

Abstract
The Mass Spec Studio package was designed to support the extraction of hydrogen-deuterium exchange and covalent labeling data for a range of mass spectrometry (MS)-based workflows, to integrate with restraint-driven protein modeling activities. In this report, we present an extension of the underlying Studio framework and provide a plug-in for crosslink (XL) detection. To accommodate flexibility in XL methods and applications, while maintaining efficient data processing, the plug-in employs a peptide library reduction strategy via a presearch of the tandem-MS data. We demonstrate that prescoring linear unmodified peptide tags using a probabilistic approach substantially reduces search space by requiring both crosslinked peptides to generate sparse data attributable to their linear forms. The method demonstrates highly sensitive crosslink peptide identification with a low false positive rate. Integration with a Haddock plug-in provides a resource that can combine multiple sources of data for protein modeling activities. We generated a structural model of porcine transferrin bound to TbpB, a membrane-bound receptor essential for iron acquisition in Actinobacillus pleuropneumoniae. Using mutational data and crosslinking restraints, we confirm the mechanism by which TbpB recognizes the iron-loaded form of transferrin, and note the requirement for disparate sources of restraint data for accurate model construction. The software plugin is freely available at www.msstudio.ca.
Integrative methods in structural biology use data from disparate sources to generate accurate models of large protein structures and assemblies (1). In this way, the reach of classical structure providers such as x-ray crystallography and NMR can be extended. Biophysical data with an underlying spatial component can be combined with “building block” structures in a molecular modeling framework, to generate high-fidelity models of systems of impressive size and complexity (2–5). Mass spectrometry can provide rich sets of data in support these activities, in the form of topographical footprints (covalent labeling, CL) 1 (6–8), conformational dynamics (hydrogen/deuterium exchange, HX) (9, 10) and distance restraints (crosslinking, XL) (11–13). We have built informatics routines within the Mass Spec Studio framework to mine restraints from both CL and HX data (14), to support such data-driven molecular modeling activities. In this study, we describe a new plug-in built into the Studio for identifying crosslinks from LC-MS/MS data sets.
Advances in instrumentation, methods and cross-linking protocols have generated renewed interest in what is an older technique. However, useful informatics routines are essential for gaining access to quality crosslinking information as site identification is not a trivial problem (15). Some noteworthy tools that have emerged in the last few years include xQuest (16), Merox (17), Stavrox (18), Sim-XL (19), pLink (20), XlinkX (21), and XiQ (22). The proliferation of such tools is a strong indication that new XL reagents and methods require dynamic software development to accommodate the needs of challenging structural applications. For example, multiplexed structural analysis from whole proteomes (21), a requirement for richer sets of “molecular rulers” for de novo structure determination (23), and integration with other sources of structural data are three application areas that are not yet well served by available tools.
It would be useful to develop packages that collect and integrate concepts that have demonstrated some utility in the detection of crosslinks (24), and support the easy addition of new concepts. In our view, there are a number of features that should be bundled into a single solution. Any source of MS data should be accommodated, regardless of the instrument vendor or style of experiment. Both low and high resolution data-dependent LC-MS/MS data have been collected in crosslinking experiments, on both FT-MS and TOF-based instruments (11, 25). Many software applications are restricted to the analysis of processed and/or converted data files, which seems to us unnecessary with the increasing willingness of vendors to supply file readers directly or through Proteowizard (26). Raw data should be handled natively where possible, as it provides the greatest opportunity for error detection and results validation. Chromatographic data and the precursor ion profiles should be available to support the identification exercise, or at least the validation exercise. The output should be easily navigated for rapid hit validation, and readily harvested for integration with visualization and modeling activities (14).
Further, although isotopic labeling has high value in the validation exercise (16, 27), a very large number of useful reagents are not available in labeled format, and labeling is not a strict requirement for accurate linkage detection (28). Finally, and perhaps most importantly, robust probabilistic scoring algorithms should be implemented (29), and software design should promote the easy inclusion of alternative methods as they become available. Simple fragment-counting may be useful, but it does not always promote sensitive site identification. In the wider context of scoring, database reduction strategies are necessary to address the n2 time complexity of searching for crosslinked peptides, which becomes particularly acute when using nonspecific crosslinker chemistries and nonspecific digestion enzymes (24, 28). Methods that do not complicate the experimental workflow or compromise the validity of a probabilistic scoring function are needed. Crosslinkers that are cleavable in the gas phase through CID or ETD are emerging that have been used to reduce time complexity (21, 30), but these currently come with platform restrictions and limitations on the choice of reagents. Concepts that treat the second peptide in a linked pair as an open modification are particularly useful in site identification (28, 31–33). Integrating and extending these concepts within a wider platform will be required to meet the challenges associated with nonspecific labeling and digestion protocols, however.
In this contribution we present a crosslinking plug-in for the flexible Mass Spec Studio framework, which combines useful components for each of the feature categories discussed above. We extend the database reduction concept to limit dependences on precursor ion m/z, and provide a collection of tools for rapid results validation using the raw data. Finally, we illustrate how the XL-MS data can be combined in the Studio with other structural data to support modeling activities.
EXPERIMENTAL PROCEDURES
Software design
The Mass Spec Studio was designed to capture the growing number of experimental methods for structural mass spectrometry, and the diverse computational strategies for identifying and quantifying structural restraints for modeling purposes (14). A large number of the core activities associated with mining LC-MS/MS data are shared by most label-detection applications, therefore we designed a composite application where loosely coupled components allow effective use of shared tools, as well as straightforward development of future extensions and new pluggable content. The crosslinking software package was built as a plug-in, using an updated version of the Studio framework (v2). As with v1 of the framework, plug-ins are either individual components or collections of components, consisting of object libraries, processing algorithms, user interface (UI) elements and experiment types. During software start-up, each component is dynamically loaded and assembled into the main application. Several enhancements to the v1 framework facilitate per-session component customization, dependence management, as well as version control. Updated versions of AvalonDock and Prism boost Mass Spec Studio's capacity to conform to the latest software design practices and patterns, for scalable and sustainable .NET applications.
