

Abstract
Background
Characterising programs of gene regulation by studying individual protein-DNA and protein-protein interactions would require a large volume of high-resolution proteomics data, and such data are not yet available. Instead, many gene regulatory network (GRN) techniques have been developed, which leverage the wealth of transcriptomic data generated by recent consortia to study indirect, gene-level relationships between transcriptional regulators. Despite the popularity of such methods, previous methods of GRN inference exhibit limitations that we highlight and address through the lens of information theory.
Results
We introduce new model-free and non-linear information theoretic measures for the inference of GRNs and other biological networks from continuous-valued data. Although previous tools have implemented mutual information as a means of inferring pairwise associations, they either introduce statistical bias through discretisation or are limited to modelling undirected relationships. Our approach overcomes both of these limitations, as demonstrated by a substantial improvement in empirical performance for a set of 160 GRNs of varying size and topology.
Conclusions
The information theoretic measures described in this study yield substantial improvements over previous approaches (e.g. ARACNE) and have been implemented in the latest release of NAIL (Network Analysis and Inference Library). However, despite the theoretical and empirical advantages of these new measures, they do not circumvent the fundamental limitation of indeterminacy exhibited across this class of biological networks. These methods have presently found value in computational neurobiology, and will likely gain traction for GRN analysis as the volume and quality of temporal transcriptomics data continues to improve.
Keywords: Gene regulatory network, Transcriptional regulation, Gene expression
Background
Although it is well-established that networks of molecular interactions underlie critical cellular functions including development, differentiation and homeostasis, accurate reconstruction of network topologies using only gene expression data is a difficult problem that has received much attention in recent years [1–3]. Gene regulatory networks (GRNs) assume that active regulatory interactions can be captured as weighted, pairwise associations between genes and, accordingly, that complex interactions (e.g. between RNAs and proteins) may be mapped onto this level.
In 2007, the Dialogue for Reverse Engineering Assessments and Methods (DREAM) challenge was launched to promote and advance research in network-based analyses of biological data [4, 5]. Early DREAM challenges focused primarily on simulated data-sets, whereby a ‘true’ network topology was used to generate artificial gene expression data [6, 7]. Entrants developed algorithms to reconstruct this network from the expression data alone, with performance evaluated empirically against the supposed true network. Subsequent DREAM challenges have introduced experimental data, with mRNA transcript abundance quantified using qPCR (quantitative polymerase chain reaction), microarray and now high-throughput RNA-seq technologies. Although more biologically relevant than simulated data, ‘true’ network topologies for these systems remain fragmentary approximations of gene regulatory interactions and are thus inappropriate for benchmarking [5].
The University of Melbourne’s Systems Biology Laboratory has previously developed software (NAIL, Network Analysis and Inference Library [8, 9]) for inferring, analysing and visualising GRNs to assist with our efforts in transcriptional regulatory modelling [9–13]. As noted in a recent review by Novère [14], NAIL integrates several previously-published methods of GRN analysis. Most of these approaches apply a variant of Pearson’s correlation coefficient to assign edge-weights to each pair of genes, X and Y:
where and are the sample mean and standard deviation of X, respectively. Due to the nature of the GRN evaluation metrics (described later), arbitrary thresholds do not need to be assigned to identify edge weights accepted as representing physical biological interactions.
Although correlation is a straightforward method for assigning network edge weights, it has several fundamental limitations. Firstly, Pearson’s r assumes that X and Y are normally-distributed and thus it can only identify linear relationships, which can be unsuitable in the context of qPCR, microarray or RNA-seq-quantified transcript abundance. Rank-based correlation metrics such as Spearman’s ρ and Kendall’s τ coefficients are often applied to partially correct for this issue. Secondly, correlation is a symmetric measure (r X,Y=r Y,X) and thus it can not infer the directionality or causality of biological interactions, even when applied to the analysis of appropriate time-series or gene knock-down data.
