

Abstract
The vesicular monoamine transporter 2 (VMAT2) packages neurotransmitters for release during neurotransmission and sequesters toxicants into vesicles to prevent neuronal damage. In mice, low VMAT2 levels causes catecholaminergic cell loss and behaviors resembling Parkinson’s disease, while high levels of VMAT2 increase dopamine release and protect against dopaminergic toxicants. However, comparisons across these VMAT2 mouse genotypes were impossible due to the differing genetic background strains of the animals. Following back-crossing to a C57BL/6 line, we confirmed that mice with approximately 95% lower VMAT2 levels compared with wild-type (VMAT2-LO) display significantly reduced vesicular uptake, progressive dopaminergic terminal loss with aging, and exacerbated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity. Conversely, VMAT2-overexpressing mice (VMAT2-HI) are protected from the loss of striatal terminals following MPTP treatment. We also provide evidence that enhanced vesicular filling in the VMAT2-HI mice modifies the handling of newly synthesized dopamine, indicated by changes in indirect measures of extracellular dopamine clearance. These results confirm the role of VMAT2 in the protection of vulnerable nigrostriatal dopamine neurons and may also provide new insight into the side effects of L-DOPA treatments in Parkinson’s disease.
Keywords: VMAT2, vesicle, MPTP, dopamine, DAT, L-DOPA.
The vesicular monoamine transporter 2 (VMAT2; SLC18A2) is located on both synaptic and dense core vesicles to package monoamine neurotransmitters: dopamine, norepinephrine, serotonin, and histamine. The packaging of cytosolic dopamine by VMAT2 is particularly important because dopamine left unpackaged is susceptible to the formation of reactive oxygen species and dopamine-quinones (Sulzer and Zecca, 1999). Thus, through its sequestration of cytosolic dopamine, VMAT2 opposes this endogenous source of neurotoxicity. As a 12-transmembrane H+-ATPase antiporter, VMAT2 is evolutionarily related to a family of toxin extruding antiporters, which explains its ability to also sequester neuronal toxicants from the cytosol into vesicles. In fact, the initial cloning of VMAT was the result of the protein’s ability to confer resistance to the dopaminergic toxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Erickson et al., 1992; Liu et al., 1992a,b). It has been suggested that VMAT2 modifies neurotoxicity through two potential mechanisms: direct toxicant sequestration into the vesicle, as is the case with MPTP, and dopamine displacement by a reduction in VMAT2 function (Guillot and Miller, 2009). Environmental toxicants such as pesticides, brominated flame retardants, and polychlorinated biphenyl compounds have been shown to disrupt vesicular uptake via VMAT2 (Bemis and Seegal, 2004; Chaudhry et al., 2008; Mariussen and Fonnum, 2001; Richardson and Miller, 2004; Sanchez-Ramos et al., 1998) and also have been linked to several disease etiologies (Ascherio et al., 2006; Caudle et al., 2012; Hatcher-Martin et al., 2012; Priyadarshi et al., 2001; Steenland et al., 2013).
Genetic knockout of VMAT2 in mice is fatal (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997). We previously reported that mice with a 95% reduction in VMAT2 expression compared with wild-type animals (VMAT2-deficient mice) exhibit progressive catecholaminergic degeneration, dysregulated dopamine homeostasis, α-synuclein accumulation, and both motor and nonmotor behavioral deficits reminiscent of Parkinson’s disease (Alter et al., 2016; Caudle et al., 2007; Taylor et al., 2009, 2014). Reduced VMAT2 levels in mice also increases neuronal vulnerability to a variety of toxic compounds, including MPTP and methamphetamine (Fumagalli et al., 1999; Gainetdinov et al., 1998; Guillot et al., 2008; Mooslehner et al., 2001; Staal et al., 2000; Takahashi et al., 1997). Most recently, mice with elevated VMAT2 levels were shown to have enhanced dopaminergic output and resistance to both MPTP and methamphetamine (Lohr et al., 2014, 2015). Based on emerging evidence from human populations on the importance of VMAT2 in degenerative diseases like Parkinson’s disease (Brighina et al., 2013; Glatt et al., 2006; Pifl et al., 2014; Rilstone et al., 2013), we and others have been increasingly interested in assessing vesicular filling on a functional continuum in a mouse model.
