

Abstract
Predicting viral evolution has proven to be a particularly difficult task, mainly owing to our incomplete knowledge of some of the fundamental principles that drive it. Recently, valuable information has been provided about mutation and recombination rates, the role of genetic drift and the distribution of mutational, epistatic and pleiotropic fitness effects. However, information about the topography of virus' adaptive landscapes is still scarce, and to our knowledge no data has been reported so far on how its ruggedness may condition virus' evolvability. Here, we show that populations of an RNA virus move efficiently on a rugged landscape and scape from the basin of attraction of a local optimum. We have evolved a set of Tobacco etch virus genotypes located at increasing distances from a local adaptive optimum in a highly rugged fitness landscape, and we observed that few evolved lineages remained trapped in the local optimum, while many others explored distant regions of the landscape. Most of the diversification in fitness among the evolved lineages was explained by adaptation, while historical contingency and chance events contribution was less important. Our results demonstrate that the ruggedness of adaptive landscapes is not an impediment for RNA viruses to efficiently explore remote parts of it.
Keywords: adaptive landscapes, adaptation, contingency, experimental evolution, stochastic escape, virus evolution
1. Background
In its most visually appealing three-dimensional version, the adaptive or fitness landscape metaphor describes the process of evolution as the movement of populations in a surface in which height is proportional to fitness. High-fitness peaks are separated by valleys, that contain genotypes of low fitness, or by flat surfaces in which genotypes of equal fitness drift neutrally. Evolution can be seen as a walk in this mountainous landscape and adaptation always implies moving populations from low- to high-fitness peaks [1–3]. In reality, the landscape is much more complex; highly dimensional and not static but fluctuating [4,5]. The shape of the landscape plays a major role in the evolutionary process. If there is a single adaptive peak, then different evolving populations will eventually reach the same solution. By contrast, if multiple peaks exist, i.e. the landscape is rugged, different populations may reach different peaks, depending on the peak accessibility relative to their starting position. In other words, evolution may be predictable in as much as the number of accessible trajectories from a particular point in the landscape is limited [3]. Indeed, the number of peaks in a landscape ultimately depends on whether mutations interact multiplicatively or epistatically in determining fitness [3]. Especially, if epistasis takes the form of sign [6] or reciprocal sign [7], landscapes are highly rugged and the number of accessible pathways will be limited [8–10].
Despite tremendous experimental efforts, the amount of information available on basic evolutionary parameters for RNA viruses remains limited [11–15]. A pervasive observation for RNA viruses is that mutations interact epistatically [16–23], with many pairs fulfilling the conditions of sign and reciprocal sign epistasis [21–23], thus, suggesting that rugged adaptive landscapes shall be the norm for RNA viruses. In a series of recent studies, we have characterized a portion of the fitness landscape underlying the adaptation of Tobacco etch virus (TEV; genus Potyvirus, family Potyviridae) to its experimental host Arabidopsis thaliana L. ecotype Ler-0. The ancestral tobacco-adapted and the evolved Arabidopsis-adapted, hereafter TEV-At17, isolates differ in their fitness and symptoms in both hosts as well as in the way they interact with Arabidopsis' regulome [24]. TEV-At17 shows approximately 10-fold higher infectivity, two logs greater accumulation and induced visible and more severe symptoms, including stunting, etching and leaf malformation. The set of up- and downregulated genes upon infection with TEV-At17 isolate was almost three times larger than those altered by the ancestral TEV [24]. An analysis of the enriched biological processes whose expression was altered revealed that TEV-At17 downregulated developmental and metabolic processes, innate immunity and responses to abiotic stresses and to infection [24]. All these differences are caused by only six mutations, in five different cistrons, fixed in TEV-At17 compared with the ancestral one [24]. Symptoms and increases in virus accumulation were triggered by non-synonymous mutations VPg/L2013F, and non-synonymous mutations P3/A1047 V and 6K1/T1210M additively exacerbated the severity of symptoms in the presence of mutation VPg/L2013F but had no effect in its absence. The VPg protein plays a central role in TEV production and spread by triggering CAP-induced translation via interaction with the eukaryotic translation initiation factor 4E [25].
The TEV genome is composed by a (+)ssRNA molecule of 9539 nucleotides that encodes for 11 multifunctional peptides [26]. For such a genome length, the size of the adaptive landscape is 49539 genotypes; within which, only 64 correspond to combinations of the six mutations that made TEV-At17 different from its tobacco-adapted antecessor [24]. Lalić & Elena [22] characterized the topography of the empirical fitness landscape defined by five of these six mutations (table 1). To do so, we created by site-directed mutagenesis on a cDNA infectious clone containing the ancestral TEV genome all 25 = 32 possible genotypes contained in this small portion of the fitness landscape [22]. This resulting empirical landscape was very rugged in Arabidopsis (figure 1), characterized by the existence of two disconnected fitness peaks that are accessible throughout certain mutational paths and surrounded by holes corresponding to non-viable genotypes. The maximum fitness peak of this local landscape corresponds to the genotype that carries non-synonymous mutation P3/C3140U and synonymous mutation NIa-Pro/C6906U (table 1). Hereafter and for the sake of abbreviating the terminology, genotypes are going to be represented as binary strings in which a 0 will represent the wild-type alleles and a 1 the mutant allele. By using this terminology, the ancestral tobacco-adapted genotype can be represented as 00000 while the TEV-At17 genotype carrying all five mutations by 11111. The global optimum can then be written as 01001.
