

Abstract
Metabolic reprogramming, in which altered utilization of glucose and glutamine supports rapid growth, is a hallmark of most cancers. Mutations in the oncogenes KRAS and BRAF drive metabolic reprogramming through enhanced glucose uptake, but the broader impact of these mutations on pathways of carbon metabolism is unknown. Global shotgun proteomic analysis of isogenic DLD-1 and RKO colon cancer cell lines expressing mutant and wild type KRAS or BRAF, respectively, failed to identify significant differences (at least 2-fold) in metabolic protein abundance. However, a multiplexed parallel reaction monitoring (PRM) strategy targeting 73 metabolic proteins identified significant protein abundance increases of 1.25–twofold in glycolysis, the nonoxidative pentose phosphate pathway, glutamine metabolism, and the phosphoserine biosynthetic pathway in cells with KRAS G13D mutations or BRAF V600E mutations. These alterations corresponded to mutant KRAS and BRAF-dependent increases in glucose uptake and lactate production. Metabolic reprogramming and glucose conversion to lactate in RKO cells were proportional to levels of BRAF V600E protein. In DLD-1 cells, these effects were independent of the ratio of KRAS G13D to KRAS wild type protein. A study of 8 KRAS wild type and 8 KRAS mutant human colon tumors confirmed the association of increased expression of glycolytic and glutamine metabolic proteins with KRAS mutant status. Metabolic reprogramming is driven largely by modest (<2-fold) alterations in protein expression, which are not readily detected by the global profiling methods most commonly employed in proteomic studies. The results indicate the superiority of more precise, multiplexed, pathway-targeted analyses to study functional proteome systems. Data are available through MassIVE Accession MSV000079486 at ftp://MSV000079486@massive.ucsd.edu.
Cancers typically exhibit a metabolic phenotype distinct from that of normal tissues. Warburg first reported that cancers have increased glucose consumption with concurrently elevated lactate production compared with normal tissues (the “Warburg effect”) (1). Since this initial observation, reprogramming of central carbon metabolism (“metabolic reprogramming”) has been characterized as an essential adaptation for tumor growth (2–5).
Mutations in KRAS or BRAF appear to play important roles in regulating metabolic reprogramming in multiple cancers (6–16). These proteins play key roles in the EGFR signaling pathway and oncogenic mutations in either protein can drive downstream activation of this pathway even in the absence of upstream EGFR activation (17–20). KRAS and BRAF mutations are thought to contribute to cancer development by driving proliferation of cells with initiating mutations (21, 22).
KRAS and BRAF may contribute to cancer phenotypes through metabolic reprogramming. The colorectal cancer cell lines DLD-1 and RKO, which have oncogenic mutations in KRAS and BRAF, respectively, display increased expression of the primary glucose transporter, SLC2A1 (commonly known as GLUT1), and exhibit a Warburg effect phenotype (6). Pancreatic ductal adenocarcinomas driven by a KRAS G12D mutation exhibit increased glucose utilization in the nonoxidative pentose phosphate pathway (PPP) 1 (9) and are highly dependent upon glutamine metabolism for tumor growth (8). BRAF V600E mutations in melanoma cells decreased expression of tricarboxylic acid (TCA) cycle enzymes and mitochondrial oxidative phosphorylation, and these effects are reversed by a BRAF V600E-selective kinase inhibitor (23). These studies demonstrate significant roles of oncogenic KRAS or BRAF in metabolic reprogramming. Transcriptome analyses of multiple cancers revealed several patterns of alterations for genes encoding metabolic enzymes (24). Although transcriptome profiles were not associated with specific mutations, the data suggested mutations may drive distinct programs of metabolism gene expression.
Multiplexed protein quantitation by multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) are powerful tools for systems characterization (25–39). These multiplexed assays can interrogate coordinated expression of proteins in functional protein networks, such β-catenin signaling (35), nuclear factor-κB signaling (36), protein expression changes because of EGFR signaling (37, 38), and phosphotyrosine quantitation in EGFR signaling (39). Drabovich et al. utilized a single MRM assay to quantify 134 proteotypic peptides from 76 proteins involved in glycolysis, the TCA cycle, the PPP and related reactions in a single MRM assay to analyze metabolic protein expression changes during hypoxia (40).
Here we used both global shotgun proteome profiling and a multiplexed PRM to quantify 73 proteins in central carbon metabolism Figure 1 to assess protein-level impact of oncogenic KRAS and BRAF in isogenic colorectal cancer cell models. Whereas global profiling failed to detect significant protein alterations associated with metabolic reprogramming, more precise PRM measurements revealed alterations in multiple carbon metabolism pathway alterations driven by oncogenic KRAS and BRAF. These observations were reproduced in MRM analyses of KRAS mutant and wild type primary human colorectal tumors. The data indicate that precise, targeted analyses are essential to detect the relatively modest protein abundance alterations that underlie metabolic reprogramming in cancers.
Fig. 1.

Proteins monitored by metabolic panel. Proteins monitored by PRM/MRM are grouped into glycolysis, TCA, PPP, glutamine metabolism, and phosphoserine biosynthesis. Proteins monitored in this panel are enclosed in a black border, whereas the metabolites used by these enzymes are in black font only. Peptides monitored by PRM for the proteins listed here are listed in supplemental Table S1, while the corresponding peptides and transitions monitored by MRM are listed in supplemental Table S2.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Sep-pak C18 desalting cartridges and XBridge C18 5 μm 4.6 × 250 mm columns were from Waters (Milford, MA). ReproSil C18-AQ resin (3 μm particle size) was purchased from Dr. Maisch, Gmbh (Ammerbuch-Entringen, Germany). Picofrit self-pack columns, 75 μm ID, 10 μm ID tip, were from New Objective (Woburn, MA). Bovine six protein equimolar digest was purchased from Bruker-Michrom, Inc. (Auburn, CA). Trypsin (Trypsin Gold) was from Promega (Madison, WI). Synthetic and 13C,15N lysine or arginine labeled peptides were purchased from New England Peptides (Gardner, MA). Labeled peptides were of greater than 99% isotopic purity and greater than 95% chemical purity; absolute concentration was determined by amino acid analysis.
Cell Culture
DLD-1 parental cells (DLD-1 Par), DLD-1 KRAS (G13D/-) cells (DLD-1 Mut), and DLD-1 KRAS (±) cells (DLD-1 WT) (catalog numbers HD 105–040, HD 105–043, and HD PAR-086), RKO parental cells (RKO Par), RKO BRAF (V600E/−/−) cells (RKO Mut), and RKO BRAF (+/−/−) cells (RKO WT) (catalog numbers HD 106–004, HD 106–003, and HD PAR-009) were from Horizon Discovery (Cambridge, UK). DLD-1 cell lines were maintained in RPMI 1640 growth media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CA) and 0.1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). RKO cell lines were maintained in McCoy's 5A growth media (Invitrogen) supplemented with 10% fetal bovine serum and 0.1% penicillin-streptomycin. All cell lines were split 1:10 every 3–5 days or before cells reached 80% confluency. Cells were reseeded from flasks into 15 cm plates 2 days prior to preparation for analyses and were harvested before reaching 75% confluency. Three replicate cultures of each cell line were analyzed by RNA-Seq to verify that the cells expressed the expected mutant or wild type KRAS and BRAF sequences. These analyses demonstrated that the cell lines expressed the expected wild type and mutant sequences (supplemental Fig. S1).
Cells were harvested by scraping on ice using cold phosphate buffered saline (PBS, Invitrogen) supplemented with 1:100 Halt Protease Inhibitor Mixture (Thermo Fisher Scientific, Waltham, MA). The cells were pelleted by centrifugation at 1000 × g at 4 °C for 5 min, washed with an additional 1 ml of PBS and pelleted a second time at 1000 × g at 4 °C for 5 min. Excess PBS was removed from the pellets and pellets were flash frozen in dry ice and ethanol.
Glucose and Lactate Analyses
Cell lines were grown to 60% confluence in 75 cm2 flasks, and were split into eight wells of a 96-well plate. Cells were plated so that there would be ∼5000 cells per well for each collection time point. Medium was collected from the cells at each doubling time and one-half doubling time for each respective cell line. Cell number was estimated using the cell proliferation reagent WST-1 (Sigma, St. Louis, MO). Media samples were analyzed on a YSI 2300 glucose and lactate biochemical analyzer (YSI Life Science, Yellow Springs, OH). Glucose and lactate standards were run every five samples to ensure instrument calibration and accuracy.