Despite widespread practical utility of GRN analysis, recent studies have made broad-stroked dismissals of the field on theoretical bases [15]. Instead, we take a two-fold approach of (a) building upon previous inference methods by leveraging the latest information theoretic advancements, and (b) discussing alternative modelling approaches that better suit some scenarios. Our measures have been implemented in post-publication versions of NAIL [8, 9], and we refer the reader to [16] for our general commentary on the conundrum of reconciling theoretical versus empirical performance bounds.
Methods
This section describes how recent innovations in information theory may be effectively applied to infer network connectivity from continuous-valued biological data. Although described in the context of GRN inference, the following techniques can be applied to many other domains (e.g. inference of neural, proteomic or metabolomic networks).
Information theoretic measures for biological network inference
The fundamental measure of information theory is Shannon entropy [17] (measured in bits), which captures the expected uncertainty associated with any measurement, x, of a random variable, X:
where p(x) is the (marginal) probability distribution of X. In order to model continuous-valued mRNA transcript levels, it is necessary to instead consider the closely-related differential entropy of X (measured in nats) [18]:
where f(x) is the probability density function (PDF) of X, f(x)>0. This definition can be extended to quantify the joint entropy of X and Y:
where f(x,y) is the joint PDF of X and Y, and D⊆X×Y such that the marginal and joint PDFs of X and Y are strictly positive. The mutual information of X and Y can then be defined in terms of these two measures:
which is interpreted as the symmetric quantity of information ‘shared’ by X and Y, I D(X;Y)=I D(Y;X). Mutual information makes no assumptions regarding the distribution or linearity of relationships between transcript abundance values.
Mutual information (MI) has been applied to assigning edge weights in previous GRN studies, with MI-based network inference tools including minet [19], relevance networks [20], MRNET [21] and earlier iterations of NAIL [8]. However, these tools have generally applied variations of binning algorithms to discretise X and Y and allow for the calculation of discrete mutual information:
where and are predictors of the marginal and joint distributions of X and Y, typically implemented as simply the empirical (sampled) distribution of gene expression values. Despite the introduction of several bias-correction techniques [22–24] and the observation that for discretisations X Δ and Y Δ with bin width Δ [25], it is well-established that discretisation is a suboptimal method for handling empirical distributions of continuous-valued data [26–28]. Although earlier studies have proposed continuous estimation schemes for gene expression data, the focus has been on temporal interpolation (i.e. correcting for non-uniform sampling or missing observations [29]) rather than the quantization error introduced by previous information theoretical approaches. In the following sections we propose and describe several methods of continuous MI estimation that specifically address the latter class of errors.
Mutual information estimators for continuous-valued data
The simplest method of continuous-valued MI estimation is the Gaussian distribution model, under which multivariate joint entropy can be expressed as [25]:
where is the matrix of M expression values for N genes, and Σ X is the covariance matrix of X. The pairwise MI between genes X and Y can then be calculated as I D(X; Y)=H(X)+H(Y)−H(X,Y). Although this approach is computationally efficient and extends well to large GRNs, it reintroduces the assumption of normally-distributed and linearly-associated variables and is thus inappropriate for modelling qPCR, microarray or RNA-seq gene expression data.
An alternative to the Gaussian distribution model is to estimate the marginal and joint PDFs of X using a kernel function, K(·):
| 1 |
where h is the kernel bandwidth and X i is the i-th row of X; e.g. 〈X i;Y i〉 for the calculation of pairwise MI between genes X and Y. Two methods of kernel estimation have been introduced into the latest version of NAIL [9], including the uniform kernel:
| 2 |
where 1 {·} is the indicator function; and the Gaussian kernel, as implemented in the popular ARACNE package [30]:
Unlike the Gaussian distribution model, kernel density estimation allows for the model-free identification of non-linear relationships between gene expression levels. However, it is both statistically biased and sensitive to the selection of kernel bandwidth [31, 32]. To provide a bias-corrected and robust method for continuous-valued MI estimation, we instead implement and evaluate two variants of the Kraskov, Stögbauer and Grassberger (KSG) algorithm [33].