Table 1.
The five point mutations used to generate the five genotypes included in this study. (Additional details on these mutations can be found elsewhere [22,24].)
| label | mutation | gene | amino acid change |
|---|---|---|---|
| 10000 | U357C | P1 | synonymous |
| 01000 | C3140U | P3 | A1047V |
| 00100 | C3629U | 6K1 | T1210M |
| 00010 | C6037U | VPg | L2013F |
| 00001 | C6906U | NIa-Pro | synonymous |
Figure 1.

The empirical topography of the local fitness landscape describing adaptation of a tobacco-adapted TEV to its novel host Arabidopsis thaliana Ler-0. Each bit string represents a viral genotype, with 0s meaning wild-type alleles and 1s meaning mutant alleles. Genotypes are ordered top-down starting from the ancestral wild-type virus (00000) and finishing with the genotype that carries the five mutations analysed in this study (11111). Each row represents genotypes with equal numbers of mutations. Genotypes marked in red are those with fitness lower than the ancestral (including lethals), genotypes in black have equal fitness than wild-type, and genotypes marked in green are significantly fitter than the tobacco-adapted wild-type [22]. Solid green lines represent likely adaptive walks (always connecting beneficial genotypes) and dashed black lines pathways that involve genotypes not significantly better than the ancestral (neutral pathways). The landscape is highly epistatic and contains two disconnected adaptive peaks corresponding to genotypes 01001 and 00110 (see table 1 for details on each mutation). Peak 01001 corresponds to the maximum fitness of this local landscape. (Online version in colour.)
A concept intimately linked to the shape of the fitness landscape is that of evolutionary contingency, i.e. the effect of past evolutionary events on the evolution of populations in the future [2]. This is a highly relevant question in the context of evolutionary theory that roots back to the late Steven J. Gould ‘replying life's tape’ gedanken experiment [27]. Gould argued that evolution would be not repeatable and that re-starting evolution from some past-time point would lead to a pathway radically different. How repeatable is virus evolution? How strongly does it depend on past-history events? What factors, if any, may help to better predict the outcome of evolution? Here, we sought to answer these questions in the context of adaptation of TEV to A. thaliana. More precisely, to explore the effect that ruggedness and the precise location of populations on the landscape (i.e. contingency) have on the evolvability of TEV populations, we have chosen five viable genotypes from the landscape shown in figure 1. Genotypes 01000, 00001 and 01101 are located at one mutational step from the local maximum fitness peak 01001 (figure 1); genotype 00010 is located at three mutational steps from this local maximum but only one step away from the second lower peak (00110; figure 1). Genotype 10110 is five mutational steps away from the local maximum (figure 1). We hypothesize that, if historical contingency plays a major role, then genotypes closer to the local maximum will be trapped into its basin of attraction and further evolution will probably result in reaching the 01001 peak by fixing the missing mutations. By contrast, genotypes located farther away will have less chance of reaching the 01001 peak and may move into different regions of the global landscape. To test these predictions, we generated five independent evolution lineages from each of the five genotypes. Evolution was done by serial passages in Arabidopsis ecotype Ler-0 as described in Agudelo-Romero et al. [24]. After five passages, we evaluated the fitness of the 25 evolved lineages, as well as of the five ancestral genotypes, all relative to the 01001 local optimum genotype.
2. Material and methods
(a). Generation of viral genotypes
All five TEV genotypes used in this study were constructed by successive rounds of site-directed mutagenesis starting from template plasmid pMTEV that contains a full copy of the genome of a TEV isolate from tobacco (GenBank accession DQ986288) [28], using mutagenic primers [22] with specific single-nucleotide mismatches and Phusion® High-Fidelity DNA polymerase (Finnzymes) following the instructions provided by the manufacturer. The PCR-mutagenesis profile consisted of 30 s denaturation at 98°C, followed by 30 cycles of 10 s at 98°C, 30 s at 60°C and 3 min at 72°C, ending with 10 min elongation at 72°C. Next, the PCR-mutagenesis products were incubated with DpnI (Fermentas) for 2 h at 37°C to digest the methylated parental DNA template. Escherichia coli DH5α electrocompetent cells were transformed with 2 µl of these reactions products and plated out on Luria-Bertani (LB) agar supplemented with 100 µg ml−1 ampicillin. Bacterial colonies were inoculated in 8 ml LB-ampicillin liquid medium and grown for 16 h in an orbital shaker (37°C, 225 rpm). Plasmid preparations were carried out using Pure YieldTM Plasmid Maxiprep System (Promega) and following the manufacturer's instructions. Incorporation of mutation was confirmed by sequencing an approximately 800 bp fragment circumventing the mutagenized nucleotide. The plasmid DNA was BglII linearized and in vitro transcribed using mMESSAGE mMACHINE® SP6 Kit (Ambion) in order to obtain infectious RNA of each virus genotype [29].