Sample preparation and basic reverse phase liquid chromatography (bRPLC)
Frozen cell pellets were suspended in a 1:1 (v/v) mixture of 100 mm ammonium bicarbonate (Sigma, St. Louis, MO), pH 8.0 and 2,2,2-trifluoroethanol (Acros Organics. Pittsburg, PA), supplemented with 1:100 HALT protease inhibitor mixture. Suspensions were sonicated 3 times for 15 s and were placed on ice for at least 1 min between sonication cycles. Protein concentration was determined with the bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA) and 250 μg of protein was taken for analysis. Samples were reduced with 40 mm tris-carboxyethylphosphine (Sigma, St. Louis, MO) and 100 mm dithiothreitol (Research Products International, Mt. Prospect, IL) at 60 °C for 30 min at 1000 rpm on an Eppendorf Thermomixer (Eppendorf, Hauppauge, NY). Samples then were cooled to room temperature and alkylated with 200 mm iodoacetamide (Sigma, St. Louis, MO) in the dark for 30 min. Samples were diluted with 50 mm ammonium bicarbonate, pH 8.0 to reduce the 2,2,2-trifluoroethanol to 10%, prior to adding trypsin at a 1:50 (w/w) ratio and incubating overnight at 37 °C with shaking. Digests were lyophilized, the lyophilized peptide samples were suspended in water prior to solid phase extraction with a Waters Sep-pak C18 desalting cartridge. Prior to use, desalting cartridges were first charged with 1 ml of acetonitrile and then equilibrated with 2 ml of water. Peptide samples were loaded onto the equilibrated column, washed once with 1 ml water, and the peptides were eluted with 70% acetonitrile containing 0.1% formic acid (Thermo Fisher Scientific, Waltham, MA). These samples were evaporated to dryness in vacuo and redissolved in 10 mm triethylammonium bicarbonate, pH 8.0. bRPLC peptide fractionation was done with an Agilent 1260 Infinity LC system equipped with an XBridge C18 5 μm 4.6 × 250 mm column. Solvent A was 10 mm triethylammonium bicarbonate (Sigma, St. Louis, MO), pH 7.4 and solvent B was 10 mm triethylammonium bicarbonate in acetonitrile. Peptides were loaded onto the column with solvent A at a flow rate of 0.5 ml/minute and were eluted at a flow rate of 0.5 ml/minute with a program in which solvent B was increased from 0% to 5% from 0 to 10 min, 5% to 35% from 10 to 70 min, 35% to 70% from 70 to 85 min, held at 70% from 85 to 95 min, and then reduced to 0% from 95 to 105 min. The eluted peptides were collected in 64 fractions, which were concatenated to eight fractions as described by Wang et al. (41). Concatenated fractions were evaporated to dryness in vacuo and the dried samples were suspended in 100 μl 3% acetonitrile with 0.1% formic acid for LC-MS/MS analysis.
Global LC-MS/MS Analyses
LC-MS/MS analyses were performed on a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with an Easy-nLC 1000 autosampler. Peptides were resolved on an PicoFrit Emitter column (11 cm x 75 μm ID, New Objective, Wortham, MA) with a 10 μm ID opening, packed with ReproSil C18-AQ resin of 3 μm particle size. Liquid chromatography was performed at room temperature with a mobile phase gradient program using 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B).
Sample solutions (2 μl) were loaded onto the column over 14 min with 100% solvent A at 0.5 μl/min, followed by an elution gradient (300 nL/min) from 2% to 5% solvent B in 5 min, 5% to 35% solvent B over 85 min, 35% to 90% solvent B in 3 min, and held at 90% solvent B for 7 min. Peptides eluting from the capillary tip were introduced into the Q Exactive Plus source via nano electrospray mode with a capillary voltage of 2.1 kV. A full scan was obtained from the eluting peptides in the range of 300–1800 m/z, with a resolution of 70,000, a max injection time of 64 ms, and an AGC target of 3e6. The full scan was then followed by 20 data-dependent MS/MS scans of the most intense ions, with a resolution of 17,500, a maximum injection time of 100 ms, an AGC target of 2e5, an isolation window of 1.4 m/z, a fixed first mass of 100 m/z, and a normalized collision energy of 27. MS/MS spectra were acquired with dynamic exclusion of previously analyzed precursors for 20 s.
Global Proteomics Data Analysis
For database searching, The “ScanSifter” algorithm (42) read MS/MS spectra stored as centroid peak lists from Thermo RAW files and transcoded them to mzData v1.05 files. Spectra that contain fewer than six peaks were not transcoded to mzData files and only MS/MS scans are written to the mzData files; MS scans were excluded. MS/MS spectra were assigned to peptides from the Human RefSeq Version 54 database (accession date of September 2012, with 34,589 protein entries, which included contaminant protein sequences) using the database search algorithms Myrimatch version 2.1.132 (43) and MS-GF+ Beta (v9979) (44). The database forward protein sequences were appended with reversed sequences to allow for false discovery rate (FDR) estimations (45, 46). Myrimatch and MS-GF+ were configured to allow for all cysteines to be modified by carboxamidomethylation as a static modification, while allowing for possible dynamic methione oxidation, with a maximum of two dynamic modifications per peptide. Candidate peptides were required to be tryptic, although missed cleavages were allowed. For Myrimatch searches, precursor mass error was set to 1.5 m/z, and fragment ion mass error was 0.5 m/z. For MS-GF+ searches, the precursor mass error was set to ±15 ppm. Identified peptides from both searches were assembled together into proteins with IDPicker version 3.1.642.0 (46). Proteins were assembled using a maximum Q value of 0.01, a minimum of 2 distinct peptides per protein, and a minimum of 5 spectra per protein in order to achieve a protein FDR of less than 5%. Indistinguishable proteins were recognized and grouped. Parsimony rules were applied to generate a minimal list of proteins that explain all of the peptides that pass the entry criteria. A minimum of two spectra per protein across all samples and both biological replicates was required for quantitative comparisons. This list of quantifiable proteins then was used to determine 1.5- and twofold differences in spectral count data in pairwise comparisons. The lists of 1.5- and twofold differentially expressed proteins were used to determine pathway enrichment using the network based enrichment algorithm WebGestalt (47), where the list of all quantifiable proteins for each data set was used as the reference data set.
Sample Preparation for PRM Analyses of Metabolic Proteins
Samples for PRM analyses were prepared in the same manner as for global proteomics, except that 200 μg of protein was used for trypsin digestion and no bRPLC fractionation was performed. Desalted peptide samples were dissolved to 0.5 μg/μl with 3% aqueous acetonitrile containing 0.1% formic acid and a mixture of three labeled reference peptide (LRP) standards (β-actin peptide U-13C,15N-Arg-GYSFTTTAER, alkaline phosphatase peptide U-13C,15N-Arg-AAQGITAPGGAR, and β-galactosidase peptide U-13C,15N-Arg-APLDNDIGVSEATR) was spiked into the samples at a final concentration of 12.5 fmol/μl.
PRM Analyses of Metabolic Proteins
PRM analyses were performed on a Q Exactive Plus mass spectrometer equipped with the same LC system and column described above. Liquid chromatography was performed at room temperature over 70 min using a gradient program of 0.1% (v/v) formic acid in water (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B). Sample solutions (2 μl) were loaded onto the column over 14 min with 100% solvent A at 0.5 μl/min, followed by an elution gradient (300 nL/min) from 2% to 5% solvent B in 5 min, 5% to 35% solvent B over 45 min, 35% to 90% solvent B in 5 min, and held at 90% solvent B for 10 min. Peptides eluting from the capillary tip were introduced into the Q Exactive Plus source via nano electrospray mode with a capillary voltage of 2.1 kV. The mass spectrometer was programmed to acquire a full MS-SIM scan followed by 14 targeted-MS2 runs. Full MS-SIM scans were collected with a resolution of 17,500, an AGC of 3e6, a maximum injection time of 64 ms, and a scan range of 380–1500 m/z. Targeted-MS2 spectra were acquired at a resolution of 17,500, a maximum injection time of 80 ms, an AGC target of 5e5, an isolation window of 2.0 m/z, a fixed first mass of 150 m/z, and a normalized collision energy of 27.
Peptide precursor ions targeted for acquisition were selected using Skyline software (48). To develop a scheduled PRM method, a “master mix” of unlabeled synthetic standards representing all target peptides was spiked into a matrix background made from the samples. This master mix sample was analyzed in an unscheduled PRM run to determine retention times and representative fragment ions. The 10 most intense fragment ions for each peptide were used to identify peptides in the synthetic master mix used for designing the scheduled run. A total of 73 proteins were monitored with at least 2 peptides each, for a total of 204 peptides monitored in each scheduled PRM run (Fig. 1 and supplemental Table S1). Three biological replicates from each cell type were analyzed in triplicate to assess both biological variation and instrument variation.