Fig. 1 provides a schematic of the Studio framework that we used to develop a robust concept in crosslink detection. The new dependence management in v2 makes sharing of common tools and components more streamlined, which simplifies the development of new plug-ins. We took advantage of this feature to refactor common elements of our existing packages (HX-MS and CL-MS) into a new “Structural Biology” (StructBio) resource package. The new framework, therefore, supports all three structural mass spectrometry experiment types. As a result, any future development of structural biology applications or upgrades can employ a common, carefully implemented and tested set of tools and resources. Such tools and resources include mass calculators, digestion/fragmentation rules, peptide identification algorithms, and molecular visualization.
Fig. 1.

The architecture of Mass Spec Studio and placement of the XL package. During start-up, components are discovered and injected (solid arrows) via the extensibility controller into the main application (a lightweight shell with minimal requirements). UI components are inserted into predefined regions within the shell, which are provided by the user interface controller. All components depend (dotted lines) on the Studio framework for common high-level elements related to mass spectrometry, and not for specific applications of mass spectrometry (e.g. reading a mass spec file format is abstracted from data processing). Common resources can be shared across applications in order to avoid re-engineering tools and features, which accelerates development of downstream components. The StructBio package is one such high level collection of resources, shared by the HX, CL and XL plug-ins.
XL Application
To develop a crosslink detection concept that supports both computational and visual validation of large data sets, we applied design concepts specifically borrowed from our HX-MS2 platform (34). This primarily involved revising our strategy for peptide library generation and augmenting our scoring metrics for crosslink detection and validation. These two elements are described below.
Library Creation
To generate a searchable library, a database of linear peptides is created, using proteins known to exist in the sample or from a whole proteome when the sample composition cannot be inferred from other data (e.g. a prior data-dependent proteomics experiment). For individual proteins, the sequence information and any associated structures can be fetched automatically from the protein data bank (PDB, www.rcsb.org). The database is expanded according to a flexible selection of fixed or variable modifications, and a suitable crosslinker is selected from a set of available options. New reagents can also be designed and implemented, using a crosslinker design wizard, with heavy and light versions if desired. Library expansion is parameterized by representing a user-defined range of possible charge states and user-selected mass modifications. This list automatically includes peptides singly labeled with the crosslinker, or so-called dead-ended peptides. Multiple data-dependent LC-MS/MS runs, from any file type, are associated with the library for subsequent processing. An option exists to reformat the data into a highly compressed binary file structure (.mssdata) for efficient storage and later use in other applications, although this is not required.
Library Reduction and Crosslink Detection
We have implemented a probabilistic scoring strategy within the Mass Spec Studio framework, behind a robust library reduction method. For library reduction, we considered a number of concepts to address the underlying n2 time complexity associated with investigating all possible pairs of peptides. In one case, the peptide library can be sorted by mass and a smaller list of peptide pairs can be assembled with entries that fall within an acceptable tolerance around the precursor ion mass (an MS-level approach) (24, 35). A classic binary search could then speed the analysis. In another case, assumptions can be made based on the acquired fragment spectra (an MS/MS-level approach). For example, one method requires that evidence of a dead-ended version of a crosslinked individual peptide should be observable (19). This idea is based on the assumption that crosslinking chemistries will always generate a distribution of all product types, with the actual crosslinking event itself occurring with a relatively low abundance. A third approach extends the idea of MS/MS-level library filtering, by requiring fragment evidence for one peptide in the MS/MS spectrum and treating the other as an open modification constrained by the precursor ion m/z (e.g. (28)).
In the Studio, we chose to develop an approach using MS/MS-level filtering, based loosely on the concept of ion tags in xQuest (16). Our approach is outlined in Fig. 2. We assume that an MS/MS spectrum of a crosslinked peptide must possess some minimum unmodified sequence information for each peptide comprising the pair, which can be scored in a probabilistic manner. That is, a population of simple sequence ions will exist for each peptide (possibly few and low intensity), akin to a peptide sequence tag (36). Ions from the unmodified portions represent one of the more abundant ion types in CID MS/MS data of crosslinked peptides (16, 37). A library of peptides is constructed that consists of all linear peptides having some minimum threshold score, which we base on the E-score concept used in OMSSA (38),
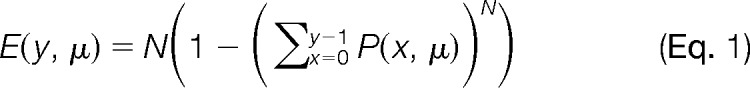 |
where P(x, μ) is a Poisson distribution: x is the number of fragment matches between an experimental MS/MS spectrum and a theoretical spectrum of a library entry, the average number of random matches is μ, y is the total number of successful fragment matches and N is the peptide library size. The E-score provides an opportunity for a probabilistic assessment of the ion series for the unmodified portions of peptide, based on an underlying Poisson model of noise in the search, which seems sensible to preserve for crosslinked peptide detection. That is, we assume that the noise characteristics of the search space for crosslinked peptides is not significantly different than linear peptide noise, which is well modeled by the Poisson distribution (38). In the initial search for library reduction, we employ a hard filter on fragment ions in the MS/MS spectrum by requiring strong evidence for monoisotopic ions (i.e. M peak >5 times the intensity of an M-1 peak, if detected). The exercise is to float E as high as possible, while preserving a time-manageable list of candidate linear peptides for the next stage of scoring.
Fig. 2.

Crosslinked peptide candidate generation step involving an MS2-based library reduction strategy. A search of the full set of possible linear peptides is conducted against each MS2 spectrum using OMSSA (38), to generate a set of peptide-spectrum matches (PSM's) for each MS2 spectrum. Any peptides with an acceptable E-score for a given spectrum are allowed to form crosslinked candidates. Two PSM's are shown for Spectrum #3 for illustration: peptide 1 and 4 are strongly represented by fragment ions from the unmodified portions of peptide and are both accepted. Once the PSM's are scored and selected, all possible candidates are generated and filtered by precursor mass. Hits can include peptides that are self-linked, crosslinked, dead-ended, or simply exist as linear peptides. Candidates undergo a refined scoring method based on a rigorous peak assignment of MS and MS2 data, and a wider set of fragment ion types.