The VMAT2-deficient mice described previously were of a mixed genetic background consisting of both C57BL/6 and 129SV strains (Caudle et al., 2007; Guillot et al., 2008; Taylor et al., 2009, 2011), whereas the VMAT2-HI mice were bred to a C57BL/6 strain. It is well established that background strain of mice greatly influences neurochemical, pharmacological, and behavioral outcomes. Thus, studying the neurochemical and toxicological effects of altered VMAT2 function in mice with varying levels of VMAT2 requires that these animals are on the same background strain. Here, we describe the first genotypic continuum of vesicular function using VMAT2-LO, wild-type, and VMAT2-HI mice congenic for a C57BL/6 background. We show that reduced vesicular function results in both progressive neurodegeneration and an increased susceptibility to the dopaminergic toxicant, MPTP. Conversely, elevated vesicular function opposes this vulnerability. Because VMAT2 dictates dopamine storage capacity, we also examined the handling of exogenously applied dopamine following L-DOPA (levodopa) application in mice with varying VMAT2 levels. We show that elevated VMAT2 alters the neurochemical handling of the excess dopamine and preserves stimulated dopamine release following a neurotoxic lesion.
MATERIALS AND METHODS
Transgenic mice
The VMAT2-deficient mice previously described by our lab (Caudle et al., 2007; Guillot et al., 2008; Taylor et al., 2009, 2014) had been outbred to reintroduce the α-synuclein gene, which had been spontaneously deleted from the original VMAT2-deficient strain (Ka1) originally described by other groups (Mooslehner et al., 2001; Specht and Schoepfer, 2001). These α-synuclein-positive VMAT2-deficient mice were maintained on a C57BL/6 and 129SV mixed genetic background at Emory University, Atlanta, Georgia. For this study, these mixed background VMAT2-deficient mice were backcrossed to Charles River C57BL/6 mice for 4 generations using a marker assisted selection (ie, “speed congenic”) approach. Mouse genomes were assessed at the DartMouseTM Speed Congenic Core Facility at Dartmouth Medical School, which uses the Illumina, Inc (San Diego, California) GoldenGate Genotyping Assay to examine 1449 single nucleotide polymorphisms (SNPs) throughout the genome. Raw SNP data were analyzed using DartMouse’s SNaP‐MapTM and Map‐SynthTM software, determining the genetic background at each SNP location for each mouse. These back-crossed VMAT2-deficient mice on the C57BL/6 background are referred to as VMAT2-LO hereafter. Mice were 1 year old upon testing, except in the aging study where mice were 22 months old. This age was chosen based on the previous characterization of VMAT2-deficient mice by our laboratory (Caudle et al., 2007).
BAC transgenic VMAT2-overexpressing mice (VMAT2-HI) were generated as previously described (Lohr et al., 2014). Briefly, bacterial artificial chromosome (BAC) RP23-292H20 contains the entire VMAT2 (Slc18a2) locus (35 kb), 100 kb upstream, and 60 kb downstream. Subsequent generations were bred to Charles River C57BL/6 breeders for these experiments. Mice were 1 year old upon testing. Wild-type littermates from both VMAT2-LO and VMAT2-HI colonies were used.
Besides the genomic insertion and spontaneous recombination event known to be present in the VMAT2-HI and VMAT2-LO mice, we attempted to eliminate any additional genetic strain confounds both through SNP array testing and repeated back-crossing of our colonies to Charles River C57Bl/6 mice. All procedures were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Emory University.
MPTP injection schedules
Male mice of the varying genotypes were injected (SC) with either MPTP-HCl (M0896, Sigma) or saline (0.9%). The lesion consisted of 2 injections of 10 mg/kg MPTP (freebase) with an interinjection interval of 10 h. Mice were sacrificed 7 days after the final dose for immunochemical and voltammetry analyses.