Analysis of PRM Data for Metabolic Proteins
Peptide transitions to be extracted for PRM were selected using the program Skyline (48). The top five most intense fragment ions were used to verify the detection of each peptide in the PRM analyses and the top three most intense fragment ions were used for peptide peak area quantitation. Peptide signals for metabolic proteins were normalized by the LRP method (49). Peptide peak areas were calculated as the sum of the peak areas for the 3 most intense fragment ions, and this summed peak area was normalized to the summed peak area for the LRP standard with the lowest coefficient of variation (CV) across all of the samples in the sample set. For quantitative comparisons between cell lines, peptides were required to have CV values below 0.25 between biological replicate experiments. For proteins with more than one quantifiable peptide, the peptide having a CV below 0.25 and the largest normalized peak area was used for quantitation. This minimizes measurement variation across the data set (49). Measurements of the other quantifiable peptides were not used for quantitative comparisons, but provided validation of single peptide-based measurements.
Targeted quantitation of KRAS and BRAF protein forms
For targeted analysis of wild type and mutant KRAS and BRAF protein forms, cells were grown as previously described (28). For KRAS analyses, DLD-1 cells were suspended in 50 mm Tris-HCl (J. T. Baker, Center Valley, PA), pH 8, 0.1% sodium dodecyl sulfate (Thermo Fisher Scientific, Waltham, MA), 150 mm sodium chloride, 0.5% sodium deoxycholate, 1% Igepal (Sigma, St. Louis, MO) supplemented with 1:100 Halt Protease, whereas for BRAF analyses, RKO cells were suspended in HEPES buffer (50 mm HEPES pH 7.5, 10 mm KCl, 150 mm NaCl,1 mm EDTA, 1 mm EGTA, 1.5 mm MgCl2, 10% glycerol, 0.1% CHAPS, and 0.01% Brij-35). Cells were lysed by sonication and protein concentration was measured using the bicinchoninic acid assay. For each sample, 50 μg of protein was loaded on a single lane of a 10-lane NuPAGE Novex 10% Bis-Tris gel (Invitrogen). Samples from 3 replicate cultures of each cell line were loaded on each gel and gel electrophoresis was performed at 180 V for 45 min. Gels were stained with SimplyBlue SafeStain (Invitrogen) and destained with deionized water overnight. For KRAS analysis, the MW 20 to 25 kDa region containing KRAS (MW 21.7 kDa) and the MW 25 to 37 kDa region (background matrix for calibration curve) were excised from the gel. For BRAF analysis, the MW 75 to 100 kDa region containing BRAF (94 kDa) and the MW 100 to 150 kDa region (background matrix for calibration curve) were excised from the gels.
Gel slices were placed in 100 μl of 100 mm ammonium bicarbonate, pH 8.0, and were reduced with 10 μl of 100 mm dithiothreitol for 20 min at 50 °C, and alkylated with 10 μl of 200 mm iodoacetamide for 20 min in the dark at room temperature. Gel slices were destained with 50% acetonitrile/50 mm ammonium bicarbonate until the gel slices were no longer blue, and the destained slices were dehydrated with 100% acetonitrile. Acetonitrile was removed from the gels in vacuo and the pieces were incubated in 50 mm ammonium bicarbonate containing 0.01 μg/μl of trypsin at 37 °C overnight with shaking. Peptides were extracted from the gel slices with 3 200 μl washes of 60% acetonitrile containing 0.1% formic acid, and were evaporated to dryness in vacuo. Stable isotope dilution (SID) standard peptides for KRAS (U-13C,15N-Lys-LVVVGAGGVGK and U-13C,15N-Lys-LVVVGAGDVGK for wild type KRAS and KRAS G13D, respectively), or BRAF (U-13C,15N-Lys-IGDFGLATVK and U-13C,15N-Lys-IGDFGLATEK for wild type BRAF and BRAF V600E, respectively) were added to each dried sample to produce concentrations of 1 fmol/μl for each labeled peptide when the samples were resuspended in 50 μl of 3% acetonitrile with 0.1% formic acid. For SID calibration curves, light peptides (LVVVGAGGVGK and LVVVGAGDVGK for KRAS SID, or IGDFGLATVK and IGDFGLATEK for BRAF SID) were added to the dried matrix background digests, to generate calibration points with light peptide concentrations of 0 amol/μl, 8 amol/μl, 40 amol/μl, 200 amol/μl, 1 fmol/μl, 5 fmol/μl, and 25 fmol/μl in 50 μl of 3% acetonitrile and 0.1% formic acid.
PRM analyses of KRAS and BRAF peptides were performed with the same LC-MS/MS system described above for PRM analyses of metabolic proteins. Scheduled run retention time windows were determined using an unscheduled run from the 1 fmol/μl calibration curve samples. Three biological replicates of each cell line were analyzed and each biological replicate was analyzed in duplicate. Calibration curve standard samples were also processed in three biological replicates, with two process replicates generated for serial dilutions to generate the calibration curve, and each sample was analyzed twice as technical replicates.
RAW files were imported into Skyline and transitions were selected according to intensity and mass accuracy. Transition peak areas were integrated using the area under the curve, and 5 transitions were used to determine peptide detection, and the single most intense transition was used for quantitation. Peak areas for light peptides were normalized to the peak areas of the corresponding heavy peptide, with the same fragment ions used for both light and heavy peptides. For the calibration curve, the ratio of the light peptide peak area to the heavy peptide peak area was plotted against the theoretical concentration of the light peptide using Quasar (50) and this calibration curve was used to calculate the concentration of endogenous light peptide in the quantitative samples. Lower limit of quantitation (LLOQ) was determined for each peptide according to the lowest calibration point measurement with a calculated concentration CV lower than 0.25. Lower limit of detection (LLOD) was 1/3 of the LLOQ. All measurements were plotted in GraphPad Prism, and Student's t test was performed to determine significant differences. The LLOQ and LLOD values for the WT KRAS peptide LVVVGAGGVGK, the G13D KRAS peptide LVVVGAGDVGK and the BRAF V600E GDFGLATEK peptide were 8 amol/μl and 2.6 amol/μl, respectively. The LLOQ and LLOD values for the BRAF WT GDFGLATVK peptide were 40 amol/μl and 13.3 amol/μl, respectively.
Analysis of Human Stage II Colon Tumor Specimens
All tumor specimens were derived from histologically confirmed Stage II colon cancer under Institutional Review Board protocol #120805. Tissue blocks were obtained from the Surgical Pathology archives at Vanderbilt University. Selection was based on complete surgical removal of the tumor. Blocks were sectioned and the sections were macrodissected to remove normal tissue, including normal epithelium and smooth muscle, such that the remainder was at least 80% tumor material, which included tumor cells and stroma. A minimum carcinoma cell percentage in the tumor material was not specified. Tumors were genotyped for KRAS, NRAS, BRAF, and PI3K mutational status with a multiplexed mutation profiling panel (supplemental Table S2) (51). Eight tumors had codon 12 mutations in KRAS (11 G12V and one G12D) and eight had wild type KRAS. Of the KRAS mutant tumors, two also had PIK3CA E545K mutations, whereas one of the KRAS wild type tumors had a NRAS Q61K mutation and one had a BRAF V600E mutation. Formalin-fixed, paraffin-embed (FFPE) samples were deparaffinized, rehydrated, reduced, alkylated and digested with trypsin as described previously by Sprung et al. (52). Digested samples (50 μg) were lyophilized and the peptide mixtures were desalted using a Waters Sep-pak C18 desalting cartridges. Desalted samples were redissolved in 100 μl of 3% aqueous acetonitrile containing 0.1% formic acid and 25 fmol/μl of each of the 3 LRP standards were spiked into each sample.
MRM Analyses of Human Stage II Colon Tumors
MRM analyses were performed on a TSQ Vantage triple quadrupole mass spectrometer equipped with an Eksigent Ultra nanoLC and microautosampler. Samples were analyzed using a scheduled experiment, where peptide retention times were determined from an unscheduled analysis of a master mix of synthetic unlabeled peptides, as described above. Sample solutions (2 μl) were loaded onto the column over 14-min in water containing 0.1% formic acid at 0.5 μl/min. Peptides were resolved on an 11 cm × 75 μm ID PicoFrit Emitter with a 10 μm ID opening, packed with ReproSil C18-AQ resin of 3 μm particle size. Liquid chromatography was performed at room temperature over 70 min with of 0.1% (v/v) formic acid in water (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B) at a flow rate of 400 nL/min. The gradient was programmed from 2% to 5% solvent B in 5 min, 5% to 35% solvent B over 45 min, 35% to 90% solvent B in 5 min, held at 90% solvent B for 5 min, and then reduced to 2% solvent B for 5 min. MRM analyses monitored 61 proteins represented by 194 peptides with five transitions per peptide for a total of 980 transitions. The MRM method was split into two separate injections, with one method monitoring 390 transitions and a second monitoring 590 transitions (supplemental Table S3).