Precursor ion mass is only introduced after this initial reduction in search space. It can be difficult for data-dependent experiment types to accurately select a monoisotopic ion, particularly as peptide mass and charge increase. We filter candidates based on the actual isotope profile in the LC-MS data, rather than relying solely on the instrument-generated precursor ion values. Bounded by a precursor mass tolerance that we set in the search, we initially accept all possible peptide candidates with an abundant isotope at the triggered mass/charge value, but ultimately require every high probability crosslinked peptide to generate an isotopic distribution that maintains a minimum goodness-of-fit to the actual data. Our acceptance criteria allows us to evaluate candidate crosslink peptides against all possible background peptides that may fall within the ion inclusion window (i.e. crosslinked peptides, linear peptides, dead-end peptides or other modified linear peptides permitted in the library construction phase).
A list of possible crosslinked peptides is assembled from the output of the mass filtering, based on linear peptides that may coexist in the MS/MS spectrum. The actual scoring phase implements a strategy adopted from X!link (39) and OMSSA (38), again using an E-score calculation, but now invoking a more rigorous evaluation of fragment ion isotopic distributions, and increasing sensitivity in two ways: (1) by permitting all other probable ion types in the identification, and (2) by including a requirement that l fragments are drawn from the top n most intense fragments,
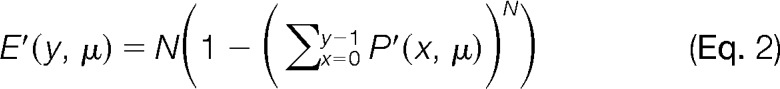 |
where the Poisson distribution is now
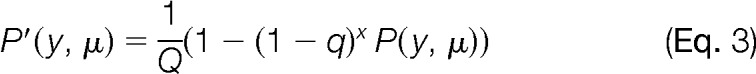 |
and the q value is
 |
where nCl is the number of combinations having l calculated peaks in the top n peaks, and vCl is the corresponding number of combinations having l calculated peaks in the full set of v peaks in the spectrum. The defaults are n = 10 and l = 3, as suggested by Lee (39); q is simply a normalization factor. Of note, the N in this calculation now derives from the reduced set of combinations emerging from the initial library reduction and mass filtering. With this approach, the contribution of the ions from the unmodified portions of sequence to the final score may actually be very small, as other fragment ion types may dominate. All that is required for library prefiltering are intensities of these ions that are detectable above the noise (here 2.5% of the base peak). We note that a filtering strategy can be implemented at this final step, which involves a recalculation of individual E-scores for each peptide retained in the hits. A recalculation is needed as more ion fragments can now be included for the assessment of each peptide, using a linearization strategy similar to one recently described (28). The user can require that each individual peptide retains a certain minimum score, to ensure that the deferred re-inspection of the fragment ion isotopic distribution does not reduce confidence. Fig. 3 shows a conceptualized version of the postprocessing peptide inspection view.
Fig. 3.
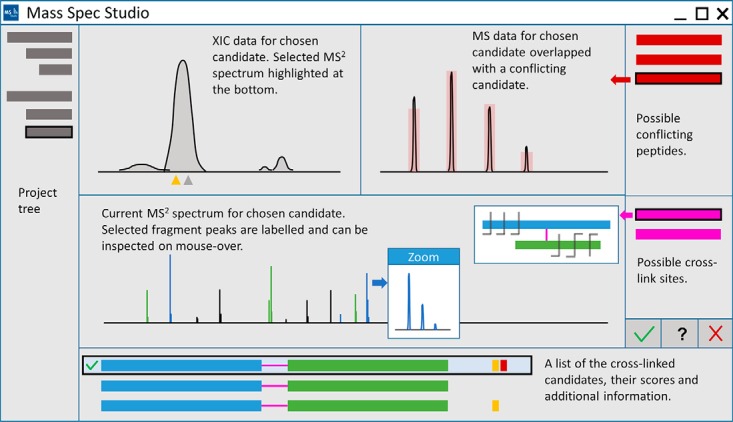
The conceptualized UI for visual inspection of crosslinks and conflict resolution. The Studio provides a UI for navigation and visual inspection of peptides. Peptides are selected from a list (bottom of view), containing information such as sequence, charge, peptide type, selection status, score and validation flags. Flags are automatically generated during processing to provide additional information regarding the quality of a crosslink candidate. Raw data is inspected using three interactive views (XIC, MS and MS2). If other candidates point to the same MS2 spectrum, they are marked as conflicts and shown in the conflict list (right of view). Selecting a conflicting crosslink candidate will overlap its information on MS2 spectrum view. Specific crosslink sites can be selected from the sites list, and displayed in a widget. A user captures a decision about the selected crosslink using the status buttons underneath the crosslink site list.
Protein Crosslinking and Digestion: Software Performance
For development purposes and initial performance testing, we crosslinked bovine serum albumin (BSA) utilizing the protocol in Lietner et al. (15), with minor modifications. Briefly, disuccinimidyl suberate (DSS) was dissolved in dimethyl sulfoxide (DMSO), to a concentration of 25 mm. DSS was added to a 1 mg/ml BSA solution (containing 75 mm KCl, 5 mm MgCl2, 20 mm HEPES, pH 8.3) to a final concentration of 1 mm. Crosslinking proceeded at 37 °C for 30 min with gentle mixing. Quenching was completed by adding ammonium bicarbonate to a final concentration of 50 mm for 20 min at 37 °C. The sample was dried down, and then dissolved and denatured in 8 m urea. Disulfide bonds were reduced using 2.5 mm Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and the resulting free cysteines alkylated using 5 mm iodoacetamide. For digestion, the sample was diluted to 1 m urea using 50 mm ammonium bicarbonate, and 1 μg of trypsin added. Sample was digested overnight at 37 °C before quenching with formic acid, to a concentration of 2% (v:v). Salt was removed using a 200 μl HyperSep SpinTip solid phase extraction tip (Thermo Scientific). After evaporation, the sample was reconstituted in 30% acetonitrile/0.1% formic acid and separated by size exclusion chromatography (SEC). SEC was completed on an Agilent 1100 chromatography system, with a Superdex Peptide PC 3.2/30 column (GE Healthcare, Mississauga, Ontario), equilibrated with SEC buffer (30% acetonitrile 0.1% formic acid). Fractions were collected every minute at a flow rate of 0.1 ml/min. All fractions were evaporated and reconstituted for LC-MS/MS.