Western blotting
Western blots were performed as previously described (Caudle et al., 2007). Primary antibodies used were polyclonal rabbit anti-VMAT2 serum (generated for the Miller lab by Covance, 1:20 000), rat anti-dopamine transporter (DAT) (Millipore, 1:5000), rabbit anti-tyrosine hydroxylase (TH) (Millipore, 1:1000), mouse anti-glial fibrillary acidic protein (GFAP) (Cell Signaling, 1:5000), and mouse anti-β-actin (Sigma, 1:5000). Actin levels were confirmed for equivalent loading. The appropriate HRP-linked secondary antibodies (Jackson ImmunoResearch, 1:5000) were used. Analysis was calibrated to coblotted standards of pooled sample from wild-type samples. Rabbit polyclonal anti-VMAT2 serum was raised against a peptide in the C-terminal region of mouse VMAT2 (CTQNNVQPYPVGDDEESESD) by Covance Custom Immunology Services.
Immunohistochemistry
Immunohistochemistry was performed as previously described (Caudle et al., 2007). Primary antibodies used were polyclonal rabbit anti-VMAT2 serum (1:50 000), rat anti-DAT (1:1000), mouse anti-GFAP (1:1000), or rabbit anti-TH (1:1000). The appropriate biotinylated secondary antibodies (Jackson ImmunoResearch, 1:200) were used. All images were acquired with NeuroLucida (MicroBrightField).
Fast-scan cyclic voltammetry in striatal slice
Slice fast-scan cyclic voltammetry (FSCV) was performed as previously described (Kile et al., 2012). A 5-recording survey of 4 different dorsal striatal release sites was taken for each animal with a 5-min rest interval between each synaptic stimulation (2.31 V). Application of waveform, stimulus, and current monitoring was controlled by TarHeel CV [University of North Carolina (UNC)] using a custom potentiostat (UEI, UNC Electronics Shop). The waveform for dopamine detection consisted of a −0.4 V holding potential versus an Ag/AgCl (In Vivo Metric) reference electrode. The applied voltage ramp goes from −0.4 V to 1.0 V and back to −0.4 V at a rate of 600 V/s at 60 Hz. Maximal release at striatal sites in a slice was averaged. Carbon-fiber microelectrodes were calibrated with dopamine standards using a flow-cell injection system. Kinetic constants were extracted using nonlinear regression analysis of release and uptake of dopamine.
L-DOPA fast-scan cyclic voltammetry
The applied voltage ramp and recording parameters were the same as described earlier. Both saline and MPTP-lesioned wild-type, VMAT2-LO, and VMAT2-HI animals were used in these experiments. Following incubation in oxygenated HEPES aCSF (30°C), 5 recordings were taken from a single stable dorsolateral striatal site for 2 different sets of stimulation parameters. Stimulation 1 was 1 pulse applied at 60 Hz (2.31 V) with 5 min of rest in between stimulations. Afterwards, stimulation set 2 was applied with 4 pulses applied at 10 Hz with 10 min of rest between stimulations. After these baseline recordings, HEPES aCSF with 100 μM L-DOPA (Sigma) and 1.7 mM ascorbate was applied to the slice for 1 h followed by a 1 h washout with HEPES aCSF. Following the washout period, recordings were taken again with both stimulation parameter sets.
[3H]-WIN binding
[3H]-WIN 35 428 (83 Ci/mmol) was obtained from PerkinElmer Life Sciences. Bilateral striata were dissected from wild-type and VMAT2-HI mice following rapid decapitation. Tissue was homogenized in homogenization buffer (4 mM HEPES, 0.32 M sucrose, pH 7.4) using a Teflon homogenizer at 1000 rpm for 10–12 strokes. Following, a 10 min spin at 48000 × g, the pellet was resuspended and the spin was repeated 3 times. The final pellet was then resuspended in DAT binding buffer at a ratio of 1 bilateral striatum per 1 ml. These crude striatal synaptosomes were incubated with a single concentration (10 nM) of [3H]-WIN 35 428 in 25 mM sodium phosphate buffer for 1 h at 4°C. Incubations were terminated by rapid vacuum filtration onto GF/B filter paper, and radioactivity was determined by liquid scintillation counting. Nonspecific binding was determined using the addition of 10 μM nomifensine, and specific binding was calculated as the total binding minus nonspecific binding (sample incubated with nomifensine). After determination of protein concentrations, binding to DAT was calculated as fmol/mg protein and expressed as raw values.