(b). Plant inoculations and evolution passages
Nicotiana tabacum cv. Xanthi NN plants were used for production of a large stock of virus particles from each of the five genotypes. Batches of eight-week old N. tabacum plants were inoculated with 5 µg of RNA of each viral genotype by abrasion of the third true leaf. Ten days post-inoculation (dpi), the whole infected plants were collected and pooled for each virus genotype. Plant tissue was frozen with liquid N2, homogenized using mortar and pestle and aliquoted in 1.5 ml tubes. Saps were prepared by adding 1 ml of 50 mM potassium phosphate buffer (pH 8.0) per gram of homogenized plant tissue, centrifuged at 4°C and 10 000g for 10 min and the upper liquid phase taken and mixed with 10% Carborundum (w/v).
To start the evolution experiment, between 15 and 25 A. thaliana Ler-0 plants at growth stage 3.5 according to Boyes scale [30] were mechanically inoculated with the previous virus preparations. Plants were maintained in a Biosafety Level-2 greenhouse at 25°C and a 16 h light period. Infection status was determined by one step RT-PCR 14 dpi as described previously [31]. Five plants infected with each genotype were selected to initiate the independent evolution lineages. Infected whole plants were collected at 21 dpi, which corresponds to the transmission time used in previous experiments [24]. In total, 200 mg of tissue were homogenized in liquid N2 using a mortar and a pestle, mixed with 1 ml of inoculation buffer (50 mM potassium phosphate pH 8.0, 10% Carborundum w/v) and used to inoculate the next set of A. thaliana Ler-0 plants. This procedure was repeated four more times, adding up to a total of five serial passages.
(c). Virus genomic RNA purification, quantification and sequencing
RNA extraction from 100 mg of tissue per plant was performed using InviTrap® Spin Plant RNA Mini Kit (Invitek) following the manufacturer's instructions. The concentration of total plant RNA extracts was adjusted to 50 ng µl−1 for each sample and the quantification of viral load was done with real time RT-qPCR [31]. Quantification amplifications were done using an ABI StepOnePlusTM Real-Time PCR System (Applied Biosystems) with the One Step SYBR® Prime ScriptTM RT-PCR kit II Perfect Real Time (Takara) as follows: 5 min at 42°C, 10 s at 95°C following 40 cycles of 5 s at 95°C and 34 s at 60°C. Quantifications were performed in duplicate for each sample.
The consensus genomic sequences were obtained as described by Aguelo-Romero et al. [24]. In short, RT was performed using M-MuLV (Thermo Scientific) reverse transcriptase and a reverse primer outside the region to be PCR-amplified for sequencing. PCR was then performed with Phusion DNA polymerase (Thermo Scientific) and appropriate sets of primers. Sanger sequencing was performed at GenoScreen (Lille, France: www.genoscreen.com) with an ABI3730XL DNA analyzer. Chromatogram visualization and contigs assembling were done with Geneious v. R9.1 (www.geneious.com).
(d). Fitness determinations
Total RNA was extracted and virus accumulation was quantified by RT-qPCR as described above as in [31]. Virus accumulation (picogram of TEV RNA per 100 ng of total plant RNA) was quantified at t = 15 dpi for the five mutant genotypes and the reference 01001 viruses. These values were then used to compute the fitness of the mutant genotypes relative to the reference 01001 genotype using the expression 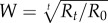 , where R0 and Rt are the ratios of accumulations estimated for the mutant and reference viruses, respectively, at inoculation and after t days of growth [29].
, where R0 and Rt are the ratios of accumulations estimated for the mutant and reference viruses, respectively, at inoculation and after t days of growth [29].
The magnitude of effects in ANOVA models was evaluated using the 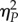 statistic that represents the proportion of total variability attributable to a given factor while controlling for the other factors. Conventionally, values
statistic that represents the proportion of total variability attributable to a given factor while controlling for the other factors. Conventionally, values 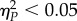 are considered as small,
are considered as small, 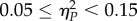 as medium and
as medium and 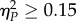 as large effects. When multiple tests of the same null hypothesis were performed, as for instance when comparing the fitness of independently evolved lineages versus the fitness of the starting genotype or the fitness of the nearest peak, significance levels were adjusted using the Holm–Bonferroni correction. In these cases, only the less significant p-values will be provided: values above this corrected cut-off will not be considered as significant. All statistical analyses have been done with SPSS v. 23 (IBM Inc.).
as large effects. When multiple tests of the same null hypothesis were performed, as for instance when comparing the fitness of independently evolved lineages versus the fitness of the starting genotype or the fitness of the nearest peak, significance levels were adjusted using the Holm–Bonferroni correction. In these cases, only the less significant p-values will be provided: values above this corrected cut-off will not be considered as significant. All statistical analyses have been done with SPSS v. 23 (IBM Inc.).