Fractions were analyzed on a nanoLC-Orbitrap Velos (Thermo Scientific, San Jose, CA). Fractions were dissolved in 0.1% formic acid and loaded on an 8 cm x 75 μm self-packed picotip column (Aeris Peptide XB-C18, 3.6 μm particle size, Phenomenex, Torrance, CA). Separation was achieved with a 30 min gradient (5–60%) of mobile phase B (97% acetonitrile with 0.1% formic acid) at 300 nl/minute. The mass spectrometer was operated in positive ion mode, using a high/high configuration (40), with MS resolution set at 60,000 (400–2000 m/z) and MS2 resolution at 7500. Up to ten of the most abundant ions were selected for fragmentation using higher energy collisional dissociation (HCD), rejecting charge states 1 and 2, and using a normalized collision energy of 40%.
Protein Crosslinking and Digestion: Crosslink Evaluation and Interaction Modeling
The new Studio plug-in was used in a crosslinking study involving the binding of porcine transferrin (78 kDa, expressed and deglycosylated as described previously (41)) to a bacterial transferrin-binding protein B (TbpB, 80 kDa, Actinobacillus pleuropneumoniae strain H49, expressed and purified also as previously described (41)). The complex was prepared by binding a slight excess of TbpB to holo-pTf in 50 mm sodium phosphate buffer (pH 7.4) with 50 mm NaCl and incubated for ∼3 h at room temperature. After incubation, the bound sample was further incubated with DSS crosslinker as above. Approximately 10 μg of total protein was separated on SDS-PAGE, and a band corresponding to the crosslinked proteins was excised and subjected to in-gel tryptic digestion using established protocols (42). The digest was analyzed by mass spectrometry as above, and the crosslinked residues identified using our XL-MS plug-in and Kojak (43). Crosslinked residues identified in the Studio were tested against, and combined with, mutational data to constrain data-driven protein docking experiments, using another plug-in available in the Studio (14), used as a customized launchpad to the Haddock webserver (44). Mutations that reduced binding strength, and used as markers of the binding interface included D360A, S625K, R509A (pTf, (41)) and F68E, K61E, E112K, and F171A (TbpB, (45)).
RESULTS AND DISCUSSION
Software Performance
The DSS-crosslinked BSA digest generated a set of 2500 MS/MS spectra enriched for larger digest fragments. The data were searched using our Mass Spec Studio XL plug-in against a random selection of E. coli proteins added as noise, up to and including the full E. coli proteome (estimated at 4300 proteins). Fig. 4A shows that processing time for the full search was a linear function of the database size, when using an E-score of 0.3 for initial library reduction. A search conducted in this fashion returned 113 unique peptide candidates (cross-linked, dead-end, self-linked and linear peptides). Hit criteria included a requirement for a high-fidelity isotopic distribution in the MS data and an observable ion chromatogram. The -ln transformed E′ score for the crosslinked peptide was set at 13 and each peptide had recalculated -ln transformed Eα/β values better than 10.
Fig. 4.

XL plug-in performance. A, Total processing time to generate a set of crosslinked peptide candidates is measured as a function of increasing the number of proteins in the root library, using an E-score threshold of 0.3. in the library prefiltering. B, Total processing time as a function of E-score threshold, with a database size of 51 proteins (BSA + 50 random E. coli proteins). C, The number of total results (identified peptides of all types) as a function of E-score threshold for search configuration as in (B). D, The same data as in (C), with the application of results filters: a threshold for crosslink score (-ln transformed E′ score of 13) and a threshold for component linear peptide score (-ln transformed E′ score of 10).
An E-score of 0.3 represents a moderately strong reduction in the initial library. To determine the sensitivity of the search speed to the E-score, we configured a scenario that partially mimics the crosslinking of a 50-protein complex, and reanalyzed the BSA data as a function of E-score in a root library consisting of BSA and 50 randomly selected E. coli protein sequences (Fig. 4B). Search times remained a linear function of E-score up to surprisingly high values, ∼80% of the maximum library size. In the linear range, calculating crosslinked peptide combinations is much faster than generating the peptide-spectrum matches (PSM's). PSM generation is linear in time complexity, but at a certain point in E-score, the time to generate the number of possible combinations begins to exceed PSM generation when the library again grows large, and time complexity transitions to n2 as expected. A high E-score correlates with very weak sequence identification power, precisely the scenario that is needed for the library reduction concept to preserve detection sensitivity for crosslinked peptides. For example, setting the E value filter to 6000 where the trend just begins to diverge from linearity retains peptide candidates needing only three or more fragments. Sensitivity would be likely be lost if a large numbers of fragments were required for both component peptides in a crosslinked pair. In a real situation representing a complex of 50 proteins, the set of MS2 spectra may be larger than the set we searched here. However, scoring has only a linear dependence on the number of spectra. A search will be bounded by ∑i=1X ni2 time complexity, where X represents the number of spectra to be evaluated, and ni the number of linear peptides that pass the initial filtering step for the ith spectrum.