Vesicular uptake
Whole brains from each genotype were homogenized in homogenization buffer (4 mM HEPES, 0.32 M sucrose, pH 7.4). Vesicular fractions were isolated using differential centrifugation as previously described (Caudle et al., 2007). Vesicular uptake was measured by [3H]-dopamine uptake (1 μM) in the vesicular fraction with nonspecific uptake (sample incubated with 10 μM tetrabenazine).
Statistical analysis
All data were analyzed in GraphPad Prism. Differences were analyzed using 2-way ANOVA (with treatment and genotype as factors) with Bonferroni post hoc tests. Outliers were defined by the Grubbs’ test for outliers (α = .05). All errors shown are SEM.
RESULTS
VMAT2-LO Mice on a C57BL/6 Strain Have Reduced Vesicular Uptake
Key pieces of raw data have been uploaded to Dryad Digital Repository (Miller et al., 2016). The newly generated VMAT2-LO mice were shown to be > 97% congenic for the desired C57BL/6 background, with significantly lower VMAT2 levels (> 90% reduction) than wild-type mice as measured by immunoblotting (Figure 1B) and reduced vesicular uptake and (Figure 1E). At 22 months of age, VMAT2-LO mice also show a significant loss of striatal levels of the DAT, a marker of dopamine terminal integrity (30% loss, LO vs WT, P < .05) (Figure 1C), replicating previous results that showed that impaired vesicular storage results in progressive degeneration of the nigrostriatal pathway (Caudle et al., 2007).
FIG. 1.

C57BL/6 vesicular monoamine transporter 2 (VMAT2)-LO mice show progressive dopaminergic degeneration with aging and reduced vesicular uptake. A–D, VMAT2-LO mice have significantly reduced VMAT2 levels and, by 22 months of age, also show significant reductions in dopamine transporter (DAT) level in the striatum, as measured by immunoblotting (n = 10). E, VMAT2-LO mice show an 80% reduction in VMAT2-mediated [3H]-dopamine uptake in isolated vesicles compared with wild-type littermates at 12 months of age (P < .01, n = 2–3).
VMAT2 Level Modulates MPTP Toxicity in Mice on a C57BL/6 Genetic Background
Male VMAT2-LO mice show exacerbated MPTP toxicity in the striatum following a 2 × 10 mg/kg MPTP dosing regimen. This increased toxicity was shown by greater losses of striatal DAT levels following MPTP in the VMAT2-LO mice compared with the other genotypes (Figure 2C). Conversely, mice with elevated VMAT2 levels show a protection from MPTP toxicity by these measures (83% vs 77% vs 48% DAT protein loss in LO, WT, HI mice, respectively). Similar trends were also seen in protection from striatal TH loss (72% vs 67% vs 49% TH protein loss, LO, WT, HI mice, respectively) (Figure 2D). Immunoblotting results were further confirmed with immunohistochemical analysis of DAT and TH in the striatum (Figs. 2E and F). A trend toward decreased DAT levels was also seen in saline-treated male VMAT2-LO mice at 12 months of age compared with wild-type mice, further suggesting a progressive degenerative phenotype.
FIG. 2.

VMAT2 level modifies vulnerability to dopamine terminal marker loss following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). A, Representative blots of VMAT2 and dopaminergic markers DAT and tyrosine hydroxylase (TH) following MPTP or saline treatment. S, saline; M, 2 × 10 mg/kg MPTP. B, VMAT2 levels vary with genotypes as expected. C and D, VMAT2-LO mice show a larger decrease in DAT and TH, compared with wild-type and VMAT2-HI mice (n = 4–6). Different letters above bars (A vs B, for example) indicate differences of at least P < .05 between those groups by a 2-way ANOVA and Bonferroni post hoc tests. Bars with the same letter above (A vs A) are not significantly different. Results are % wild-type control. E and F, Results were confirmed with immunohistochemistry for DAT and TH.
