Antibiot (Tokyo) 55:571–577. doi: 10.7164/antibiotics.55.571. [DOI] [PubMed] [Google Scholar]
- 12.Luo C, Roussel P, Dreier J, Page MG, Paetzel M. 2009. Crystallographic analysis of bacterial signal peptidase in ternary complex with arylomycin A2 and a beta-sultam inhibitor. Biochemistry 48:8976–8984. doi: 10.1021/bi9009538. [DOI] [PubMed] [Google Scholar]
- 13.Smith PA, Powers ME, Roberts TC, Romesberg FE. 2011. In vitro activities of arylomycin natural-product antibiotics against Staphylococcus epidermidis and other coagulase-negative staphylococci. Antimicrob Agents Chemother 55:1130–1134. doi: 10.1128/AAC.01459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson AD, Poteete AR, Lauer G, Sauer RT, Ackers GK, Ptashne M. 1981. Lambda repressor and cro—components of an efficient molecular switch. Nature 294:217–223. doi: 10.1038/294217a0. [DOI] [PubMed] [Google Scholar]
- 15.Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S. 2005. Switches in bacteriophage lambda development. Annu Rev Genet 39:409–429. doi: 10.1146/annurev.genet.39.073003.113656. [DOI] [PubMed] [Google Scholar]
- 16.Craney A, Romesberg FE. 2015. A putative Cro-like repressor contributes to arylomycin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 59:3066–3074. doi: 10.1128/AAC.04597-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matveeva EA, He P, Whiteheart SW. 1997. N-ethylmaleimide-sensitive fusion protein contains high and low affinity ATP-binding sites that are functionally distinct. J Biol Chem 272:26413–26418. doi: 10.1074/jbc.272.42.26413. [DOI] [PubMed] [Google Scholar]
- 18.Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 19.Craney A, Dix MM, Adhikary R, Cravatt BF, Romesberg FE. 2015. An alternative terminal step of the general secretory pathway in Staphylococcus aureus. mBio 6:e01178-15. doi: 10.1128/mBio.01178-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23:3393–3404. doi: 10.1096/fj.09-135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubin G. 2002. Extracellular proteases of Staphylococcus spp. Biol Chem 383:1075–1086. doi: 10.1515/BC.2002.116. [DOI] [PubMed] [Google Scholar]
- 22.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. doi: 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clinical and Laboratory Standards Institute. 2007. Performance standards for antimicrobial susceptibility testing; 17th informational supplement. CLSI document M100-S17 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 24.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao J, Zhong J, Fang Y, Geisinger E, Novick RP, Lambowitz AM. 2006. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 12:1271–1281. doi: 10.1261/rna.68706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Date SV, Modrusan Z, Lawrence M, Morisaki JH, Toy K, Shah IM, Kim J, Park S, Xu M, Basuino L, Chan L, Zeitschel D, Chambers HF, Tan MW, Brown EJ, Diep BA, Hazenbos WL. 2014. Global gene expression of methicillin-resistant Staphylococcus aureus USA300 during human and mouse infection. J Infect Dis 209:1542–1550. doi: 10.1093/infdis/jit668. [DOI] [PubMed] [Google Scholar]
- 27.Meijer PJ, Andersen PS, Haahr Hansen M, Steinaa L, Jensen A, Lantto J, Oleksiewicz MB, Tengbjerg K, Poulsen TR, Coljee VW, Bregenholt S, Haurum JS, Nielsen LS. 2006. Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J Mol Biol 358:764–772. doi: 10.1016/j.jmb.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 28.Meijer PJ, Nielsen LS, Lantto J, Jensen A. 2009. Human antibody repertoires. Methods Mol Biol 525:261–277. doi: 10.1007/978-1-59745-554-1_13. [DOI] [PubMed] [Google Scholar]
- 29.Hazenbos WL, Kajihara KK, Vandlen R, Morisaki JH, Lehar SM, Kwakkenbos MJ, Beaumont T, Bakker AQ, Phung Q, Swem LR, Ramakrishnan S, Kim J, Xu M, Shah IM, Diep BA, Sai T, Sebrell A, Khalfin Y, Oh A, Koth C, Lin SJ, Lee BC, Strandh M, Koefoed K, Andersen PS, Spits H, Brown EJ, Tan MW, Mariathasan S. 2013. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog 9:e1003653. doi: 10.1371/journal.ppat.1003653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods. Download
Compound 103: structure and inhibition of SpsB enzyme activity. Chemical structure of (A) compound 103 and (B) arylomycin A16. (C) Compound 103 inhibits enzymatic activity of purified SpsB with a calculated 50% inhibitory concentration (IC50) of 6.86 ± 0.27 nM. All values are the average ± SD from three independent experiments. Download
USA300 cro/cI mutant bacteria are normally virulent in mice. A/J mice were infected with S. aureus USA300 Δmcr (WT; strain GNE0023) (left two panels) or USA300 Δmcr cro/cI(T29N) (strain GNE0219) (right two panels). At 2 (D2) and 7 (D7) days after infection, the numbers of CFU in the kidneys were determined. The cro/cI mutation was not associated with a significant in vivo fitness cost as determined by the Mann-Whitney t test (P > 0.05). Dotted line, detection limit. Download