Fig. 4C illustrates the impact of the initial E-score filter on the number of actual crosslinking results generated, and this figure is the complement to Fig. 4B. Crosslinking results containing only BSA fragments increase weakly, as expected because a single protein will begin to saturate at moderate E-scores, whereas the number of E. coli results increase dramatically at high E. To return sensitivity and specificity at high E-score, we apply the filtering tools provided in the plug-in. Fig. 4D shows the filtered output of Fig. 4C. Using a -ln transformed E′ score cut-off of 13 for identification caps the number of BSA crosslinked peptides to a maximum of 207 and reduces the E. coli false positives to 4.8%. Specificity is further improved by requiring that crosslinked peptides generate a minimum of evidence for both peptides (-ln transformed Eα/β scores of 10). Together, the two cut-offs generate 0.5% false positives at an E-score of 1000 for initial library reduction. Our default value for the initial E-score is set at 10% of the total library size. This value can be increased by the user depending on the objectives of the search, but we find little benefit to decreasing the value below 10%.
To test the sensitivity and selectivity of this reduction strategy, we implemented the ranked-pair search, using the precursor ion mass in place of the E-score reduction method. This alternative method has a high sensitivity for crosslinked peptide detection. As per Choi (35) and Petrotchenko (24), all possible peptide pairs in the library plus the crosslinker residue mass were retained, bounded by the mass tolerance in the measurement of the precursor mass. All other features of the workflow were preserved. There is a large reduction in the library to be searched when high-accuracy monoisotopic masses are used as constraints for pair selection (24), but this method does not scale well when mass accuracy is reduced (Table I). With a precursor ion tolerance set to a 5 ppm window, processing time is over sevenfold greater for the matched-pair method versus the application of a high E score (80% of the library size). This effect more noticeable as the mass accuracy diminishes. The mass-pairing strategy generates a majority of candidates that derive from the random E. coli proteins used as noise in the comparison (Table I). To determine if sensitivity is lost using the plug-in's filtering approach, each reduced library was searched against the MS2 data and scored with the same E′ score method described in equation (2), using the full set of sequence ion types. All 98 unique hits discovered using the mass pairing method were found with the E-score method (supplemental Fig. S1), and an additional 15 were discovered only by the E-score method. At first glance, the 15 unique hits seem to indicate a lower selectivity of the method (false positives). However, the reduced library for the mass-pairing method is 40-fold larger than the library of the E-score approach, and entries are mostly derived from E. coli. The difference could reflect an inappropriately large search space. We note simply that the 15 unique identifications are high quality, and the strong library reduction achieved with the E-score method does not omit any reasonable hits otherwise found with the more permissive mass-pairing library reduction method. Finally, we note that a structural analysis reveals that the large majority of unique crosslinked sites we identified using the Studio plug-in (78 hits) fall within an expected Euclidean distance distribution for the DSS crosslinker (<30 Å (46)).
Table I. Impact of library reduction method and mass accuracy on performance.
| Precursor tolerance (ppm) | Time (s)a | BSA (hits) | E. coli (hits) | BSA (E′)b (hits) | E. coli (E′)b (hits) | BSA (E′, E′α/β)c (hits) | E. coli (E′, E′α/β)c (hits) | |
|---|---|---|---|---|---|---|---|---|
| E-score | 5 | 73 | 44 | 30 | 29 | 2 | 24 | 0 |
| 10 | 101 | 296 | 103 | 208 | 11 | 189 | 0 | |
| 15 | 124 | 525 | 263 | 339 | 12 | 304 | 1 | |
| Mass pairing | 5 | 535 | 468 | 3362 | 28 | 4 | 23 | 0 |
| 10 | 1287 | 2504 | 15266 | 180 | 13 | 151 | 0 | |
| 15 | 2520 | 5027 | 29615 | 265 | 18 | 215 | 0 |
a Search configured to include a library of 10 random E. coli proteins plus BSA (1701 peptides). E-score 80% of total library size. Run on a Quad-core Intel i7–3770 3.5 GHz with 16 Gb RAM, using 7 virtual threads. Candidates peptides of all types (cross-linked, self-linked, dead-end, linear).
b Results filtered using a –ln transformed E′ score of 13 for the candidate peptide.
c Results filtered using a –ln transformed E′ of 13 for the candidate peptide, with an additional –ln transformed E′α/β score of 10 for crosslinked peptides.
Detecting and Modeling Protein Interactions
We then applied the plug-in to a binding study involving transferrin (Tf) and a transferrin-binding protein B (TbpB), to determine if the sensitivity we observed with the prefiltering strategy could detect interprotein crosslinks, which tend to be less abundant than intraprotein crosslinks, and if the data could constraint an accurate docking exercise. Gram-negative bacteria capture host iron-binding proteins in order to acquire essential iron. TbpB receptors are a key element of iron uptake mechanisms for numerous pathogens from the Neisseriaceae and Pasteurellaceae (47). TbpB's exclusively recognize the iron-loaded (holo) form of transferrin, and deliver it to transferrin-binding protein A (TbpA), a TonB-dependent integral protein that serves as the channel for free iron transport across the outer membrane (48, 49). TbpB's are emerging as an important vaccine target, and they require an in-depth understanding of Tf/TbpB interactions in order to engineer antigen(s) with broad specificity. We collected DSS-based crosslinking data on porcine Tf bound to TbpB from Actinobacillus pleuropneumoniae (H49), a porcine pathogen that serves as a useful model system for human pathogens. Structures for the individual elements of the complex have been determined (pTf: 1H76 and H49 TbpB: 3HOL), and a RosettaDock model of the binary complex has been validated by extensive mutational analysis and functional studies (Fig. 5A) (41, 50). A crystallographic structure of human transferrin bound to TbpB from Neisseria meningitidis provides strong supporting evidence for the localization of the binding site (51).
Fig. 5.