Experimental Design and Statistical Rationale
For glucose consumption and lactate production rate measurements, three separate cultures were analyzed in duplicate for each cell line and was sufficient to detect at least 1.5-fold changes in these parameters. In the global analysis of the cell lines, two separate cultures were analyzed, but a single analysis of each sample was performed. This enabled detection of at least twofold differences based on spectral count data.
For PRM analyses of metabolic proteins, three separate cultures of each cell line were analyzed and each biological replicate sample was injected in triplicate. These triplicate analyses were used to determine instrument performance, as assessed with quality control samples, and to calculate coefficients of variation (CV). CVs were calculated for each peptide after LRP normalization for both biological and technical replicates and peptides with CVs of greater than 0.25 for normalized peak areas were not considered further. For peptides with normalized peak area CVs less than 0.25, we calculated intraclass correlation coefficient (ICC) values, which indicate the fraction of the measurement variation associated with differences between distinct classes (experimental groups). Each cell line was treated as a distinct class. Peptides with a CV below 0.25 and an ICC above 0.6 were used for further statistical comparisons. Peptides with a CV below 0.25, but an ICC below 0.6 were not used for statistical comparison, and were classified as not significantly different between biological classes. For peptide measurements that satisfied the above criteria, Student's t test was used to determine the significance of the measured differences. For targeted SID measurements of mutant and wild type KRAS and BRAF protein forms, three separate cultures were analyzed and each was analyzed in triplicate. Replicate injections with the SID samples were used in conjunction with quality control samples to assess instrument performance and to calculate biological sample CVs.
For MRM analyses of colon tumors, sample amounts were limiting; a single biological replicate sample was prepared from each tumor and analyzed in triplicate. ICCs were calculated for each peptide for the MRM results, where the two classes were the KRAS mutant and wild type tumors. ICC values greater that 0.7 were required for significant comparisons. For pairwise comparison of MRM data, protein measurements were grouped according to KRAS mutational status, and Student's t test was performed to determine statistical significance of differences.
RESULTS
Isogenic cell lines enable systematic study of the effects of oncogenes on cellular networks. DLD-1 parental (DLD-1 Par) colon cancer cells express one copy each of KRAS G13D and KRAS wild type (6, 21); RKO parental (RKO Par) colon cancer cells express two copies of BRAF V600E and a single copy of BRAF wild type (6). DLD-1 Mut and DLD-1 WT cells are derived by homologous recombination from DLD-1 Par cells and express only KRAS G13D or KRAS wild type, respectively. RKO Mut and RKO WT cells are derived by homologous recombination from RKO Par cells and express either a single BRAF V600E or BRAF wild type, respectively. We used these isogenic DLD-1 and RKO cell lines to determine how oncogenic KRAS and BRAF affect the expression of proteins in central carbon metabolism.
Glucose Consumption and Lactate Production in DLD-1 and RKO Cell Lines
The “Warburg effect” is functionally defined as increased glucose consumption and increased lactate production. DLD-1 Mut and DLD-1 Par cells showed nearly equivalent rates of glucose consumption, which were both significantly higher than in DLD-1 WT cells (p = 0.0004 for DLD-1 Mut versus DLD-1 WT, p = 0.0134 for DLD-1 Par versus DLD-1 WT) (Fig. 2). This result is similar to that reported previously for shorter time course measurements in the isogenic DLD-1 cell lines (6). RKO Par cells consumed glucose at a rate equivalent to that for RKO Mut cells, which was significantly higher than in RKO WT cells (p = 0.0317). RKO Mut cells consumed glucose at a higher rate than RKO WT cells, but this difference was not significant (p = 0.2723) (Fig. 2). These results were similar to previous results with shorter time course experiments (6).
Fig. 2.

Glucose consumption and lactate production rates in isogenic cell lines. Glucose consumption and lactate production rates were determined from 3 biological replicates per cell line. Growth media collected from cultured cell lines every doubling and half-doubling time was analyzed in duplicate on a YSI 2300 STAT Plus Glucose and Lactate Analyzer. Error bars represent the standard error of the mean. A and B, show glucose consumption rates in DLD-1 and RKO cells, respectively. C and D, show lactate production rates in DLD-1 and RKO cells, respectively.
Lactate production rates were equal in the DLD-1 Mut and DLD-1 Par cells, and both produced lactate at a significantly higher rate than the DLD-1 WT cells (p = 0.0012 and p = 0.002, respectively) (Fig. 2). Lactate production in the RKO cells increased significantly from RKO WT to RKO Mut to RKO Par (Fig. 2), as previously reported in short term experiments (6). The DLD-1 and RKO cell lines thus display a Warburg phenotype associated with KRAS and BRAF mutations, respectively, as previously reported.
Global Proteome Analysis of Isogenic Cell Lines
Global proteomic analysis of the DLD-1 cell lines identified inventories of nearly identical size in each of the DLD-1 and RKO cell lines. The global protein-level FDR values in the DLD-1 and RKO data sets were 2.77% and 2.67%, respectively. A core proteome of 7461 proteins was detected in all three of the DLD-1 cell lines, while a core proteome of 7410 proteins was detected in all three of the RKO cell lines (Fig. 3). The DLD-1 data sets contained 5,730 proteins quantifiable with at least two spectra per protein in each replicate analysis, whereas in the RKO data set contained 5628 quantifiable proteins (Tables S4 and S5). The combined quantifiable protein inventories were used as the reference proteomes for comparisons within each cell line group.
Fig. 3.

Overlap of protein groups expressed in isogenic cell lines. Protein inventories were from two replicate analyses per cell line. A minimum of one spectrum per protein group per cell line was required for a protein group to be shared between cell lines.
Pairwise comparisons of the global proteome data sets indicated highly similar proteomes. Proteins with ≥twofold differences between cell lines constituted ∼2–10% of the quantifiable proteome in each DLD-1 cell comparison (supplemental Figs. S2A and S2B and supplemental Table S6). Pairwise comparisons of the RKO cell lines indicated comparable differences between each. Proteins with ≥twofold differences were ∼4–8% of the quantifiable proteome in the RKO cell lines (supplemental Figs. S2C and S2D and supplemental Table S7).
We used WebGestalt (47) to map differential proteins to Gene Ontology classifications using the 5730 quantifiable proteins in the DLD-1 cell lines and the 5628 quantifiable proteins in the RKO cell lines as the reference sets, respectively. This analysis identified no significant enrichment for molecular functions or biological processes related to metabolism in either the DLD-1 or RKO models (supplemental Table S8). We note that almost all of the metabolic proteins quantified by PRM (see below) were detected and quantifiable in the global data sets. However, precision of the shotgun analysis platform is limited by well-known factors, including undersampling and run-to-run sampling variations in data-dependent analysis. Biologically significant variations of less than ∼twofold would not have been detected in the global profiles. Taken together, the results of the global analyses of the DLD-1 and RKO cell lines revealed modest impact of the mutations on the proteomes, but no detectable alterations associated with metabolism.
Targeted PRM Analysis of Isogenic Cell Lines
We hypothesized that metabolic reprogramming in the DLD-1 and RKO cell lines may be mediated by relatively small differences in abundance of metabolic proteins. To test this hypothesis, we developed a targeted, multiplexed PRM assay to specifically interrogate the expression of 73 proteins involved in glycolysis, the TCA cycle, the PPP, phosphoserine biosynthesis, and glutamine metabolism (Fig. 1). We used the LRP method for quantitation, in which all measured peptide peak areas are normalized to the peak area for a single isotope-labeled reference peptide standard (49). This PRM assay measured 204 peptides corresponding to 73 proteins in a single scheduled analysis on a Q Exactive Plus instrument. Although three LRP peptide standards (β-actin peptide U-13C,15N-Arg-GYSFTTTAER, alkaline phosphatase peptide U-13C,15N-Arg-AAQGITAPGGAR, and β-galactosidase peptide U-13C,15N-Arg-APLDNDIGVSEATR), were added to each sample and detected by PRM, all target peptide peak areas were normalized to the alkaline phosphatase AAQGITAPGGAR peptide, as it had the lowest CV in both DLD-1 (average CV of 0.112) and RKO (average CV of 0.082) data sets.
The results of the targeted PRM analyses of the DLD-1 and RKO cells are summarized in supplemental Tables S9 and S10. All cell line comparisons were based on the normalized peak areas for the target peptides. Data for each reliably detected peptide are plotted in supplemental Figs. S3 and S4. Statistically significant differences were determined by pairwise comparisons for each peptide using an unpaired, two tailed Student's t test, with a p value of less than 0.05 considered to be statistically significant. The resulting pairwise comparisons for each of these normalized peptide measurements were organized according to corresponding metabolic pathways (Figs. 4 and 5).