3. Results and discussion
To evaluate the contribution of historical events, selection and chance on the outcome of future evolution of viral populations, we selected five TEV genotypes located at different positions in the empirical fitness landscape shown in figure 1. Three lineages (01000, 00001 and 01101) were located one step away from the local fitness optimum (01001), one genotype (01001) was located at three mutational steps from the local fitness optimum but only one step away from a second lower fitness peak (00110), and one genotype (10110) was five mutational steps from the local fitness optimum. Five independent evolution lineages were founded with each one of these genotypes and evolved by five serial passages in the novel host A. thaliana. After this phase of experimental evolution, the fitness of all 25 evolved lineages was measured relative to the fitness of the local fitness optimum genotype 01001. The results of all these fitness assays are shown in figure 2. Twenty out of the 25 evolved lineages reached fitness values that were significantly greater than that of the local maximum 01001 genotype, indicated with red lines in figure 2 (two-samples t-tests; all corrected p ≤ 0.002). One lineage evolved from 01000 and another from 00010 reached fitness values indistinguishable from this local maximum (two-samples t-tests; both corrected p ≥ 0.050). Three lineages evolved from genotype 00001 and one from genotype 00010 retained fitness values that were still significantly inferior to the local maximum (two-samples t-test; all corrected p ≤ 0.001).
Figure 2.

Evolution of fitness of TEV genotypes at different distances from the local optima. Each genotype is indicated using a binary string in which 0 indicates the wild-type allele and 1 the mutant allele. The red solid line represents the fitness of the local optima (genotype 01001) and the red dashed lines represent its ±1 s.e.m. (Online version in colour.)
(a). Parallelism and divergence in fitness
Figure 2 suggests the existence of evolutionary phenotypic parallelism that depends on the starting location on the landscape. To assess the contribution of genetic divergence versus phenotypic parallelism, we used the index 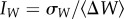 , where
, where 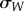 is the genetic standard deviation for fitness between the evolved lineages, a measure of genetic divergence among lineages, and
is the genetic standard deviation for fitness between the evolved lineages, a measure of genetic divergence among lineages, and 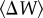 is the average change in fitness between the evolved lineages and the ancestral stage, a measure of phenotypic parallelism [32]. For lineages evolved from genotype 00001, IW > 1, indicating little parallelism in fitness relative to the observed amount of genetic divergence among lineages (table 2 and figure 2). In all other cases, IW < 1, indicating that phenotypic parallelism was larger than expected given the amount of genetic divergence among lineages (table 2 and figure 2), with parallelism being most important among lineages derived from genotype 10110 and less important among lineages evolved from genotype 00010. This suggests that, to some extent, the result of evolution actually depends on the starting point in the landscape.
is the average change in fitness between the evolved lineages and the ancestral stage, a measure of phenotypic parallelism [32]. For lineages evolved from genotype 00001, IW > 1, indicating little parallelism in fitness relative to the observed amount of genetic divergence among lineages (table 2 and figure 2). In all other cases, IW < 1, indicating that phenotypic parallelism was larger than expected given the amount of genetic divergence among lineages (table 2 and figure 2), with parallelism being most important among lineages derived from genotype 10110 and less important among lineages evolved from genotype 00010. This suggests that, to some extent, the result of evolution actually depends on the starting point in the landscape.
Table 2.
Evaluation of the relative extent of genetic divergence versus phenotypic parallelism among lineages evolved from the same starting genotypes. (Fitness is expressed relative to the local optima genotype 01001. Errors represent ±1 s.d.)
| starting genotype | initial fitness (W0) | average change in fitness 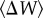
|
genetic variance among lineages 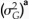
|
parallelism index (IW) |
|---|---|---|---|---|
| 01000 | 0.934 ± 0.153 | 0.570 ± 0.429 | 0.068 ± 0.043 | 0.458 ± 0.491 |
| 00010 | 0.899 ± 0.171 | 0.489 ± 0.623 | 0.184 ± 0.117 | 0.878 ± 1.397 |
| 00001 | 0.847 ± 0.124 | 0.275 ± 0.543 | 0.158 ± 0.100 | 1.446 ± 3.314 |
| 10110 | 0.875 ± 0.178 | 1.116 ± 0.254 | 0.005 ± 0.003 | 0.063 ± 0.035 |
| 01101 | 0.882 ± 0.168 | 0.955 ± 0.283 | 0.012 ± 0.008 | 0.114 ± 0.070 |
aMaximum-likelihood estimators.