For each matched observation 〈x m;y m〉 of genes X and Y, the first KSG algorithm calculates the difference in expression, 〈Δ x;Δ y〉, between that observation and its K-th nearest neighbour in X×Y. The number of neighbours within max{Δ x,Δ y} of x m and y m are then calculated in their respective marginal spaces, with n x and n y defined as the mean of these counts across all matched observations. The MI of genes X and Y can then be estimated using the first KSG algorithm:
| 3 |
where ψ(·) is the digamma function:
The second KSG algorithm calculates n x and n y by considering Δ x and Δ y separately (rather than their maximum), yielding the following alternative MI estimator:
| 4 |
which is more accurate for large values of M and thus more appropriate for large (genome-wide) GRN inference. Both of these algorithms correct for bias and have been empirically demonstrated as robust to the selection of K [33].
Extensions to information theoretic network inference
Despite MI providing a non-linear and model-free approach for quantifying pairwise associations between genes, it suffers from another fundamental limitation common of correlation-based analysis: spurious inference of fully-connected subgraphs (K-vertex cliques) caused by indirect regulation of the form g 1⋯⇔g 2⇔⋯⇔g K. Under the simplified assumption that genes g 1 and g K only interact via a single path through g 2, these cliques can be reduced to a single pathway by considering the data processing inequality (DPI) [34]:
which can be expressed concisely as ‘post-processing cannot increase information’ (colloquially, ‘garbage in, garbage out’). The DPI was first leveraged in the context of GRN inference by ARACNE [30], where it reduces connectivity of the final boolean connectivity matrix (obtained by applying a minimum MI threshold to edge weights) by removing the lowest-MI edge for all fully-connected gene triplets.
Although the DPI is proven to reconstruct correct network topology given the aforementioned assumptions [30], the existence of a single isolated pathway of regulation between any pair of genes is known to be an over-simplification of most biological regulatory processes (exemplified by signaling pathway cross-talk [35, 36]). We instead propose an alternative method of resolving GRN cliques that involves calculating MI between the transcript abundances of two genes, X and Y, conditioned on abundance of a third gene, Z:
where T⊆X×Y×Z such that all marginal and joint PDFs are strictly positive. Importantly, the conditional MI between X and Y given Z can be either smaller or larger than I D(X; Y); conditioning removes redundant information between Y and Z regarding X, but identifies synergistic information that requires both Y and Z to be known. In the earlier example of indirect regulation of the form g 1⋯⇔g 2⇔⋯⇔g K, a strong MI between g 1 and g K conditioned on g 2 would indicate additional regulatory pathways between g 1 and g K that do not involve g 2. In such scenarios, application of the DPI would be inappropriate.
Conditional MI can be estimated for continuous-valued data using a similar technique. For kernel estimation, this simply involves applying Eq. (1) to approximate the marginal PDF, f(z), and joint PDFs, f(x,y,z), f(x,z) and f(y,z). The KSG algorithms can also be applied in the following form [37, 38]:
where n xz and n yz refer to counts in X×Z and Y×Z respectively.
If one has access to uniformly-sampled time series of transcript abundance data X={…,X n−1,X n,X n+1,… } (and likewise for Y), the directional transfer of information from X to Y can be determined by conditioning their MI on past observations of Y. For a Markovian process of length k, the transfer entropy from X to Y is this defined as [32, 39]:
where Y n−k:n is the length-k history of Y preceding time n. This definition generalises to non-Markovian processes for and can be extended to calculate pairwise information transfer conditioned on a third gene (or set of genes), Z:
Transfer entropy (TE) allows directional gene regulatory associations to be inferred, which can be interpreted as capturing evidence of causal relationships. Under the erroneous assumption that gene expression values are normally distributed, TE reduces to Granger Causality [40], which has previously been applied to sparse vector autoregressive (SVAR) inference of GRNs from microarray data [41].
Data and evaluation
Network inference from pairwise MI and TE was evaluated on the benchmark collection of synthetic GRNs proposed by Mendes et al. [42]. Although synthetic data is not ideal for validation, there are very few (if any) biological systems studied in sufficient detail for confident assertions regarding their true underlying topology [5]. Instead, the ‘gold standard’ Mendes models apply a simplified, bottom-up model (multiplicative Hill kinetics [43, 44], simulated in Gepasi [45]) to approximate transcriptional activity, as illustrated in Fig. 1:
| 5 |
Fig. 1.