Fig. 4.

Pathway map of quantitative comparisons of metabolic proteins in DLD-1 cell lines. Pairwise comparisons of metabolic PRM measurements of A, DLD-1 Mut versus DLD-1 WT and B, DLD-1 Par versus DLD-1 WT. Peptides with the lowest CV and largest normalized peak area were used for quantitative comparisons and normalized peak area for all detected peptides are shown in supplemental Fig. S3. The legend for each pairwise comparison shows fold changes relative to the cell line listed first in each comparison. Proteins with a CV < 0.25, an ICC > 0.6, and a p < 0.05 and that are higher in the first cell line are shown in red (at least a twofold difference) or light red (between 1.2- and 1.9-fold difference). Proteins with a CV < 0.25, an ICC > 0.6, and a p < 0.05 and that are lower in the first cell line are shown in green (at least a 2-fold difference) or light green (between 1.2- and 1.9-fold difference). Proteins with a CV < 0.25, an ICC > 0.6, but a p > 0.05 or with a CV < 0.25 but an ICC < 0.6 are listed in gray (no difference). Proteins with a CV > 0.25 or with no detectable peak area are shown in white.
Fig. 5.

Pathway map of quantitative comparisons of metabolic proteins in RKO cell lines. Pairwise comparisons of metabolic PRM measurements of A, RKO Mut versus RKO WT and B, RKO Par versus RKO WT. Peptides with the lowest CV and largest normalized peak area were used for quantitative comparisons, and normalized peak area for all detected peptides are shown in supplemental Fig. S4. The legend for each pairwise comparison shows fold changes relative to the cell line listed first in each comparison. Proteins with a CV < 0.25, an ICC > 0.6, and a p < 0.05 and that are higher in the first cell line are shown in red (at least a twofold difference) or light red (between 1.2- and 1.9-fold difference). Proteins with a CV < 0.25, an ICC > 0.6, and a p < 0.05 and that are lower in the first cell line are shown in green (at least a 2-fold difference) or light green (between 1.2- and 1.9-fold difference). Proteins with a CV < 0.25, an ICC > 0.6, but a p > 0.05 or with a CV < 0.25 but an ICC < 0.6 are listed in gray (no difference). Proteins with a CV > 0.25 or with no detectable peak area are shown in white.
PRM Analyses of Metabolic Proteins in DLD-1 Cell Lines
PRM analyses of DLD-1 cell lines demonstrated that mutant KRAS is associated with the altered expression of proteins involved in glycolysis, glutamine metabolism, phosphoserine biosynthesis, and nonoxidative PPP (Fig. 4). Of the 20 glycolytic proteins measured in this assay, 15 were quantifiable in both DLD-1 Mut and DLD-1 Par cells;10 were significantly increased in the DLD-1 Par cells and 8 were significantly increased in the DLD-1 Mut cells compared with the DLD-1 WT cells. LDHA was most consistently 2-fold up-regulated in these pairwise comparisons (p = 0.0002 in DLD-1 Mut versus DLD-1 WT, p = 0.0005 in DLD-1 Par versus DLD-1 WT), and the glucose transporter SLC2A1 was 2-fold up-regulated in DLD-1 Mut cells (p = 0.0003) and 1.6-fold up-regulated in DLD-1 Par cells (p = 0.0003). These differences are directly consistent with the Warburg phenotype that has been observed in these cell lines. HK2, ALDOA, ENO1, and LDHB also were consistently increased between 1.2 and 1.9-fold.
Mutant KRAS also affected several proteins in the phosphoserine biosynthesis pathway. Seven of the 11 proteins analyzed were significantly elevated DLD-1 Par and 6 were significantly elevated in the DLD-1 Mut cells, all by between 1.2 and 2-fold.
The glutamine transporter SLC1A5 and the transaminase GLS were significantly increased in DLD-1 Par and DLD-1 Mut cells compared with the DLD-1 WT cells. This is consistent with previous observations that cells with increased c-Myc activity have increased glutamine utilization (53, 54). Furthermore, GOT2 and GLUD2 were significantly increased in the DLD-1 Par cells, which is consistent with previous evidence that glutamine utilization can support TCA cycle activity (8, 55, 56). ACLY, which converts cytosolic citrate to acetyl CoA for lipid biosynthesis, was increased in both DLD-1 Mut and DLD-1 Par cells. We found no consistent alterations of proteins in the TCA cycle.
We detected no significant protein alterations in the oxidative branch of the PPP (G6PD, PGLS, and PGD), but two enzymes in the nonoxidative branch, RPIA and TKT, were consistently up-regulated in the KRAS mutant cell lines by ∼3-fold and 1.5-fold, respectively (Fig. 4). These changes are consistent with a previous report that KRAS mutations in pancreatic ductal adenocarcinoma may upregulate the nonoxidative branch of the PPP to provide ribose for nucleotide synthesis (9).
PRM Analyses of Metabolic Proteins in RKO Cell Lines
PRM analyses of the RKO model system indicated that BRAF mutations trigger metabolic reprogramming similar to that observed with KRAS mutations (Fig. 5). Of the 20 glycolytic proteins measured, 15 were quantifiable in both the RKO Par and RKO Mut cells compared with the RKO WT cells (Fig. 5). Six glycolytic proteins were significantly increased in the RKO Mut cells and 8 were significantly increased in the RKO Par cells. SLC2A1, LDHA, and LDHB were more highly increased in the RKO Par cells than in the RKO Mut cells, which is consistent with the higher rates of both glucose transport and lactate production in the RKO Par cells (Fig. 2). Elevations in HK2, GAPDH, PGAM1, and ENO1 also were consistent with an up-regulation of glycolytic activity in both BRAF mutant RKO cell lines (Fig. 5).
Proteins involved in the phosphoserine biosynthesis pathway were uniformly up-regulated in BRAF mutant RKO cells. Nine of the 11 of the proteins measured were increased in the RKO Mut cells, whereas all 11 were increased in the RKO Par cells. Protein abundance increases ranged from 1.3-fold to 2.8-fold for most of these proteins, although PSAT1 was elevated by 4.9-fold in RKO Par cells relative to RKO WT.
Proteins involved in glutamine utilization were increased in BRAF mutant RKO cell lines, although levels of the glutamine transporter SLC1A5 were unaffected. GLS, GLUD1, and GLUD2 were increased in both RKO Mut and RKO Par cells, but GOT2 was only increased in RKO Par cells compared with RKO WT cells. Proteins in the TCA cycle were not affected by BRAF mutations, except for MDH2 and IDH3A in both RKO Mut and RKO Par cells. Of the quantifiable PPP proteins in RKO cells, only TALDO1 and three other proteins in the nonoxidative PPP were significantly increased in the BRAF mutant RKO cells.
A pseudo-heatmap summary view of all comparisons based on PRM analyses of DLD-1 and RKO cells is presented in supplemental Fig. S5.
Targeted Quantitative Analysis of KRAS and BRAF Protein Forms
In the DLD-1 Par cells, KRAS G13D protein was present at twice the level measured for the wild type protein (p = 0.0024) (Fig. 6), consistent with the report by Zhang et al. that cells with KRAS mutations downregulate expression of the remaining wild type KRAS gene (57). The content of mutant KRAS in the DLD-1 Mut cells was almost twice that in the parental cells, whereas the total KRAS protein varied by about 1.5-fold across the three cell lines. The wild type KRAS LVVVGAGGVGK peptide detected in the DLD-1 Mut cells and may be derived from NRAS or HRAS, which contain the same sequence, but the amount was below the LLOQ for the assay. The content of wild type KRAS in the DLD-1 WT cells was nearly fivefold higher than in the parental cells.
Fig. 6.
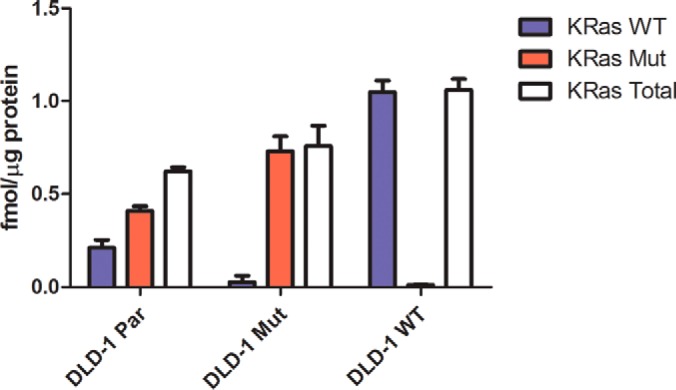
Quantitation of KRAS protein forms in DLD-1 cell lines. The KRAS wild type G13 tryptic peptide LVVVGAGGVGK and the G13D tryptic peptide LVVVGAGDVGK were measured by PRM with quantitation by SID. Integrated peptide peak areas for the single best transition for each peptide were normalized to the corresponding transition for the isotopically labeled peptide standard and an external calibration curve for each peptide was used to determine the concentration of wild type and mutant KRAS in DLD-1 cells. The LLOQ for both the wild type and mutant KRAS peptides was 8 amol. Total KRAS measurements are the sum of the amount of wild type KRAS and mutant KRAS in each biological replicate. Error bars represent standard deviation.