(b). Contributions of selection, chance and historical contingency to the outcome of evolution
Phenotypic diversification between the evolved lineages may result from the contribution of three evolutionary factors: adaptation by natural selection, chance events such as mutation and drift and historical contingency. Their contribution to the evolutionary change is still debated [33] and controversial for viruses [34–36]. To disentangle the contribution of these three factors to the above patterns of TEV fitness, we followed the statistical methods developed by Travisano et al. [33]. Figure 3a shows the fitness of each independent lineage before and after the five passages of experimental evolution. The observed pattern is consistent with historical effects (differences in ancestral fitness), which are mostly erased after evolution. To formalize the contributions of adaptation, chance and history, we estimated the change in grand mean fitness 0.681 ± 0.430 (1 s.d.), which reflects adaptation, and by doing a nested ANOVA (table 3) we estimated the variance components corresponding to chance (differences among lineages within genotypes) and history (differences among genotypes). Figure 3b shows the relative contributions of the above-mentioned factors. After five passages of experimental evolution, the grand mean fitness of the 25 populations has significantly increased (z-test; p < 0.001), thus suggesting that a further adaptation took place. The effect of history was still significant, although smaller than the effect of chance (table 3; compare the 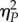 values). This observation is mainly owing to the presence of the four aforementioned lineages whose fitness was not improved relative to the reference genotype 01001 at the local fitness maximum (figure 2 and dots close to the red line in figure 3a). The effect of chance was highly significant (table 3) and had a great impact on the observed fitness variability. These results are congruent to the previously described weak influence of the past experimental evolutionary histories into future adaptation of TEV to another, novel host [35].
values). This observation is mainly owing to the presence of the four aforementioned lineages whose fitness was not improved relative to the reference genotype 01001 at the local fitness maximum (figure 2 and dots close to the red line in figure 3a). The effect of chance was highly significant (table 3) and had a great impact on the observed fitness variability. These results are congruent to the previously described weak influence of the past experimental evolutionary histories into future adaptation of TEV to another, novel host [35].
Figure 3.
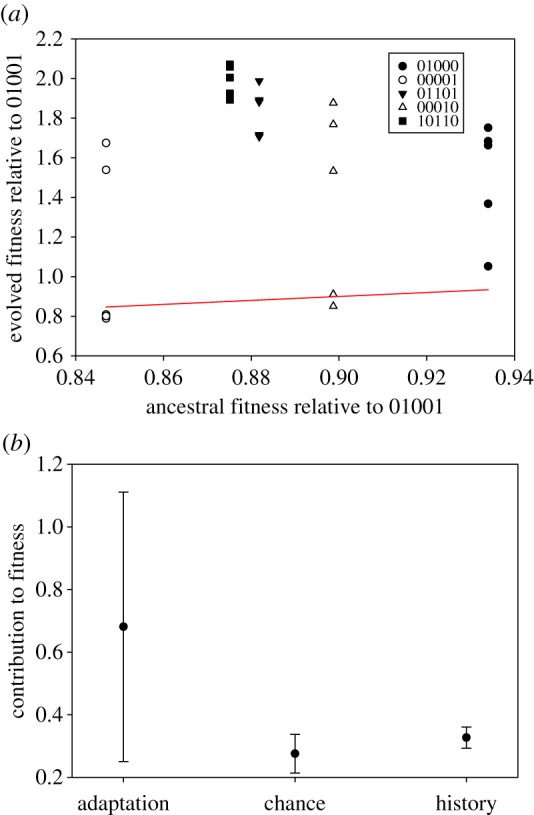
Disentangling the effects of adaptation, chance and history in the evolution and diversification of TEV experimental populations evolved in Arabidopsis thaliana Ler-0. (a) Derived versus ancestral values for mean fitness in 25 experimental TEV populations after five serial passages in A. thaliana Ler-0 plants. The red line corresponds to the hypothesis of fitness differences only explained by historical effects with no contribution of chance and adaptation. Each genotype is represented by different symbols (legend). (b) Relative contributions of adaptation, chance, and history to mean fitness evolution. Error bars represent ±1 s.d. (Online version in colour.)
Table 3.
Nested analysis of variance to estimate the contribution of chance and history to the observed pattern of diversification in viral fitness.
| source of variation | SSa | d.f. | F | p-values | 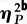 |
1−βc | variance componentd | s.d. of variance componentd |
|---|---|---|---|---|---|---|---|---|
| intercept | 122.963 | 1 | 574.703 | <0.001 | 0.966 | 1 | ||
| genotype (history) | 4.865 | 4 | 5.684 | 0.003 | 0.532 | 0.942 | 0.0759 | 0.0619 |
| lineage (chance) | 4.279 | 20 | 771.342 | <0.001 | 0.998 | 1 | 0.1068 | 0.0338 |
| error | 0.007 | 25 | 2.770×10−4 | 7.845×10−5 |
aType III sum of squares.
bPartial η2 measures the magnitude of the effect.
cStatistical power of the test.
dMaximum-likelihood estimators of the corresponding variance component and their s.d.