VMAT2 Level Modifies L-DOPA-Induced Increases in Dopamine Release
At baseline, VMAT2 level dictates the amplitude of stimulated dopamine release (Figs. 3A–C). VMAT2-LO mice show the smallest baseline dopamine release (0.47 μM DAmax), and VMAT2-HI mice show the largest release (2.52 μM DAmax), with dopamine release levels in wild-type mice falling in between the genotypes (1.75 μM DAmax). We were interested in using our VMAT2 genotypic continuum to assess the uppermost limits of vesicular function by flooding the system with an exogenous dopamine source. Following an acute bath L-DOPA application and washout (Figure 3D), VMAT2-LO mice show the lowest peak dopamine release (0.9 μM DAmax), VMAT2-HI mice show the largest peak release (4.1 μM DAmax), and wild-type mice fall in between the 2 other genotypes (3.0 μM DAmax) (Figs. 3A–C). Interestingly, VMAT2-LO mice show the largest percent increase in peak dopamine release with a 91% increase in DAmax following L-DOPA, compared with baseline. Wild-type mice show a significant 73% increase in stimulated dopamine release when L-DOPA is applied, and VMAT2-HI mice show the smallest increase in release with a 64% increase in stimulated dopamine release compared with baseline.
FIG. 3.

VMAT2 level modifies stimulated dopamine (1p) release and dopamine clearance following L-DOPA application. A–C, At baseline, VMAT2-LO mice show the smallest dopamine release (n = 3), and VMAT2-HI mice show the largest release (n = 6), compared with wild-type mice (n = 6). Following L-DOPA application (D), all 3 genotypes showed increases in DAmax compared with baseline values, however the fall time of the release traces appeared to be particularly affected between the genotypes. E and F, VMAT2-HI mice show a significant decrease in both the rate constant, tau, and half-width, all of which reflects the steeper fall of the release traces in this genotype compared with wild-type and VMAT2-LO mice. Different letters above the bars indicate differences of at least P < .05 as indicated by a 2-way ANOVA and Bonferroni post hoc tests.
VMAT2 Level Modifies Movement of Extracellular Dopamine Following L-DOPA Application
In addition to differences in peak dopamine release following L-DOPA application and washout, the VMAT2 genotypes also showed changes in the levels and the movement of extracellular dopamine via indirect measures. We estimated the levels of extracellular dopamine following stimulation after L-DOPA application based on the area under the curve (AUC) of the release trace in the varying VMAT2 genotypes (Figure 3). VMAT2-LO mice show the smallest L-DOPA-induced increase in AUC with a 1028% increase from baseline, presumably due their low storage capacity (baseline = 0.793 ± 0.043 μM*s; L-DOPA = 8.952 ± 0.502 μM*s). Surprisingly, L-DOPA treatment induced a smaller 1149% increase in the AUC in VMAT2-HI mice (baseline = 1.676 ± 0.0247 μM*s; L-DOPA = 15.099 ± 4.327 μM*s) compared with the 2523% increase in wild-type mice (baseline = 0.907 ± 0.085 μM*s; L-DOPA = 23.793 ± 8.035 μM*s). These results suggest that the VMAT2-HI mice, though having a greater peak dopamine release, actually have less extracellular dopamine in this L-DOPA treatment paradigm.
To address this difference, we next examined the extracellular handling of the newly synthesized dopamine as indicated by the rate constant tau, which is the time at which the trace falls to approximately 63.3% of its maximum. Tau was significantly smaller in the VMAT2-HI mice than that of both VMAT2-LO and wild-type mice (LO = 16.49 ± 2.58 s, WT = 8.59 ± 2.74 s, HI = 2.86 ± 0.96 s; P < .01) (Figure 3E). The size of the release trace and clearance of extracellular dopamine can also be reflected by the measure of half-width of the trace, which is the time that it takes for the trace to reach half of its total width. The half-width is also significantly smaller in the VMAT2-HI mice following L-DOPA compared with the other genotypes (LO = 14.67 ± 1.84 s, WT = 6.74 ± 1.08 s, HI = 3.31 ± 0.83 s; P < .01) (Figure 3F). Both the tau measures and the half-width measures indicate faster extracellular dopamine clearance in animals with elevated vesicular function. The VMAT2-HI mice also showed this increased peak release and faster extracellular dopamine clearance following L-DOPA application and washout when larger stimulation parameters (4 pulses, 10 Hz) were used (data not shown).