RKO cell lines expressed levels of BRAF proteins ∼25-fold lower than KRAS proteins in DLD-1 cells (Fig. 7). Measured values were near the LLOQ for the assay. RKO Par cells expressed approximately the same level of wild type and mutant BRAF V600E proteins. RKO Mut cells expressed approximately half the amount of BRAF V600E mutant protein compared with the RKO Par cells (p = 0.0023), whereas RKO WT cells expressed the same amount of wild type protein as RKO Par cells (Fig. 7). Thus, BRAF protein forms in RKO cells were proportional to respective allelic compositions.
Fig. 7.
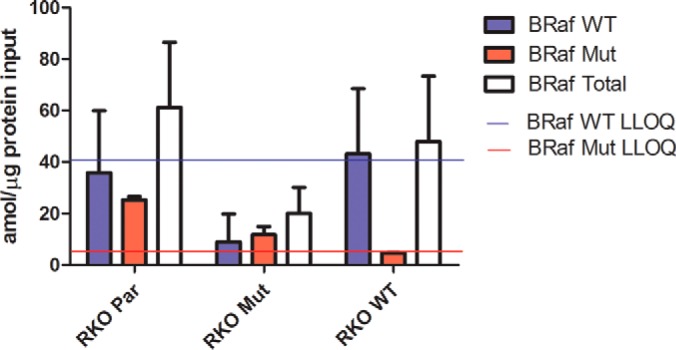
Quantitation of BRAF protein forms in RKO cell lines. The BRAF wild type V600 tryptic peptide IGDFGLATVK and the V600E tryptic peptide IGDFGLATEK tryptic peptide were measured by PRM with quantitation by SID. Integrated peptide peak areas for the single best transition for each peptide were normalized to the corresponding transition for the isotopically labeled peptide standard, and an external calibration curve was used to determine the concentration of wild type and mutant BRAF in RKO cells. The solid blue line indicates the LLOQ for the wild type BRAF peptide (40 amol), and the solid red line indicates the LLOQ for the mutant BRAF peptide (8 amol). Total BRAF is the sum of the amount of wild type BRAF and mutant BRAF in each biological replicate. Error bars represent standard deviation.
Targeted MRM Analysis of Primary Human Tumors
To evaluate the impact of KRAS mutation status on metabolic protein pathways, we analyzed a set of 16 stage II human colon cancers. KRAS mutations are observed in ∼43% of colon cancers (58). The tumors were classified for KRAS mutational status with a multiplexed mutation profiling panel that also detected NRAS, BRAF, and PIK3CA mutations (Table S2) (51). Eight tumors had codon 12 mutations in KRAS (11 G12V and one G12D) and eight had only wild type KRAS. Two of the KRAS mutant tumors also had PIK3CA E545K mutations, one of the KRAS wild type tumors had a NRAS Q61K mutation and one had a BRAF V600E mutation.
We performed MRM analyses for 61 metabolic proteins with quantitative normalization by the LRP method. The proteins in the serine biosynthesis pathway were not included in this MRM study because the tumor analyses were performed prior to the cell model studies and the serine biosynthesis pathway assay module had not yet been developed. We later attempted to analyze these proteins in stored MS-ready samples, but were unable to detect any of them. Additional samples of these tumors were not available for a full workup and reanalysis. Thus, the tumor analyses report measurements for 61 proteins, rather than 73 proteins. The results of the MRM Skyline output of the targeted MRM analysis of the stage II human tumors are summarized in supplemental Tables S11 and S12. The LRP-normalized values for each protein were averaged within the KRAS mutant and KRAS wild type groups and fold-change differences between the groups depicted are from comparisons of the averages (supplemental Fig. S6 and S7). Profiles for individual tumors are shown in Fig. 8 and supplemental Fig. S8, and the fold differences represent comparisons of the LRP-normalized value for the protein in each tumor to the global average of all LRP-normalized values for the protein across the 16 tumor data set.
Fig. 8.

Summary of MRM measurements in human stage II colorectal cancers. Normalized peptide measurement from each sample was compared with the mean normalized peak area for each individual peptide from all tumor samples. These comparisons were then classified as either no difference from the mean (gray color), 1.25- to 1.5-fold above or below the mean (light red and light green, respectively), greater than 1.5-fold above or below the mean (dark red or dark green, respectively), or not detected (white). Proteins are organized into glycolysis, TCA, PPP, glutamine metabolism, and other metabolic enzymes.
Our MRM analyses detected a majority of the 61 targeted proteins in most of the pathways studied. We detected most of the targeted proteins in glycolysis, the PPP, glutamine transport and utilization, the TCA cycle and citrate utilization. Eleven proteins were significantly higher in the KRAS mutant tumors compared with the KRAS wild type tumors. Measured differences ranged between 1.2- to 1.9-fold higher for all proteins except for SLC2A1, which was, on average, 2.3-fold higher in the KRAS mutant tumors (p = 0.0050) (supplemental Fig. S7). No proteins were significantly lower in the KRAS mutant tumors compared with the KRAS wild type tumors. Most of the detected differences were in the expression of the glucose and glutamine transporters SLC2A1 and SLC1A5, respectively and enzymes of glycolysis (ALDOA, GAPDH, PGK2, PGAM1, PKM2, and PKLR) and the TCA cycle (IDH2, MDH2, and CS). There were substantial variations between tumors, which suggested that each tumor displayed a distinct pattern of metabolic reprogramming (Fig. 8 and supplemental Fig. S8). Among the KRAS wild type tumors, one tumor (tumor C, Fig. 8 and supplemental Fig. S8A) had a BRAF V600E mutation, yet expressed no metabolic proteins at levels above the mean. Another (tumor I, Fig. 8 and supplemental Fig. S8G) had a NRAS Q61K mutation and displayed below average expression of SLC2A1 and SLC1A5 and above average expression for only TALDO and LDHA.
DISCUSSION
The purpose of this study was to characterize the proteomic changes in metabolic pathways that accompany a Warburg effect phenotype driven by expression of oncogenic KRAS and BRAF in colorectal cancer cells and tumors. Our study produced three significant findings. First, metabolic reprogramming driven by oncogenic KRAS and BRAF is manifested by protein alterations in glycolysis, serine biosynthesis, the PPP and in glutamine utilization. We observed a common set of alterations in KRAS and BRAF mutant cells and in KRAS mutant human colon tumors. Second, RKO cellular content of BRAF V600E protein correlated directly with magnitude of the Warburg effect, as measured by glucose uptake and lactate production and with protein abundance changes. On the other hand, these features were not proportional to KRAS G13D content in DLD-1 cells. Third, the protein abundance changes associated with metabolic reprogramming were relatively modest—in most cases twofold or less. These changes were detected only with multiplexed, targeted assay panel for metabolic proteins. This finding is of broader significance for the proteomics field, as it suggests that physiologically significant protein alterations may be detectable with higher precision analyses than can be achieved through global profiling strategies.
Yun et al. demonstrated that isogenic colorectal cancer cells with either oncogenic KRAS or oncogenic BRAF have an increased expression of SLC2A1, increased glucose consumption rate and increased lactate production rate (6). Our work demonstrates that mutant KRAS and mutant BRAF broadly impacts glycolysis, phosphoserine biosynthesis, glutamine metabolism, and the nonoxidative PPP. In colorectal cancer development, these mutations are associated with increased proliferation rates, but can also contribute to cancer phenotype by reprogramming metabolism to support critical biosynthetic needs. This finding is consistent with a broad body of work linking multiple oncogene mutations to metabolic reprogramming and altered glucose metabolic fluxes (8–11, 13–15, 54, 55). For example, mouse pancreatic ductal adenocarcinomas driven by KRAS G12D have increased glucose utilization in the nonoxidative pentose phosphate pathway (9). These same KRAS G12D driven pancreatic adenocarcinomas also have an increased utilization of glutamine to replenish TCA cycle intermediates (8). Thyroid cancers with a BRAF V600E mutation have significantly increased glucose transport compared with similar thyroid cancers without BRAF mutations (15). Lastly, melanoma cells have an increased glycolytic flux of glucose to serine and glycine because of increased PHGDH expression (59, 60).