(c). Genotypic diversification
The question now is whether contingency is still detectable at the genotypic level, supporting our previous hypothesis that the proximity to the local fitness maximum would condition the set of mutations fixed afterwards. To do so, we have obtained the population consensus sequence of the 25 evolved lineages. Noteworthy, a population consensus sequence represents an ‘average genotype’ in which only mutations over a given frequency in the population would be detected, although they may not have real existence in the population. Figure 4 shows a schematic representation of the mutation fixed within each lineage. None of the lineages starting from genotype 01000 fixed the mutation 00001 that would bring them into the local fitness optima; even lineage 4 that had a fitness value indistinguishable from the fitness optima (see above), has different mutations. Interestingly, lineages 4 and 5 had both fixed the same pair of mutations: P1/G418A that results in P1/G92S conservative amino acid change and synonymous mutation NIb/G7668A. A more complex situation happens with lineages evolved from genotype 00001. Lineages 1 and 2 had fixed mutation 01000, although lineage 1 had reverted mutation 00001 to the ancestral allele. Likewise, lineage 3 has also reverted mutation 00001 to the ancestral allele. Thus, only lineage 2 has evolved to become 01001 and represents a clear case of evolutionary determinism at the genomic level. However, its fitness was significantly greater than the local maximum fitness, probably owing to fitness effects associated with the additional mutation fixed at the coat protein (CP) (figure 4). Interestingly, lineages 3 and 5 both fixed mutation 00010. Finally, lineages 1 and 4 had also fixed mutation G9444A in the 3′ untranslated region of the genome. None of the lineages evolved from genotype 01101 had reverted mutation 00100 to the ancestral allele. No convergent mutations have been fixed in these lineages either. None of the lineages starting from genotype 00010 had neither reverted this mutation nor fixed mutations 01000 or 00001. Instead, three examples of new convergent mutations have been seen. First, lineages 2 and 4 have both fixed mutation P3/U2537C that results in amino acid changes P3/I798T (non-polar to polar). Second, lineages 1 and 2 have fixed mutation CP/G8556C that results in replacement CP/K2804N (basic by polar). More interestingly, lineages 1, 2, 4 and 5 have all fixed mutation CP/G9103U that results in replacement CP/A2987S (non-polar to polar). Finally, all evolved lineages starting from genotype 10110 had fixed synonymous mutation CI/A3975U. In addition, lineages 1, 2, 4 and 5 have fixed mutation VPg/A6175C, which also results in a strong change VPg/N2011H (polar by basic) replacement. Therefore, we conclude that the presence of a given mutation, or set of mutations, in the mutational path towards a local adaptive peak, do not determine the fixation of the remaining mutations. By contrast, in 24 out of 25 cases, lineages fixed new sets of mutations, exploring distant parts of the landscape. Still, some level of parallelism at the molecular level has been detected, with some mutations being fixed in more than one lineage.
Figure 4.

Mutations fixed on the viral lineages after five additional passages of experimental evolution in A. thaliana Ler-0. The different cistrons in the TEV genome are indicated in red in the top of the figure. Mutations originally present in each genotype are indicated by green cells. Black cells indicate new mutations fixed during the evolution experiment. (Online version in colour.)
4. Conclusion
In the context of our ongoing work to understand the evolutionary mechanisms driving the adaptation of emerging RNA viruses to novel hosts right after they spill over from their reservoirs, here, we have described the results of experiments specifically designed to test the effect of adaptive landscape's ruggedness in the evolutionary fate of emerging RNA viruses. This is, to our knowledge, the first time such specific endeavour has been taken for a plant RNA virus. Our results suggest that, regardless of the proximity of the viral populations to local adaptive peaks, chances exist that they may move away from the basin of attraction of the nearby peak and explore new regions of the landscape. We have observed that these movements are mostly predicted by adaptation, whereas chance events and historical contingency had a minor yet significant contribution; the latter being particularly important in some evolutionary lineages. Parallelism at the genotypic and phenotypic levels has been observed. Its contribution relative to the extent of genetic variation was dependent on the starting genotype, although this dependence was not correlated to the proximity of the starting genotype to the local fitness peak.