No Change in Total DAT Levels in the VMAT2-HI Mice
Both of the measures mentioned earlier indicate faster extracellular dopamine clearance in the VMAT2-HI mice following L-DOPA application and washout. To examine the mechanism behind this phenomenon, we examined expression levels of the main mediator of synaptic dopamine uptake, the DAT, in both wild-type and VMAT2-HI mice. We measured total DAT levels in the striatum and show no change in the total DAT levels as measured by immunoblotting (Figure 4C) and synaptosomal levels of the DAT as measured by [3H]-WIN-35 428 binding (Figure 4E).
FIG. 4.

There are no changes in DAT expression or binding in synaptosomes prepared from wild-type and VMAT2-HI mice. A, Representative immunoblotting of VMAT2 and dopaminergic markers DAT and TH between wild-type and VMAT2-HI mice from synaptosomal preparations from wild-type and VMAT2-HI mice (n = 4). B, VMAT2 level is significantly increased in the VMAT2-HI mice. C and D, There are no differences in the expression levels of DAT and TH between wild-type and VMAT2-HI mice. E, Using isolated synaptosomal preparations from bilateral dissected striatal homogenates, WIN binding showed no significant differences in DAT level between genotypes (n = 4).
VMAT2 Level Modifies Stimulated Dopamine Release Following MPTP Treatment
We next examined the effects of varying VMAT2 levels on stimulated dopamine release both with and without a toxic MPTP lesion. Following a 2 × 10 mg/kg MPTP lesion, all of the VMAT2 genotypes show significant decreases in peak stimulated dopamine release compared with controls mice (approximately 60% decrease in DAmax compared with saline-treated for all genotypes) (Figure 5). However, VMAT2-HI mice show a significantly preserved peak dopamine release following this MPTP dosing regimen (0.21 μM vs. 0.69 μM vs. 1.06 μM DAmax in LO, WT, HI mice, respectively; P < .05). Finally, VMAT2 appears to have minimal effect on L-DOPA response following MPTP lesion.
FIG. 5.
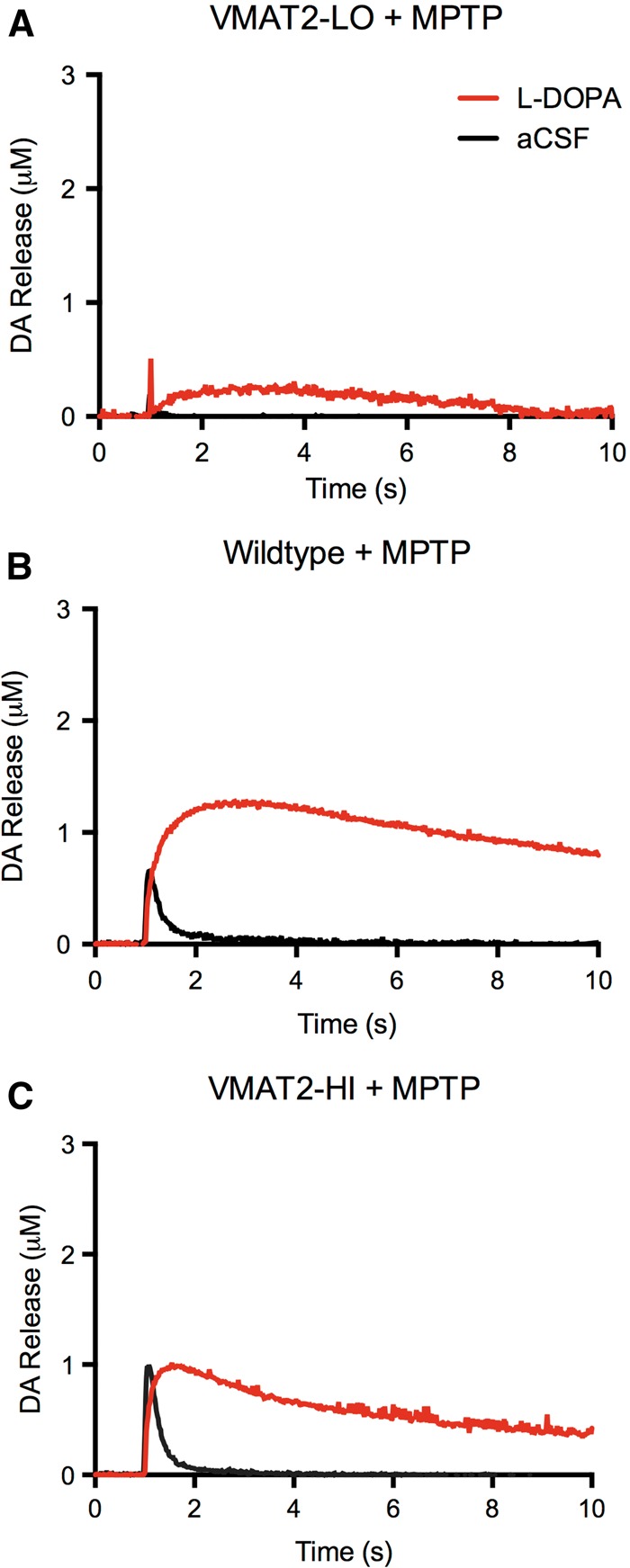
VMAT2 level alters dopamine release but has minimal effect on exogenous dopamine handling in lesioned slices. Following a 2 × 10 mg/kg MPTP lesion, all VMAT2 genotypes show significant decreases in peak stimulated dopamine release compared with controls mice (approximately 60% decrease in DAmax compared with baselines from Fig. 3). VMAT2-HI mice show a significantly preserved peak dopamine release following this dosing regimen (P < .05, n = 4).
DISCUSSION