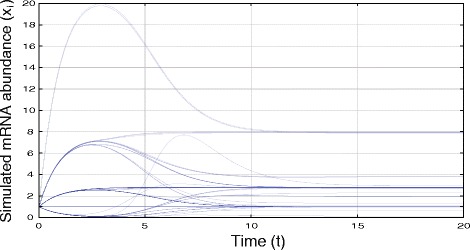
Transcriptional activity of each gene in a Century-series (100 node) scale-free Mendes network [42], simulated using multiplicative Hill kinetics as defined in 5. Each time-series was simulated until convergence (d x/d t=0) using Gepasi [45], from which gene-level correlation, MI or TE can be calculated for GRN approximation
where x i is the ‘concentration’ of gene product i, N A and N I are the number of activators and inhibitors (with concentrations A and I) and K represents the concentration at which activating/inhibiting effects are half their saturated value. The efficiency of mRNA transcription and degradation for the i-th gene are parameterised by α i and β i respectively, and n controls the sigmoidicity of the function.
The Mendes models allow benchmarking across several classes of network topologies, including including Erdős-Rényi [46] (random), Watts-Strogatz [47] (small-world) and Albert-Barabási [48] (scale-free) networks (examples provided in Fig. 2). As there is growing evidence that scale-free networks are appropriate for describing metabolic and transcriptomic interactions [49–51], 50 ‘Century’ (100-node) and 5 ‘Jumbo’ (1000-node) scale-free Mendes networks are considered [42], along with a variety of random and small-world topologies.
Fig. 2.
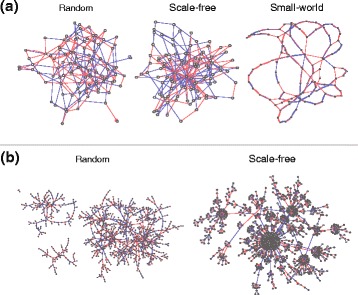
Examples of the Mendes synthetic GRNs used to benchmark the performance of the information theoretic measures proposed in this article [42], with blue and red edges representing activating and inhibiting interactions respectively. Erdős-Rényi [46] (random), Watts-Strogatz [47] (small-world) and Albert-Barabási [48] (scale-free) topologies were considered from both the (a) ‘Century’ (100-node) and (b) ‘Jumbo’ (1000-node) series. Of these topologies, there is growing evidence that scale-free networks most accurately represent the organisation of metabolic and transcriptomic regulatory systems [49–51]
The performance of each inferred GRN is evaluated using the Area Under the receiver operator characteristic (ROC) Curve (AUC) metric [52]:
where X k is the false positive rate and Y k is the true positive weight for the k-th output in the ranked list of predicted edge weights; i.e. the AUC is the trapezoidal Riemann sum of the ROC curve.
Results and discussion
In this Section we compare the performance of the directed and undirected information theoretic measures described, using the gold-standard Mendes suite of benchmark GRNs [42]. For kernel-based MI estimation we use the popular ARACNE implementation, the results of which are consistent with our implementation in NAIL [9]. Average performances (AUC) and standard deviations are presented in Table 1.
Table 1.