A question that puzzles us is why historical contingency did not have a stronger impact on evolution; especially for populations that were only one mutational step away from the local adaptive peak? There are two, non-mutually exclusive, possibilities. First, the empirical landscape restricted to combinations of only five mutations [22] was not the only possible solution for TEV to adapt to A. thaliana, yet other, perhaps even better, options existed that were not explored in the original evolution experiment [24]. In this regard, it is worth noting that the original adaptation experiment resulted in a single, successful evolutionary lineage, that is to say, with no evidence of reproducibility for this particular adaptive walk. A second, more tantalizing possibility is that close to a local optimum the adaptive dynamics slows down and the probability of a stochastic escape becomes comparable to that of an adaptive process. Then, the population wanders around in genotypic space, starting a new adaptive walk after every successful escape [37,38]. In any case, these results once again confirm the tremendous adaptive potential and evolvability of RNA viruses, probing them as good supporters of Gould vision about the unpredictability of evolution. Gathering information on the structure and topology of adaptive landscapes for RNA viruses and how they modulate virus evolution may be central for developing new antiviral strategies, personalized clinical treatments and predicting and containing the emerging diseases of viral aetiology.
Acknowledgements
We thank Francisca de la Iglesia for taking care of all the plant logistics and for conducting the evolution experiments and Paula Agudo for technical assistance.
Data accessibility
Data available on Dryad (http://dx.doi.org/10.5061/dryad.17s38).
Authors' contributions
H.C. performed the fitness assays, sequenced the evolved lineages and analysed the sequence data. J.L. produced the mutant genotypes by site-directed mutagenesis and tested their viability. S.F.E. conceived and designed the study, analysed the data and wrote the paper.
Competing interests
We have no competing interests.
Funding
This project was funded by grant nos. BFU2012-30805 and BFU2015-65037P from the Spanish Ministry of Economy and Competitiveness (MINECO), PROMETEOII/2014/021 from Generalitat Valenciana and EvoEvo (ICT610427) from the European Commission 7th Framework Program to S.F.E. H.C. was supported by contract BES2013-065595 from MINECO. J.L. was supported by a JAE-pre contract from CSIC.
References
- 1.Wright S. 1932. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc. 6th Int. Congress Genet. 1, 356–366. [Google Scholar]
- 2.Colegrave N, Buckling A. 2005. Microbial experiments on adaptive landscapes. Bioessays 27, 1167–1173. ( 10.1002/bies.20292) [DOI] [PubMed] [Google Scholar]
- 3.De Visser JAGM, Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. ( 10.1038/nrg3744) [DOI] [PubMed] [Google Scholar]
- 4.Mustnen V, Lässig M. 2009. From fitness landscapes to seascapes: non-equilibrium dynamics of selection and adaptation. Trends Genet. 25, 111–119. ( 10.1016/j.tig.2009.01.002) [DOI] [PubMed] [Google Scholar]
- 5.Ogbunugafor CB, Wylie CS, Kiakite I, Weinreich DM, Hartl DL. 2016. Adaptive landscape by environment interactions dictate evolutionary dynamics in models of drug resistance. PLoS Comput. Biol. 12, e1004710 ( 10.1371/journal.pcbi.1004710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinreich DM, Watson RA, Chao L. 2005. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution 59, 1165–1174. [PubMed] [Google Scholar]
- 7.Poelwijk FJ, Tanase-Nicola S, Kiviet DJ, Tans SJ. 2011. Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J. Theor. Biol. 272, 141–144. ( 10.1016/j.jtbi.2010.12.015) [DOI] [PubMed] [Google Scholar]
- 8.Kauffman SA, Levin S. 1987. Towards a general theory of adaptive walks on rugged landscapes. J. Theor. Biol. 128, 11–45. ( 10.1016/S0022-5193(87)80029-2) [DOI] [PubMed] [Google Scholar]
- 9.Szendro IG, Schenk MF, Franke J, Krug J, de Visser JAGM. 2013. Quantitative analysis of empirical fitness landscapes. J. Stat. Mech. 2013, P01005 ( 10.1088/1742-5468/2013/01/P01005) [DOI] [Google Scholar]
- 10.Weinreich DM, Lan Y, Wylie SC, Heckendorn RB. 2013. Should evolutionary geneticists worry about higher-order epistasis? Curr. Opin. Genet. Dev. 23, 100–7007. ( 10.1016/j.gde.2013.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes EC. 2009. The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Syst. 40, 353–372. ( 10.1146/annurev.ecolsys.110308.120248) [DOI] [Google Scholar]
- 12.Elena SF, Lalić J. 2013. Plant RNA virus fitness predictability: contribution of genetic and environmental factors. Plant Pathol. 62, S10–S18. ( 10.1111/ppa.12102) [DOI] [Google Scholar]
- 13.Wasik BR, Turner PE. 2013. On the biological success of viruses. Annu. Rev. Microbiol. 67, 519–541. ( 10.1146/annurev-micro-090110-102833) [DOI] [PubMed] [Google Scholar]