Our analysis of a small cohort of human stage II colon tumors confirmed that KRAS is associated with the same protein changes seen in the cell lines. Because the specimens we analyzed were FFPE tissues, we were not able to verify a Warburg effect phenotype through glucose uptake or lactate production measurements. Although the pattern of changes was considerably less distinct in the tumors, all of the tumors with up-regulated SLC2A1 and/or SLC1A5 together with up-regulation of glycolytic enzymes had KRAS mutations (Fig. 8 and S7). It would be reasonable to expect that metabolic reprogramming in colon cancers may reflect the influences of not only KRAS, BRAF, and other oncogenic mutations, but also other cancer-associated genomic features. This hypothesis is consistent with the observation that different tumor types have distinct metabolic profiles (24).
Our SID measurements enabled quantitative comparison of both mutant and wild type KRAS and BRAF protein forms in the DLD-1 and RKO cell models, respectively. In the DLD-1 Mut cells, the mutant KRAS protein amount was twice that measured in the DLD-1 Par cells, yet the metabolic reprogramming profiles of the cell lines were similar, which suggests that metabolic reprogramming does not strongly depend on the absolute cellular level of mutant KRAS. On the other hand, DLD-1 WT cells expressed only wild type protein, but at a level almost twice the combined mutant plus wild type KRAS level in DLD-1 Par cells. Thus, even a high expression of wild type KRAS protein is not itself able to induce metabolic reprogramming. This observation is interesting in light of recent work by Young et al., who demonstrated that both the mutant and wild type KRAS alleles play distinct roles in regulating signaling through the epidermal growth factor pathway (61).
In the RKO cell model, both glucose uptake and lactate production progressively increased in comparing the RKO WT, RKO Mut and RKO Par cells, which express zero, one and two mutant BRAF alleles, respectively. Although BRAF protein levels were close to the LLOQ, the level of mutant protein in the RKO Par cells was approximately twice that in the RKO Mut cells (Fig. 7). The wild type BRAF protein in the RKO WT cells was present at approximately the same level as the wild type protein in the RKO Par cells. Relative expression of both mutant and wild type BRAF protein thus matched the allelic composition of the cell lines and dosage of mutant BRAF at the protein level drove the degree of metabolic reprogramming.
An interesting question is why increasing BRAF mutant protein in RKO cells drives increasing metabolic protein expression, glucose uptake and lactate production, whereas the these are unaffected in DLD-1 cells by the amount of mutant KRAS protein. The answer may lie in the complexity of KRAS-driven signaling. KRAS drives not only EGFR/MAPK signaling, but also PI3K/AKT signaling (62), which also impact metabolic reprogramming (63, 64). As we noted above, Young et al. (ref. 62) showed that both mutant and wild type KRAS play distinct roles in EGFR signaling. Kerr et al. recently reported that the copy number of G12V KRAS genes drives distinct profiles of metabolic reprogramming in lung cancer (65).
This study illustrates the power of multiplexed, targeted protein quantitation for focused study of a multiprotein system. Global proteomic profiles, which can typically detect ∼twofold abundance differences, detected over 5600 quantifiable proteins in the DLD-1 and RKO cell models, but the data revealed no KRAS or BRAF mutation-dependent differences in pathways of central carbon metabolism. This was not because of failure to detect and quantify metabolic proteins, as essentially all the proteins we subsequently quantified by PRM were detected and quantifiable in our global profiling data set. It is particularly interesting that KRAS and BRAF, which are oncogenic drivers of several cancers, produced relatively modest effects on protein abundance, as we have observed previously (25). Application of a more precise, targeted PRM platform achieved measurements of modest, yet significant abundance changes of less than twofold, which reflected a Warburg phenotype in both the DLD-1 and RKO cell models. The superiority of a multiplexed, targeted platform over global proteome profiling to detect these changes provides an instructive example that may be important in other contexts for proteome analysis.
Our data associate a consistent set of protein abundance changes with a Warburg effect phenotype in colorectal cancer. Whereas metabolite profiles are subject to perturbation by ischemia associated with tissue collection, protein abundance is stable and thus provides a quantifiable signature of metabolic reprogramming (52, 66). This approach should thus be of broad utility to investigate the relationship between cancer metabolic phenotypes, metastasis and response to therapies.
Supplementary Material
Footnotes
Author contributions: J.E.H., L.J.Z., R.J.S., J.D.Y., and D.C.L. designed research; J.E.H. and I.A.T. performed research; J.E.H., X.W., R.J.S., and M.L. analyzed data; J.E.H., L.J.Z., R.J.S., J.D.Y., and D.C.L. wrote the paper.
* This work was supported by National Institutes of Health Grant U24CA159988 from the Clinical Proteomic Tumor Analysis Consortium. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PPP
- pentose phosphate pathway
- bRPLC
- basic reverse phase liquid chromatography
- CV
- coefficient of variation
- DLD-1 Mut
- DLD-1 KRAS (G13D/-) cells
- DLD-1 Par
- DLD-1 parental KRAS (G13D/+) cells
- DLD-1 WT
- DLD-1 KRAS (+/-) cells
- EGFR
- epidermal growth factor receptor
- FDR
- false discovery rate
- FFPE
- formalin-fixed, paraffin-embedded
- ICC
- intraclass correlation coefficient
- LC
- liquid chromatography
- LLOD
- lower limit of detection
- LLOQ
- lower limit of quantitation
- LRP
- labeled reference peptide
- MRM
- multiple reaction monitoring
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- PBS
- phosphate buffered saline
- PRM
- parallel reaction monitoring
- RKO Mut
- RKO BRAF (V600E/-/-) cells
- RKO Par
- RKO parental BRAF (V600E/V600E/+) cells
- RKO WT
- RKO BRAF (+/-/-) cells
- SID
- stable isotope dilution.
REFERENCES
- 1. Warburg O., Wind F., and Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deberardinis R. J., Sayed N., Ditsworth D., and Thompson C. B. (2008) Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsu P. P., and Sabatini D. M. (2008) Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707 [DOI] [PubMed] [Google Scholar]
- 6. Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J. K., Markowitz S., Zhou S., Diaz L. A., Velculescu V. E., Lengauer C., Kinzler K. W., Vogelstein B., and Papadopoulos N. (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., and Kimmelman A. C. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ying H., Kimmelman A. C., Lyssiotis C. A., Hua S., Chu G. C., Fletcher-Sananikone E., Locasale J. W., Son J., Zhang H., Coloff J. L., Yan H., Wang W., Chen S., Viale A., Zheng H., Paik J. H., Lim C., Guimaraes A. R., Martin E. S., Chang J., Hezel A. F., Perry S. R., Hu J., Gan B., Xiao Y., Asara J. M., Weissleder R., Wang Y. A., Chin L., Cantley L. C., and DePinho R. A. (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamphorst J. J., Cross J. R., Fan J., de Stanchina E., Mathew R., White E. P., Thompson C. B., and Rabinowitz J. D. (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U.S.A. 110, 8882–8887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCleland M. L., Adler A. S., Deming L., Cosino E., Lee L., Blackwood E. M., Solon M., Tao J., Li L., Shames D., Jackson E., Forrest W. F., and Firestein R. (2013) Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin. Cancer Res. 19, 773–784 [DOI] [PubMed] [Google Scholar]
- 12. Patra K. C., Wang Q., Bhaskar P. T., Miller L., Wang Z., Wheaton W., Chandel N., Laakso M., Muller W. J., Allen E. L., Jha A. K., Smolen G. A., Clasquin M. F., Robey R. B., and Hay N. (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baenke F., Chaneton B., Smith M., Van Den Broek N., Hogan K., Tang H., Viros A., Martin M., Galbraith L., Girotti M. R., Dhomen N., Gottlieb E., and Marais R. (2015) Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol. Oncol. 10, 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang H. B., Fan J., Lin R., Elf S., Ji Q., Zhao L., Jin L., Seo J. H., Shan C., Arbiser J. L., Cohen C., Brat D., Miziorko H. M., Kim E., Abdel-Wahab O., Merghoub T., Fröhling S., Scholl C., Tamayo P., Barbie D. A., Zhou L., Pollack B. P., Fisher K., Kudchadkar R. R., Lawson D. H., Sica G., Rossi M., Lonial S., Khoury H. J., Khuri F. R., Lee B. H., Boggon T. J., He C., Kang S., and Chen J. (2015) Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol. Cell 59, 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagarajah J., Ho A. L., Tuttle R. M., Weber W. A., and Grewal R. K. (2015) Correlation of BRAFV600E mutation and glucose metabolism in thyroid cancer patients: An 18F-FDG PET study. J. Nucl. Med. 56, 662–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White E. (2013) Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 27, 2065–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schubbert S., Shannon K., and Bollag G. (2007) Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308 [DOI] [PubMed] [Google Scholar]