Performance of MI and TE-based methods of GRN inference, presented as the mean AUC (and standard deviation) across a variety of random [46], small-world [47] and scale-free [48] networks from the Mendes ‘Century’ and ‘Jumbo’ collections [42]
| Collection | Networks | Nodes | Edges | Topology | AUC (Mutual Information) | AUC (Transfer Entropy) | ||
|---|---|---|---|---|---|---|---|---|
| Kernel (ARACNE [30]) | KSG | Kernel | KSG | |||||
| CenturyRND | 50 | 100 | 200 | Random | 0.514 | 0.478 | 0.589 | 0.603 |
| (0.030) | (0.028) | (0.024) | (0.027) | |||||
| CenturySF | 50 | 100 | 200 | Scale-free | 0.475 | 0.505 | 0.526 | 0.561 |
| (0.036) | (0.033) | (0.030) | (0.030) | |||||
| CenturySW | 50 | 100 | 200 | Small-world | 0.477 | 0.471 | 0.602 | 0.598 |
| (0.035) | (0.035) | (0.028) | (0.030) | |||||
| JumboRND | 5 | 1000 | 1000 | Random | 0.473 | 0.439 | 0.540 | 0.564 |
| (0.014) | (0.013) | (0.006) | (0.009) | |||||
| JumboSF | 5 | 1000 | 1000 | Scale-free | 0.526 | 0.577 | 0.606 | 0.649 |
| (0.007) | (0.010) | (0.007) | (0.012) | |||||
Kernel-based methods apply the uniform kernel (see (2)) with bandwidth h=0.1. For KSG-based methods, KSG algorithm 1 (better suited to small networks, see (3)) was applied to ‘Century’ data and algorithm 2 (see (4)) to ‘Jumbo’ data, both with K=4 [33] and assuming length-1 Markovian processes. Gene expression time-series were simulated until convergence (d x/d t=0) using Gepasi with default parameters [45]
It is evident that the performance of MI-based inference of undirected GRNs is comparable to random guessing (Taxble 1, theoretic A U C=0.5). Application of the DPI yielded no significant improvement for any MI estimator. These results are consistent with recent studies which found that the most sophisticated GRN algorithms perform no better than simple correlation-based inference, due to the fundamental limitation of considering only pairwise expression relationships. A detailed analysis by Maetschke et al. demonstrated that the utility of these techniques is limited to small networks with star-like topologies and that exclusively contain activating or inhibiting interactions [11]. Several common regulatory network motifs have since been identified that are particularly difficult to infer [30]. Moreover, Krishnan et al. have provided a theoretical explanation as to why many non-trivial GRNs are unable to be reverse-engineered from expression data alone; i.e. multiple dissimilar networks produce indistinguishable abundance profiles due to latent protein-mediated effects [15].
The inclusion of directed information transfer to extend GRN inference yielded improved performance across all networks, with all TE-based methods performing significantly better than random (presumably because these measures are better able to capture activation and inhibition relationships, which are inherently directional). These methods outperformed other Mendes-benchmarked algorithms applying variants of correlation or MI estimation [11, 20, 30], and thus both kernel and KSG-estimated TE have been implemented for causal network inference in the latest version of NAIL [9]. To our knowledge, this is the most comprehensive set of information theoretic tools available for biological network inference. NAIL is available to download from https://sourceforge.net/projects/nailsystemsbiology/.
Conclusions
Previous GRN inference frameworks have implemented mutual information as a means of inferring pairwise gene-level associations (e.g. minet [19], relevance networks [20], MRNET [21] and ARACNE [30]). However, these tools either introduce statistical bias through discretisation of expression data or are limited to modelling undirected relationships. In this article, we have proposed and evaluated new model-free and non-linear information theoretic measures that circumvent these limitations, leading to substantial improvement in empirical performance across a benchmark set of 160 synthetic GRNs.
Although NAIL is the first GRN toolkit to incorporate the measures described in this article, it does not overcome another fundamental limitation of previous models; i.e. unambiguous network reconstruction requires that the number of time samples must be greater than the number of genes, and even the highest time resolution data-sets fall short by several orders-of-magnitude. To explore transcriptional regulation in the context of current data availability, we refer the reader to the emerging body of literature surround predictive gene expression modelling [53–55]. This class of top-down modelling leverages transcriptomic and epigenetic data as independent observations of an underlying regulatory function, thus circumventing the issue of indeterminacy inherent to GRN analysis.
Despite conflicting reports of the utility of GRNs between theoretical and empirical studies [16], we believe that this class of network inference will continue to be of widespread value for exploring fundamental regulatory processes. Moreover, the methods described in this paper can be readily applied to computational neuroscience [56, 57] and other fields of complex systems theory [58, 59]. We encourage researchers to investigate how such network abstractions can be applied to their class of biological problems.