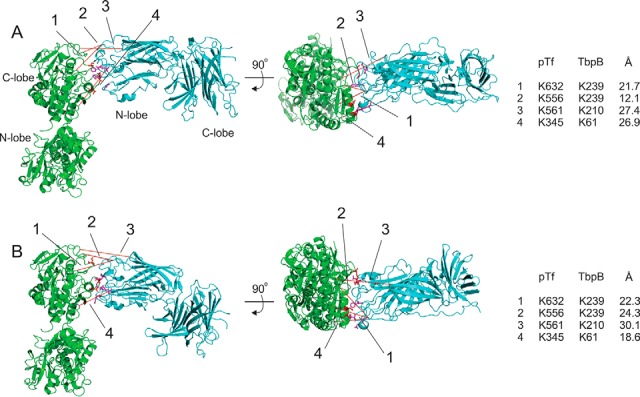
Models of the pTf:TbpB interaction. A, Docked model of 1H76 (pTf) and 3HOL (TbpB) using modified routines in RosettaDock, as published and validated through mutational analysis of key residues in the binding interface (41, 50). Crosslinks detected by the Studio plug-in are highlighted in red bars, with a linking scheme (and distances) shown on the right. Side and top views presented. B, As in (A), but using the crosslinking data and published mutational data in a Haddock docking exercise, launched through the Studio. Linking scheme also shown on right. For (A) and (B), pTf is shown in green and TbpB in cyan and the lobes are labeled for each protein. All figures generated in PyMol.
We identified 64 crosslinked peptides (41 unique ones, after removal of multiple charge states) using the probabilistic criteria presented above (i.e. a transformed E-score of >13). In this context, the score corresponds to a calculated false discovery rate (FDR) of 0.5%, here based on a decoy database search, where the input sequences were randomized. As additional validation criteria, individual linked peptides were required to have a transformed score greater than ∼10. Most of the crosslinked peptides are of the intraprotein variety (37, corresponding to 33 unique residue pairs) and fall with a spatial distribution expected for the crosslinker (supplemental Table S1). Four interprotein crosslinks were detected, which could be tested against the existing model for the pTf:TbpB structure. One of the hits has an excellent E-score but it contains a short three-residue beta peptide that scores poorly in our validation approach. However, all four interprotein crosslinks are consistent with the published model (Fig. 5A), and fall within Euclidean distances of 28 Å. For comparison purposes, we used Kojak on the same data set, constrained to a 1% FDR based on Percolator (43). Only one of the four interprotein crosslinks was found using the recommended settings for the program: the crosslink involving the short peptide. A second crosslinked peptide was found, but it fell well outside the expected distance range and pointed to a residue in the distal N-terminal domain of pTf. Kojak found more intraprotein crosslinks than the Studio plug-in (62 unique residue pairs, from 78 unique peptides), so the lower number does not appear to arise from lower detection sensitivity. The results are compared in supplemental Table S1 and supplemental Fig. S2 for this interaction.
Finally, we revised the Studio's molecular docking plug-in to support crosslinking data, and we tested if the crosslinking data was sufficient to generate a pTf:TbpB structural model similar to the established model in Fig. 5A. The plug-in provides a tailored portal to the Haddock webserver, allowing users to input crosslinked residues and the PDB files associated with the structural elements to be docked. The plug-in currently supports the inclusion of differential CL-MS and HX-MS data, useful for interface mapping (14). These data are translated into ambiguous interaction restraints (AIRs) and incorporated into the potential energy function that governs the docking exercise. Here, we added a utility to incorporate CX-MS data as unambiguous restraints (UIRs), as the linked residues are known. Three rounds of docking were initiated. In the first round, only the four identified interprotein crosslinks were used, and the docked model, while locating the correct binding domains and basic orientation, did not return an accurate pose (supplemental Fig. S3). In the second round, the mutational data alone was incorporated as a restraint set, using each mutation as an active residue and defining passive residues within 5 Å of an active residue (44). The binding model was in good agreement with the established model (supplemental Fig. S3), highlighting the value of interface data in directed docking studies. The addition of the crosslinking information to the mutational data in the third docking round served to incrementally improve docking scores over the mutational data alone, and produce a large cluster of solutions that emulates the Rosetta model (Fig. 5B, RMSD of 5.0Å using pTf C-lobe and TbpB N-lobe). Thus, although the crosslinking data could locate the axis of binding and correctly orient the two proteins around it, interface data was shown to have higher value in developing the molecular topology of the interface. We suspect that crosslinking data is limited as a singular source of modeling data as the precision of the “molecular ruler” is currently not very high. In this study, we imposed a 5–25 Å Cα-Cα length restraint, based on a reasonable span of the length distribution for DSS (46). The ambiguity in length could not be overcome with the low numbers of interprotein crosslinks we detected in this study, but we anticipate that shorter crosslinkers with alternative chemistries will add value to such exercises.
With the incorporation of restraints from two data sources, the Haddock-generated model shows an interaction between TbpB and the two C-lobes of pTf, as expected (41). These lobes close around Fe3+ in the iron-loaded holo form, bringing them into registry with the TbpB binding surface. As crystallographic structures of the heterodimers have proven difficult to generate, data-driven modeling of human transferrin with the many strain-specific TbpB receptors from human pathogens may provide the capacity and accuracy to guide antigen engineering efforts.
Conclusion and Perspective
The Studio application for XL-MS provides a solution for database size reduction, based on a simple assumption that does not negatively influence sensitivity for site determination. By requiring every putative crosslinked peptide to present a modest subset of fragment ions arising from a linear segment, we show that database size can be strongly reduced without sacrificing quality hits. This finding appears consistent with earlier explorations of the problem. Chalkley has pointed out that confidence in the detection of crosslinked peptides is tied to the poorest scoring peptide in the pair (29), and Lee has shown that higher quality results are possible when hit lists are filtered for each peptide independently (39). Although it may seem surprising that strong filtering can be achieved using a very weak E-score threshold in the reduction step, it should be noted that a fragment spectrum obviously must contain evidence for a pair of linear peptides. The additional library reduction achieved with this step is considerable.