Here, we show that VMAT2 level modifies vulnerability to MPTP intoxication and dopamine handling in mice of the same C57BL/6 genetic background. These mice of varying VMAT2 levels provide important tools to assess vesicular function on a continuum in vivo.
VMAT2 Mediates MPTP Toxicity
Previous work has shown that VMAT2 acts as a modifier of dopaminergic toxicity (Guillot and Miller, 2009). Using our newly back-crossed VMAT2-LO mice, we confirmed that lower VMAT2 levels reduce vesicular uptake and cause progressive terminal loss with aging (Figure 1). VMAT2-LO mice also exhibit significant decreases in DAT levels following a low 2 × 10 mg/kg MPTP dose, whereas VMAT2-HI mice show a preservation of these protein levels (Figure 2). Furthermore, stimulated dopamine release appears to be significantly decreased in the VMAT2-LO mice following MPTP treatment to an almost undetectable level (Figure 5). Although wild-type and VMAT2-HI mice also have decreased stimulated dopamine release following an MPTP lesion, the peak dopamine release in these genotypes remains significantly greater than that of the VMAT2-LO mice (Figure 5), with VMAT2-HI mice maintaining the highest dopamine release after MPTP treatment. These data provide evidence that VMAT2 modifies MPTP toxicity in a protein level-dependent manner and that these indicators of degeneration correlate to neurotransmitter output as measured by voltammetry. These results also confirm previously published reports on the negative effects of reduced vesicular function on toxicological outcomes (Caudle et al., 2007; Fumagalli et al., 1999; Gainetdinov et al., 1998; Guillot et al., 2008; Takahashi et al., 1997) and beneficial effects of elevated VMAT2 levels (Lohr et al., 2014, 2015). Furthermore, the background strain of the VMAT2-LO, wild-type, and VMAT2-HI mice will provide additional power when assessing VMAT2 function in future neurochemical assays and behavioral studies.
VMAT2 Modifies L-DOPA-Derived Dopamine Handling
This study also suggests that augmented vesicular function may modify the handling of L-DOPA-derived dopamine in a striatal slice. VMAT2 level determines peak dopamine release at baseline (Figure 3), supporting previous reports on the importance of vesicular filling on neurochemical output (Colliver et al., 2000; Fon et al., 1997; Lohr et al., 2014; Pothos, 2002; Pothos et al., 2000; Sulzer and Pothos, 2000). Following L-DOPA treatment, all of the VMAT2 genotypes show large increases in their peak dopamine release compared with baseline release. As expected, the VMAT2-LO mice have the smallest peak release after L-DOPA treatment, and the VMAT2-HI mice have the largest peak dopamine release (Figure 3).