Acknowledgements
This work was supported by an Australian Postgraduate Award [DMB]; the Australian Federal and Victoria State Governments and the Australian Research Council through the ICT Centre of Excellence program, National ICT Australia (NICTA) [DMB]; and the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036) [EJC]. The views expressed herein are those of the authors and are not necessarily those of NICTA or the Australian Research Council.
Authors’ contributions
Analysis and interpretation of data: DMB and EJC. Study design and concept: DMB and EJC. Software development and data processing: DMB. Drafting the paper: DMB. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
David M. Budden, Email: budden@csail.mit.edu
Edmund J. Crampin, Email: edmund.crampin@unimelb.edu.au
References
- 1.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8(1):565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pe’er D, Hacohen N. Principles and strategies for developing network models in cancer. Cell. 2011;144(6):864–73. doi: 10.1016/j.cell.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Kellis M, Collins JJ, Stolovitzky G, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stolovitzky G, Monroe D, Califano A. Dialogue on reverse-engineering assessment and methods. Ann N Y Acad Sci. 2007;1115(1):1–22. doi: 10.1196/annals.1407.021. [DOI] [PubMed] [Google Scholar]
- 6.Prill RJ, Marbach D, Saez-Rodriguez J, Sorger PK, Alexopoulos LG, Xue X, Clarke ND, Altan-Bonnet G, Stolovitzky G. Towards a rigorous assessment of systems biology models: the DREAM3 challenges. PloS ONE. 2010;5(2):9202. doi: 10.1371/journal.pone.0009202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stolovitzky G, Prill RJ, Califano A. Lessons from the DREAM2 challenges. Ann N Y Acad Sci. 2009;1158(1):159–95. doi: 10.1111/j.1749-6632.2009.04497.x. [DOI] [PubMed] [Google Scholar]
- 8.Hurley D, Araki H, Tamada Y, Dunmore B, Sanders D, Humphreys S, Affara M, Imoto S, Yasuda K, Tomiyasu Y, et al. Gene network inference and visualization tools for biologists: application to new human transcriptome datasets. Nucleic Acids Res. 2012;40(6):2377–98. doi: 10.1093/nar/gkr902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurley DG, Cursons J, Wang YK, Budden DM, Crampin EJ, et al. NAIL, a software toolset for inferring, analyzing and visualizing regulatory networks. Bioinformatics. 2015;31(2):277–8. doi: 10.1093/bioinformatics/btu612. [DOI] [PubMed] [Google Scholar]
- 10.Madhamshettiwar PB, Maetschke SR, Davis MJ, Reverter A, Ragan MA. Gene regulatory network inference: evaluation and application to ovarian cancer allows the prioritization of drug targets. Genome Med. 2012;4(5):1–16. doi: 10.1186/gm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maetschke SR, Madhamshettiwar PB, Davis MJ, Ragan MA. Supervised, semi-supervised and unsupervised inference of gene regulatory networks. Brief Bioinformatics. 2013;15(2):195–211. doi: 10.1093/bib/bbt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Hurley D, Schnell S. Integration of steady-state and temporal gene expression data for the inference of gene regulatory networks. PloS ONE. 2013;8(8):72103. doi: 10.1371/journal.pone.0072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wildenhain J, Crampin E. Reconstructing gene regulatory networks: from random to scale-free connectivity. IEE Proc Syst Biol. 2006;153(4):247–56. doi: 10.1049/ip-syb:20050092. [DOI] [PubMed] [Google Scholar]