Embedding this library reduction approach within a generic software platform provides access to this strategy using data from any instrument type, without file conversion. It provides a range of tools to visualize and validate the output, using strategies tested in HX-MS2 applications. However, our current format for probability-based scoring may not be as valid for certain crosslink strategies where affinity enrichment is applied, and where the crosslinkers themselves generate extensive fragments. The Studio XL plug-in supports the detection of reporter ions that are unique to the crosslinker, but these are not incorporated into the scoring algorithm. Such data may be better applied as a filter of the results in the current package. Our strategy for a probabilistic determination will be most useful when the searched data is dominated by linear peptide “noise,” which we suggest is the usual situation unless strong affinity enrichment methods are applied. The speed that we recover by implementing an MS2-based reduction strategy will permit much larger searches, including multi-protein complexes and proteome-wide analyses, without requiring the use of specialized crosslinkers. Cleavable crosslinkers will add confidence to the identifications of linked peptides, but as they need specialized fragmentation strategies, we present this strategy as a more generic alternative. Perhaps most importantly, the acceleration of performance that we obtained using this simple reduction strategy should permit the use of nonspecific reagents and even nonspecific digestion enzymes, which we suspect will be necessary to support robust molecular modeling activities.
Supplementary Material
Footnotes
Author contributions: V.S., N.O., A.B.S., and D.C.S. designed research; V.S., A.R., M.H., and D.C.S. performed research; N.O. and A.B.S. contributed new reagents or analytic tools; V.S., A.R., M.H., N.O., and D.C.S. analyzed data; V.S. and D.C.S. wrote the paper.
* The work was supported by an NSERC Discovery Grant RGPIN/298351-2010, the Canada Foundation for Innovation (DCS) and the University of Calgary.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CL
- covalent labeling
- BSA
- bovine serum albumin
- DSS
- disuccinimidyl suberate
- FDR
- false discovery rate
- HX
- hydrogen-deuterium exchange
- MEF
- microsoft extensibility framework
- PSM
- peptide spectrum match
- ppm
- parts per million
- pTf
- porcine transferrin
- SEC
- size-exclusion chromatography
- TpbA
- tranferrin-binding protein A
- TbpB
- tranferrin-binding protein B
- UI
- user interface
- XIC
- extracted ion chromatogram
- XL
- cross-linking.
REFERENCES
- 1. Webb B., Lasker K., Velazquez-Muriel J., Schneidman-Duhovny D., Pellarin R., Bonomi M., Greenberg C., Raveh B., Tjioe E., Russel D., and Sali A. (2014) Modeling of proteins and their assemblies with the Integrative Modeling Platform. Methods Mol. Biol. 1091, 277–295 [DOI] [PubMed] [Google Scholar]
- 2. Kim S. J., Fernandez-Martinez J., Sampathkumar P., Martel A., Matsui T., Tsuruta H., Weiss T. M., Shi Y., Markina-Inarrairaegui A., Bonanno J. B., Sauder J. M., Burley S. K., Chait B. T., Almo S. C., Rout M. P., and Sali A. (2014) Integrative structure-function mapping of the nucleoporin Nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Mol. Cell. Proteomics 13, 2911–2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fernandez-Martinez J., Phillips J., Sekedat M. D., Diaz-Avalos R., Velazquez-Muriel J., Franke J. D., Williams R., Stokes D. L., Chait B. T., Sali A., and Rout M. P. (2012) Structure-function mapping of a heptameric module in the nuclear pore complex. J. Cell Biol. 196, 419–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Sali A., and Rout M. P. (2007) The molecular architecture of the nuclear pore complex. Nature 450, 695–701 [DOI] [PubMed] [Google Scholar]
- 5. Greber B. J., Bieri P., Leibundgut M., Leitner A., Aebersold R., Boehringer D., and Ban N. (2015) Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 348, 303–308 [DOI] [PubMed] [Google Scholar]
- 6. Kaur P., Tomechko S. E., Kiselar J., Shi W., Deperalta G., Wecksler A. T., Gokulrangan G., Ling V., and Chance M. R. (2015) Characterizing monoclonal antibody structure by carboxyl group footprinting. MAbs 7, 540–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou Y., and Vachet R. W. (2013) Covalent labeling with isotopically encoded reagents for faster structural analysis of proteins by mass spectrometry. Anal. Chem. 85, 9664–9670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stocks B. B., Rezvanpour A., Shaw G. S., and Konermann L. (2011) Temporal development of protein structure during S100A11 folding and dimerization probed by oxidative labeling and mass spectrometry. J. Mol. Biol. 409, 669–679 [DOI] [PubMed] [Google Scholar]
- 9. Underbakke E. S., Iavarone A. T., Chalmers M. J., Pascal B. D., Novick S., Griffin P. R., and Marletta M. A. (2014) Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure 22, 602–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rand K. D., Zehl M., and Jorgensen T. J. (2014) Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: overcoming gas-phase hydrogen/deuterium scrambling. Acc. Chem. Re.s 47, 3018–3027 [DOI] [PubMed] [Google Scholar]
- 11. Sinz A., Arlt C., Chorev D., and Sharon M. (2015) Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology. Protein Sci. 24, 1193–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sinz A. (2006) Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom. Rev. 25, 663–682 [DOI] [PubMed] [Google Scholar]
- 13. Rappsilber J. (2011) The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 173, 530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rey M., Sarpe V., Burns K. M., Buse J., Baker C. A., van Dijk M., Wordeman L., Bonvin A. M., and Schriemer D. C. (2014) Mass spec studio for integrative structural biology. Structure 22, 1538–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., and Aebersold R. (2010) Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol. Cell. Proteomics 9, 1634–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rinner O., Seebacher J., Walzthoeni T., Mueller L. N., Beck M., Schmidt A., Mueller M., and Aebersold R. (2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods 5, 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Götze M., Pettelkau J., Fritzsche R., Ihling C. H., Schäfer M., and Sinz A. (2015) Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom. 26, 83–97 [DOI] [PubMed] [Google Scholar]
- 18. Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C. H., Krauth F., Fritzsche R., Kühn U., and Sinz A. (2012) StavroX—a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 23, 76–87 [DOI] [PubMed] [Google Scholar]