- 18. Lavoie H., and Therrien M. (2015) Regulation of RAF protein kinases in ERK signaling. Nat. Rev. Mol. Cell Biol. 16, 281–298 [DOI] [PubMed] [Google Scholar]
- 19. Cantwell-Dorris E. R., O'Leary J. J., and Sheils O. M. (2011) BRAFV600E: implications for carcinogenesis and molecular therapy. Mol. Cancer Ther 10, 385–394 [DOI] [PubMed] [Google Scholar]
- 20. Wellbrock C., Karasarides M., and Marais R. (2004) The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875–885 [DOI] [PubMed] [Google Scholar]
- 21. Shirasawa S., Furuse M., Yokoyama N., and Sasazuki T. (1993) Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260, 85–88 [DOI] [PubMed] [Google Scholar]
- 22. Chang E. H., Furth M. E., Scolnick E. M., and Lowy D. R. (1982) Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature 297, 479–483 [DOI] [PubMed] [Google Scholar]
- 23. Haq R., Shoag J., Andreu-Perez P., Yokoyama S., Edelman H., Rowe G. C., Frederick D. T., Hurley A. D., Nellore A., Kung A. L., Wargo J. A., Song J. S., Fisher D. E., Arany Z., and Widlund H. R. (2013) Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 23, 302–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu J., Locasale J. W., Bielas J. H., O'Sullivan J., Sheahan K., Cantley L. C., Vander Heiden M. G., and Vitkup D. (2013) Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 31, 522–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., and Liebler D. C. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gallien S., Duriez E., Crone C., Kellmann M., Moehring T., and Domon B. (2012) Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallien S., Kim S. Y., and Domon B. (2015) Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol. Cell. Proteomics 14, 1630–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Halvey P. J., Ferrone C. R., and Liebler D. C. (2012) GeLC-MRM quantitation of mutant KRAS oncoprotein in complex biological samples. J. Proteome Res. 11, 3908–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., and Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., and Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 31. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schilling B., MacLean B., Held J. M., Sahu A. K., Rardin M. J., Sorensen D. J., Peters T., Wolfe A. J., Hunter C. L., MacCoss M. J., and Gibson B. W. (2015) Multiplexed, scheduled, high-resolution parallel reaction monitoring on a full scan QqTOF instrument with integrated data-dependent and targeted mass spectrometric workflows. Anal. Chem. 87, 10222–10229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suomi T., Corthals G. L., Nevalainen O. S., and Elo L. L. (2015) Using peptide-level proteomics data for detecting differentially expressed proteins. J. Proteome Res. 14, 4564–4570 [DOI] [PubMed] [Google Scholar]
- 34. Zhang G., Fang B., Liu R. Z., Lin H., Kinose F., Bai Y., Oguz U., Remily-Wood E. R., Li J., Altiok S., Eschrich S., Koomen J., and Haura E. B. (2011) Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J. Proteome Res. 10, 305–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y., Gruidl M., Remily-Wood E., Liu R. Z., Eschrich S., Lloyd M., Nasir A., Bui M. M., Huang E., Shibata D., Yeatman T., and Koomen J. M. (2010) Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J. Proteome Res. 9, 4215–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiang Y., Remily-Wood E. R., Oliveira V., Yarde D., He L., Cheng J. Q., Mathews L., Boucher K., Cubitt C., Perez L., Gauthier T. J., Eschrich S. A., Shain K. H., Dalton W. S., Hazlehurst L., and Koomen J. M. (2011) Monitoring a nuclear factor-κB signature of drug resistance in multiple myeloma. Mol. Cell. Proteomics 10, M110.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Myers M. V., Manning H. C., Coffey R. J., and Liebler D. C. (2012) Protein expression signatures for inhibition of epidermal growth factor receptor-mediated signaling. Mol. Cell. Proteomics 11, M111.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmidt C., Lenz C., Grote M., Lührmann R., and Urlaub H. (2010) Determination of protein stoichiometry within protein complexes using absolute quantification and multiple reaction monitoring. Anal. Chem. 82, 2784–2796 [DOI] [PubMed] [Google Scholar]
- 39. Wolf-Yadlin A., Hautaniemi S., Lauffenburger D. A., and White F. M. (2007) Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 104, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drabovich A. P., Pavlou M. P., Dimitromanolakis A., and Diamandis E. P. (2012) Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Mol. Cell. Proteomics 11, 422–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y., Yang F., Gritsenko M. A., Clauss T., Liu T., Shen Y., Monroe M. E., Lopez-Ferrer D., Reno T., Moore R. J., Klemke R. L., Camp D. G. 2nd, and Smith R. D. (2011) Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabb D. L., Fernando C. G., and Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim S., and Pevzner P. A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang B., Chambers M. C., and Tabb D. L. (2007) Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., and Tabb D. L. (2009) IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J., Duncan D., Shi Z., and Zhang B. (2013) WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 41, W77–W83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H., Liu Q., Zimmerman L. J., Ham A. J., Slebos R. J., Rahman J., Kikuchi T., Massion P. P., Carbone D. P., Billheimer D., and Liebler D. C. (2011) Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol. Cell. Proteomics 10, M110.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mani D. R., Abbatiello S. E., and Carr S. A. (2012) Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics 13, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Su Z., Dias-Santagata D., Duke M., Hutchinson K., Lin Y. L., Borger D. R., Chung C. H., Massion P. P., Vnencak-Jones C. L., Iafrate A. J., and Pao W. (2011) A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J. Mol. Diagn. 13, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sprung R. W., Martinez M. A., Carpenter K. L., Ham A. J., Washington M. K., Arteaga C. L., Sanders M. E., and Liebler D. C. (2012) Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed, paraffin-embedded tissue. J. Proteome Res. 11, 3498–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao P., Tchernyshyov I., Chang T. C., Lee Y. S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., and Dang C. V. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu X., Li J., Sun X., Guo Y., Chu D., Wei L., Li X., Yang G., Liu X., Yao L., Zhang J., and Shen L. (2015) Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget 6, 26161–26176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., Zimmerman L. J., Liebler D. C., Slebos R. J., Lorkiewicz P. K., Higashi R. M., Fan T. W., and Dang C. V. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., and Stephanopoulos G. (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Z., Wang Y., Vikis H. G., Johnson L., Liu G., Li J., Anderson M. W., Sills R. C., Hong H. L., Devereux T. R., Jacks T., Guan K. L., and You M. (2001) Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 29, 25–33 [DOI] [PubMed] [Google Scholar]
- 58. (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Locasale J. W., Grassian A. R., Melman T., Lyssiotis C. A., Mattaini K. R., Bass A. J., Heffron G., Metallo C. M., Muranen T., Sharfi H., Sasaki A. T., Anastasiou D., Mullarky E., Vokes N. I., Sasaki M., Beroukhim R., Stephanopoulos G., Ligon A. H., Meyerson M., Richardson A. L., Chin L., Wagner G., Asara J. M., Brugge J. S., Cantley L. C., and Vander Heiden M. G. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Possemato R., Marks K. M., Shaul Y. D., Pacold M. E., Kim D., Birsoy K., Sethumadhavan S., Woo H. K., Jang H. G., Jha A. K., Chen W. W., Barrett F. G., Stransky N., Tsun Z. Y., Cowley G. S., Barretina J., Kalaany N. Y., Hsu P. P., Ottina K., Chan A. M., Yuan B., Garraway L. A., Root D. E., Mino-Kenudson M., Brachtel E. F., Driggers E. M., and Sabatini D. M. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Young A., Lou D., and McCormick F. (2013) Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 3, 112–123 [DOI] [PubMed] [Google Scholar]
- 62. Rajalingam K., Schreck R., Rapp U. R., and Albert S. (2007) Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 1773, 1177–1195 [DOI] [PubMed] [Google Scholar]
- 63. Rathmell J. C., Fox C. J., Plas D. R., Hammerman P. S., Cinalli R. M., and Thompson C. B. (2003) Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 23, 7315–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Riley J. K., Carayannopoulos M. O., Wyman A. H., Chi M., and Moley K. H. (2006) Phosphatidylinositol 3-kinase activity is critical for glucose metabolism and embryo survival in murine blastocysts. J. Biol. Chem. 281, 6010–6019 [DOI] [PubMed] [Google Scholar]
- 65. Kerr E. M., Gaude E., Turrell F. K., Frezza C., and Martins C. P. (2016) Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sprung R. W., Brock J. W., Tanksley J. P., Li M., Washington M. K., Slebos R. J., and Liebler D. C. (2009) Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell. Proteomics 8, 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.