- 14.Le Novère N. Quantitative and logic modelling of molecular and gene networks. Nat Rev Genet. 2015;16(3):146–58. doi: 10.1038/nrg3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan A, Giuliani A, Tomita M. Indeterminacy of reverse engineering of gene regulatory networks: the curse of gene elasticity. PLoS ONE. 2007;2(6):562. doi: 10.1371/journal.pone.0000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budden DM, Jones M. Cautionary tales of inapproximability. J Comput Biol.(in press). [DOI] [PubMed]
- 17.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27(3):379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- 18.Lazo AC, Rathie PN. On the entropy of continuous probability distributions. Inf Theory IEEE Trans. 1978;24(1):120–2. doi: 10.1109/TIT.1978.1055832. [DOI] [Google Scholar]
- 19.Meyer PE, Lafitte F, Bontempi G. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics. 2008;9(1):461. doi: 10.1186/1471-2105-9-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. In: Pacific Symposium on Biocomputing, vol. 5, World Scientific;2000;5:415–426. [DOI] [PubMed]
- 21.Meyer PE, Kontos K, Lafitte F, Bontempi G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J Bioinform Syst Biol. 2007;2007:8–8. doi: 10.1155/2007/79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paninski L. Estimation of entropy and mutual information. Neural Comput. 2003;15(6):1191–253. doi: 10.1162/089976603321780272. [DOI] [Google Scholar]
- 23.Schäfer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat Appl Genet Mol Biol. 2005;4(1):1–30. doi: 10.2202/1544-6115.1175. [DOI] [PubMed] [Google Scholar]
- 24.Schürmann T, Grassberger P. Entropy estimation of symbol sequences. Chaos. 1996;6(3):414–27. doi: 10.1063/1.166191. [DOI] [PubMed] [Google Scholar]
- 25.Cover TM, Thomas JA. Elements of information theory: John Wiley & Sons; 2012.
- 26.Ross BC. Mutual information between discrete and continuous data sets. PloS ONE. 2014;9(2):87357. doi: 10.1371/journal.pone.0087357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roulston MS. Estimating the errors on measured entropy and mutual information. Physica D Nonlinear Phenom. 1999;125(3):285–94. doi: 10.1016/S0167-2789(98)00269-3. [DOI] [Google Scholar]
- 28.Seok J, Kang YS. Mutual information between discrete variables with many categories using recursive adaptive partitioning. Sci Rep. 2015;5:1–10. doi: 10.1038/srep10981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bar-Joseph Z, Gerber GK, Gifford DK, Jaakkola TS, Simon I. Continuous representations of time-series gene expression data. J Comput Biol. 2003;10(3-4):341–56. doi: 10.1089/10665270360688057. [DOI] [PubMed] [Google Scholar]
- 30.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaiser A, Schreiber T. Information transfer in continuous processes. Physica D Nonlinear Phenom. 2002;166(1):43–62. doi: 10.1016/S0167-2789(02)00432-3. [DOI] [Google Scholar]
- 32.Schreiber T. Measuring information transfer. Phys Rev Lett. 2000;85(2):461. doi: 10.1103/PhysRevLett.85.461. [DOI] [PubMed] [Google Scholar]
- 33.Kraskov A, Stögbauer H, Grassberger P. Estimating mutual information. Phys Rev E. 2004;69(6):066138. doi: 10.1103/PhysRevE.69.066138. [DOI] [PubMed] [Google Scholar]
- 34.Beaudry NJ, Renner R. An intuitive proof of the data processing inequality. Quantum Inf Comput. 2012;12(5-6):432–41. [Google Scholar]
- 35.Guo X, Wang XF. Signaling cross-talk between TGF- β/BMP and other pathways. Cell Res. 2009;19(1):71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF- κB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 37.Frenzel S, Pompe B. Partial mutual information for coupling analysis of multivariate time series. Phys Rev Lett. 2007;99(20):204101. doi: 10.1103/PhysRevLett.99.204101. [DOI] [PubMed] [Google Scholar]
- 38.Gómez-Herrero G, Wu W, Rutanen K, Soriano MC, Pipa G, Vicente R. Assessing coupling dynamics from an ensemble of time series. 2010. https://arxiv.org/pdf/1008.0539.pdf.