Inactivation of the putative ABC transporter sensitizes S. aureus to SpsB inhibition in vivo. (A) Neutropenic mice were given an intramuscular injection with 2 × 105 CFU of S. aureus and treated by continuous infusion with PBS (0) or with two different doses of compound 103 analog, which inhibits SpsB, reaching a plasma concentration at steady state (Css) of either 2 or 8 µg/ml. One day after infection, the numbers of viable bacteria in the thighs were determined. The SAUSA300_0352(K44A) point mutant with inactive ABC transporter in a WT cro/cI background showed a lower bacterial burden in the thigh muscle than did its parental WT strain. The MICs of the compound 103 analog were equal for both strains (0.25 µg/ml). The dotted line represents the number of bacteria in the inoculum. (B) The SAUSA300_0352(K44A) mutant shows no difference from the WT strain (GNE0023) in growth rate when cultured at 37°C in MHB either in the absence (left) or presence (right) of 25% normal human serum (NHS). (C) The ability to form biofilm in vitro was similar for both strains. WT, S. aureus USA300 WT (strain GNE0023); K44A, S. aureus USA300 SAUSA300_0352(K44A) mutant (strain GNE0174). Download
SpsB-independent protein secretion. Supernatants of S. aureus USA300 spsB::Tgn cro/cI(M1V) double mutant (strain GNE0191) and of WT USA300 Δmcr (strain GNE0023) were harvested from 18-h cultures grown at 37°C in MHB and analyzed by mass spectrometry and gel electrophoresis. (A) Logogram showing an alternative cleavage site identified by mass spectrometry in proteins secreted independently of SpsB in the spsB cro/cI double mutant (strain GNE0191). The alternative cleavage site appeared at various positions between 13 and 22 (N terminal to the signal peptide) in 12 of 25 secreted proteins (listed in Table S4 in the supplemental material). (B) Gel image of secreted proteins. Supernatants were separated by ultrafiltration using 10,000 nominal molecular weight limit (NMWL) cellulose filters into flowthrough and 20-fold-concentrated retentate fractions. Retentates (estimated to be 25 µg/lane) were fractionated by SDS-PAGE, and the gel was stained with Coomassie blue G-250. Lane 1, MW marker; lane 2, WT USA300 Δmcr (strain GNE0023); lane 3, USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191). This image shows that the secretion profile of the spsB cro/cI double mutant strain is generally different from that of the WT. Download
cro/cI nucleotide and amino acid changes leading to reduced sensitivity to compound 103. Shown are all mutations found inside or upstream of the SAUSA300_0350 (cro/cI) gene. Each of these mutations resulted in an increase in the MIC for compound 103 by at least 16-fold compared to the MIC for USA300 WT.
cro/cI(M1V) mutant of S. aureus USA300 exhibits unaltered sensitivity to antibiotics of various classes.
Genes that are overexpressed by at least twofold in the cro/cI(M1V) mutant of USA300 (strain GNE0117) compared to their expression in WT USA300 by RNA sequencing. The essential role of the clearly outstanding top four genes, which are organized in an operon with cro/cI itself, in resistance to compound 103 was confirmed experimentally by mutagenesis analysis (this study). For more details, see the text and Fig. 2A
SpsB-independent processing of secreted proteins. Proteins secreted into the culture supernatants of S. aureus WT USA300 Δmcr (strain GNE0023) and USA300 Δmcr spsB::Tgn cro/cI(M1V) (strain GNE0191), cultured for 18 h at 37°C in MHB, were analyzed by mass spectrometry. Proteins detected in the supernatants of USA300 Δmcr spsB::Tgn cro/cI(M1V) were secreted independently of SpsB. SpsB cleavage sites in proteins secreted by WT USA300 Δmcr were confirmed either by the presence of a peptide whose N terminus matched the right flanking sequence in the trypsin digest of secreted proteins retained by a 10-kDa filter or by a peptide with the matching C-terminal sequence identified in the filter’s flowthrough. The observed N- or C-terminal flanking residues are indicated in boldface. For the first 12 proteins in this list, we predicted a consensus cleavage motif (see Fig. S4A in the supplemental material).
Bacterial strains and plasmids.
Primers and probes used for qRT-PCR analysis of gene expression. Shown are the sequences of the forward and reverse primers and probes used to determine the expression of the corresponding genes, indicated in the left column, by qRT-PCR (see Fig. 3B).