- 19. Lima D. B., de Lima T. B., Balbuena T. S., Neves-Ferreira A. G., Barbosa V. C., Gozzo F. C., and Carvalho P. C. (2015) SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J. Proteomics 129, 51–55 [DOI] [PubMed] [Google Scholar]
- 20. Gao Q., Xue S., Doneanu C. E., Shaffer S. A., Goodlett D. R., and Nelson S. D. (2006) Pro-CrossLink. Software tool for protein cross-linking and mass spectrometry. Anal. Chem. 78, 2145–2149 [DOI] [PubMed] [Google Scholar]
- 21. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 22. Fischer L., Chen Z. A., and Rappsilber J. (2013) Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteomics 88, 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Politis A., Stengel F., Hall Z., Hernandez H., Leitner A., Walzthoeni T., Robinson C. V., and Aebersold R. (2014) A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods 11, 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrotchenko E. V., and Borchers C. H. (2014) Application of a fast sorting algorithm to the assignment of mass spectrometric cross-linking data. Proteomics 14, 1987–1989 [DOI] [PubMed] [Google Scholar]
- 25. Soderberg C. A., Lambert W., Kjellstrom S., Wiegandt A., Wulff R. P., Mansson C., Rutsdottir G., and Emanuelsson C. (2012) Detection of crosslinks within and between proteins by LC-MALDI-TOFTOF and the software FINDX to reduce the MSMS-data to acquire for validation. PLoS ONE 7, e38927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holman J. D., Tabb D. L., and Mallick P. (2014) Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr. Protoc. Bioinformatics 46, 13 24, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petrotchenko E. V., and Borchers C. H. (2010) ICC-CLASS: isotopically-coded cleavable crosslinking analysis software suite. BMC Bioinformatics 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giese S. H., Fischer L., and Rappsilber J. (2016) A study into the CID behavior of cross-linked peptides. Mol. Cell. Proteomics 15, 1094–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gotze M., Pettelkau J., Fritzsche R., Ihling C. H., Schafer M., and Sinz A. (2015) Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom. 26, 83–97 [DOI] [PubMed] [Google Scholar]
- 31. Chu F., Baker P. R., Burlingame A. L., and Chalkley R. J. (2010) Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol. Cell. Proteomics 9, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J., Anania V. G., Knott J., Rush J., Lill J. R., Bourne P. E., and Bandeira N. (2014) Combinatorial approach for large-scale identification of linked peptides from tandem mass spectrometry spectra. Mol. Cell. Proteomics 13, 1128–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh P., Shaffer S. A., Scherl A., Holman C., Pfuetzner R. A., Larson Freeman T. J., Miller S. I., Hernandez P., Appel R. D., and Goodlett D. R. (2008) Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal. Chem. 80, 8799–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burns K. M., Sarpe V., Wagenbach M., Wordeman L., and Schriemer D. C. (2015) HX-MS2 for high performance conformational analysis of complex protein states. Protein Sci. 24, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi S., Jeong J., Na S., Lee H. S., Kim H. Y., Lee K. J., and Paek E. (2010) New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 9, 626–635 [DOI] [PubMed] [Google Scholar]
- 36. Mann M., and Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 37. Yang B., Wu Y. J., Zhu M., Fan S. B., Lin J., Zhang K., Li S., Chi H., Li Y. X., Chen H. F., Luo S. K., Ding Y. H., Wang L. H., Hao Z., Xiu L. Y., Chen S., Ye K., He S. M., and Dong M. Q. (2012) Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 [DOI] [PubMed] [Google Scholar]
- 38. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., and Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 39. Lee Y. J. (2009) Probability-based shotgun cross-linking sites analysis. J. Am. Soc. Mass Spectrom. 20, 1896–1899 [DOI] [PubMed] [Google Scholar]
- 40. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Muller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., and Makarov A. (2012) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, O111 013698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silva L. P., Yu R., Calmettes C., Yang X., Moraes T. F., Schryvers A. B., and Schriemer D. C. (2011) Conserved interaction between transferrin and transferrin-binding proteins from porcine pathogens. J. Biol. Chem. 286, 21353–21360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 43. Hoopmann M. R., Zelter A., Johnson R. S., Riffle M., MacCoss M. J., Davis T. N., and Moritz R. L. (2015) Kojak: efficient analysis of chemically cross-linked protein complexes. J. Proteome Res. 14, 2190–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Zundert G. C., Rodrigues J. P., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquiond A. S., van Dijk M., de Vries S. J., and Bonvin A. M. (2016) The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 428, 720–725 [DOI] [PubMed] [Google Scholar]
- 45. Moraes T. F., Yu R. H., Strynadka N. C., and Schryvers A. B. (2009) Insights into the bacterial transferrin receptor: the structure of transferrin-binding protein B from Actinobacillus pleuropneumoniae. Mol. Cell 35, 523–533 [DOI] [PubMed] [Google Scholar]
- 46. Merkley E. D., Rysavy S., Kahraman A., Hafen R. P., Daggett V., and Adkins J. N. (2014) Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 23, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schryvers A. B., and Stojiljkovic I. (1999) Iron acquisition systems in the pathogenic Neisseria. Mol. Microbiol. 32, 1117–1123 [DOI] [PubMed] [Google Scholar]
- 48. Gray-Owen S. D., and Schryvers A. B. (1996) Bacterial transferrin and lactoferrin receptors. Trends Microbiol. 4, 185–191 [DOI] [PubMed] [Google Scholar]
- 49. Silva L. P., Yu R. H., Calmettes C., Yang X., Moraes T. F., Schriemer D. C., and Schryvers A. B. (2012) Steric and allosteric factors prevent simultaneous binding of transferrin-binding proteins A and B to transferrin. Biochem. J. 444, 189–197 [DOI] [PubMed] [Google Scholar]
- 50. Calmettes C., Yu R. H., Silva L. P., Curran D., Schriemer D. C., Schryvers A. B., and Moraes T. F. (2011) Structural variations within the transferrin binding site on transferrin-binding protein B, TbpB. J. Biol. Chem. 286, 12683–12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Calmettes C., Alcantara J., Yu R. H., Schryvers A. B., and Moraes T. F. (2012) The structural basis of transferrin sequestration by transferrin-binding protein B. Nat. Struct. Mol. Biol. 19, 358–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.