- 14.Elena SF, Sanjuán R. 2007. Virus evolution: insights from an experimental approach. Annu. Rev. Ecol. Evol. Syst. 38, 27–52. ( 10.1146/annurev.ecolsys.38.091206.095637) [DOI] [Google Scholar]
- 15.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76, 159–216. ( 10.1128/MMBR.05023-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burch CL, Chao L. 1999. Evolution by small steps and rugged landscapes in the RNA virus ϕ6. Genetics 151, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burch CL, Chao L. 2004. Epistasis and its relationship to canalization in the RNA virus ϕ6. Genetics 167, 559–567. ( 10.1534/genetics.103.021196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ. 2004. Evidence for positive epistasis in HIV-1. Science 306, 1547–1550. ( 10.1126/science.1101786) [DOI] [PubMed] [Google Scholar]
- 19.Sanjuán R, Moya A, Elena SF. 2004. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Natl Acad. Sci. USA 101, 15 376–15 379. ( 10.1073/pnas.0404125101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. 2010. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics 185, 293–303. ( 10.1534/genetics.109.112458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalić J, Elena SF. 2012. Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 109, 71–77. ( 10.1038/hdy.2012.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lalić J, Elena SF. 2015. The impact of high-order epistasis in the within-host fitness of a positive-sense plant RNA virus. J. Evol. Biol. 28, 2236–2247. ( 10.1111/jeb.12748) [DOI] [PubMed] [Google Scholar]
- 23.Hillung J, Cuevas JM, Elena SF. 2015. Evaluating the within-host fitness effects of mutations fixed during virus adaptation to different ecotypes of a new host. Phil. Trans. R. Soc. B 370, 20140292 ( 10.1098/rstb.2014.0292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agudelo-Romero P, Carbonell P, Pérez-Amador MA, Elena SF. 2008. Virus adaptation by manipulation of host's gene expression. PLoS ONE 3, e2397 ( 10.1371/journal.pone.0002397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robaglia C, Caranta C. 2006. Translation initiation factors: a weak link in plant RNA virus infection. Trends Plant Sci. 11, 40–45. ( 10.1016/j.tplants.2005.11.004) [DOI] [PubMed] [Google Scholar]
- 26.Revers F, García JA. 2015. Molecular biology of potyviruses. Adv. Virus Res. 92, 101–199. ( 10.1016/bs.aivir.2014.11.006) [DOI] [PubMed] [Google Scholar]
- 27.Gould SJ. 1989. Wonderful life: the burgess shale and the nature of history. New York, NY: WW Norton & Company. [Google Scholar]
- 28.Bedoya L, Daròs JA. 2010. Stability of Tobacco etch virus infectious clones in plasmid vectors. Virus Res. 149, 234–240. ( 10.1016/j.virusres.2010.02.004) [DOI] [PubMed] [Google Scholar]
- 29.Carrasco P, de la Iglesia F, Elena SF. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco etch virus. J. Virol. 81, 12 979–12 984. ( 10.1128/JVI.00524-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyes DC, Zayed AM, Ascenzi R, McCaskill MJ, Hoffman NE, Davis KR, Görlach J. 2001. Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13, 1499–1510. ( 10.1105/tpc.13.7.1499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lalić J, Agudelo-Romero P, Carrasco P, Elena SF. 2010. Adaptation of tobacco etch potyvirus to a susceptible ecotype of Arabidopsis thaliana capacitates it for systemic infection of resistant ecotypes. Phil. Trans. R. Soc. B 365, 1997–2008. ( 10.1098/rstb.2010.0044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasi F, Travisano M, Lenski RE. 1994. Long-term experimental evolution in Escherichia coli. II. Changes in life-history traits during adaptation to a seasonal environment. Am. Nat. 144, 432–456. ( 10.1086/285685) [DOI] [Google Scholar]
- 33.Travisano M, Mongold JA, Bennet AF, Lenski RE. 1995. Experimental tests of the roles of adaptation, chance, and history in evolution. Science 267, 87–90. ( 10.1126/science.7809610) [DOI] [PubMed] [Google Scholar]
- 34.Meyer JR, Dobias DT, Weitz JS, Barrick JE, Quick RT, Lenski RE. 2012. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335, 428–432. ( 10.1126/science.1214449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bedhomme S, Lafforgue G, Elena SF. 2013. Genotypic but not phenotypic historical contingency revealed by viral experimental evolution. BMC Evol. Biol. 13, 46 ( 10.1186/1471-2148-13-46) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morley VJ, Mendiola SY, Turner PE. 2015. Rate of novel host invasion affects adaptability of evolving RNA virus lineages. Proc. R. Soc. B 282, 20150801 ( 10.1098/rspb.2015.0801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinreich DM, Chao L. 2005. Rapid evolutionary escape by large populations from local fitness peaks is likely in nature. Evolution 59, 1175–1182. ( 10.1111/j.0014-3820.2005.tb01769.x) [DOI] [PubMed] [Google Scholar]
- 38.Jain K, Krug J. 2007. Deterministic and stochastic regimes of asexual evolution on rugged fitness landscapes. Genetics 175, 1275–1288. ( 10.1534/genetics.106.067165) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on Dryad (http://dx.doi.org/10.5061/dryad.17s38).