- 39.Prokopenko M, Lizier JT. Transfer entropy and transient limits of computation. Sci Rep. 2014;4:1–7. doi: 10.1038/srep05394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnett L, Barrett AB, Seth AK. Granger causality and transfer entropy are equivalent for gaussian variables. Phys Rev Lett. 2009;103(23):238701. doi: 10.1103/PhysRevLett.103.238701. [DOI] [PubMed] [Google Scholar]
- 41.Fujita A, Sato JR, Garay-Malpartida HM, Yamaguchi R, Miyano S, Sogayar MC, Ferreira CE. Modeling gene expression regulatory networks with the sparse vector autoregressive model. BMC Syst Biol. 2007;1(1):39. doi: 10.1186/1752-0509-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendes P, Sha W, Ye K. Artificial gene networks for objective comparison of analysis algorithms. Bioinformatics. 2003;19(suppl 2):122–9. doi: 10.1093/bioinformatics/btg1069. [DOI] [PubMed] [Google Scholar]
- 43.Hill AV. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol (London) 1910;40:4–7. [Google Scholar]
- 44.Hofmeyr J-HS, Cornish-Bowden H. The reversible hill equation: how to incorporate cooperative enzymes into metabolic models. Comput Appl Biosci. 1997;13(4):377–85. doi: 10.1093/bioinformatics/13.4.377. [DOI] [PubMed] [Google Scholar]
- 45.Mendes P. GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput Appl Biosci. 1993;9(5):563–71. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 46.Rényi A, Erdős P. On random graphs. Publ Math. 1959;6(290-297):5. [Google Scholar]
- 47.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393(6684):440–2. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 48.Barabási AL, Albert R. Emergence of scaling in random networks. Science. 1999;286(5439):509–12. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 49.Featherstone DE, Broadie K. Wrestling with pleiotropy: genomic and topological analysis of the yeast gene expression network. Bioessays. 2002;24(3):267–74. doi: 10.1002/bies.10054. [DOI] [PubMed] [Google Scholar]
- 50.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási AL. The large-scale organization of metabolic networks. Nature. 2000;407(6804):651–4. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 51.Newman ME. The structure and function of complex networks. SIAM Rev. 2003;45(2):167–256. doi: 10.1137/S003614450342480. [DOI] [Google Scholar]
- 52.Hand DJ, Till RJ. A simple generalisation of the area under the ROC curve for multiple class classification problems. Mach Learn. 2001;45(2):171–86. doi: 10.1023/A:1010920819831. [DOI] [Google Scholar]
- 53.Budden DM, Hurley DG, Cursons J, Markham JF, Davis MJ, Crampin EJ. Predicting expression: the complementary power of histone modification and transcription factor binding data. Epigenetics Chromatin. 2014;7(1):1–12. doi: 10.1186/1756-8935-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Budden DM, Hurley DG, Crampin EJ. Predictive modelling of gene expression from transcriptional regulatory elements. Brief Bioinform. 2014;16(4):616–28. doi: 10.1093/bib/bbu034. [DOI] [PubMed] [Google Scholar]
- 55.Budden DM, Hurley DG, Crampin E. Modelling the conditional regulatory activity of methylated and bivalent promoters. Epigenetics Chromatin. 2015;8(1):1–10. doi: 10.1186/s13072-015-0013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindner M, Vicente R, Priesemann V, Wibral M. TRENTOOL: A MATLAB open source toolbox to analyse information flow in time series data with transfer entropy. BMC Neurosci. 2011;12(1):119. doi: 10.1186/1471-2202-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lizier JT, Heinzle J, Horstmann A, Haynes JD, Prokopenko M. Multivariate information-theoretic measures reveal directed information structure and task relevant changes in fmri connectivity. J Comput. 2011;30(1):85–107. doi: 10.1007/s10827-010-0271-2. [DOI] [PubMed] [Google Scholar]
- 58.Barnett L, Bossomaier T. Transfer entropy as a log-likelihood ratio. Phys Rev Lett. 2012;109(13):138105. doi: 10.1103/PhysRevLett.109.138105. [DOI] [PubMed] [Google Scholar]
- 59.Boedecker J, Obst O, Lizier JT, Mayer NM, Asada M. Information processing in echo state networks at the edge of chaos. Theory Biosci. 2012;131(3):205–13. doi: 10.1007/s12064-011-0146-8. [DOI] [PubMed] [Google Scholar]