Surprisingly, the VMAT2-HI mice actually have a smaller total area under the release curve compared with wild-type mice. This led us to analyze the kinetics of the release traces, which reflect the clearance of dopamine from the extracellular space following stimulation. It has previously been shown that VMAT2-HI mice have enhanced stimulated dopamine release at baseline (Lohr et al., 2014). In this baseline state, the VMAT2-HI mice have no change in the DAT-mediated uptake of dopamine back into the presynaptic neuron following stimulation as measured by tau or half-width. However, it appears that VMAT2-HI mice have modified DAT-mediated synaptic dopamine uptake after the application of L-DOPA. VMAT2-LO mice have the slowest extracellular dopamine clearance, whereas VMAT2-HI mice have faster clearance of extracellular dopamine by these measures (Figure 3). This change in dopamine handling in the VMAT2-HI mice preserves a more typical time course of dopamine release and reuptake, similar to baseline release traces. Based on our verification of the unchanged DAT levels in the VMAT2-HI mice (Figure 4), we attribute change in tau and half-width to alterations of the movement of dopamine through the DAT.
Implications for L-DOPA-Induced Dyskinesias
Several therapeutic strategies have been used to enhance dopamine neurotransmitter signaling in diseases of deficient dopamine like Parkinson’s disease. Although L-DOPA treatment does not halt the progression of the neurodegeneration in Parkinson’s disease, it remains the best intervention to restore movement in affected individuals (Cheshire et al., 2015). However, L-DOPA’s complicated pharmacokinetics, fluctuations in endogenous dopamine levels throughout the circadian period, and deleterious side effects add additional layers of complexity to L-DOPA therapy. The most notable side effect of L-DOPA treatment is uncontrolled movements termed L-DOPA-induced dyskinesias (LIDs) (Jenner, 2008). There are a variety of hypotheses to describe the mechanism by which LIDs occur, including temporal dysregulation of dopamine release and uptake into serotonin neurons following the loss of dopaminergic innervation in the putamen (Cenci, 2014; Crittenden and Graybiel, 2011; Jenner, 2008). These changes may result in massive sensitization of postsynaptic receptors over time. In the presence of a huge flood of dopamine derived from L-DOPA treatment, these receptors would become excessively activated, perhaps resulting in the aberrant motor movements of dyskinesias. Perhaps most promisingly, it has been suggested that LIDs are due to unregulated dopamine release from formerly serotonin-releasing neurons (Carta et al., 2008; Cheshire et al., 2015; Mosharov et al., 2014). In the absence of striatal dopaminergic innervation in Parkinson's disease, serotonergic terminals store L-DOPA. Because serotonergic terminals lack the proper synaptic inputs and neuromodulatory connections that dopamine neurons previously held in the basal ganglia, this aberrant transmitter release may result in uncoordinated neurotransmission and, perhaps, dyskinesias. Though it is unclear as to exactly how or why these dyskinetic effects appear in Parkinson’s patients over time, most would agree that LIDs develop due to the improper handling of dopamine in a brain that lacks the neurons that formerly produced the transmitter (Cenci, 2014). A method to protect dopamine-producing neurons and, perhaps, allow them to better handle and store large floods of dopamine may be an avenue to modify the severity of LIDs.
Possible Mechanisms of for Altered Extracellular Dopamine Uptake
There are 3 possible mechanisms that may explain the ability of vesicular function via VMAT2 expression to modify extracellular dopamine levels in the presence of excess dopamine. First, DAT function may be dictated by a concentration gradient across the plasma membrane. Following L-DOPA treatment, a large amount of newly synthesized dopamine is likely held within the neuron. VMAT2-HI mice store this newly synthesized dopamine, leaving a low concentration of cytosolic dopamine. After electrical stimulation, a flood of extracellular dopamine is released into the extracellular space. VMAT2-HI mice would have a very high extracellular dopamine content (large release), and relatively low cytosolic dopamine concentration (large storage capacity), creating a greater driving force across the DAT and faster dopamine clearance. Wild-type mice would have a greater amount of cytoplasmic dopamine because of their lower vesicular capacity. Thus, the difference between the cytoplasmic and extracellular dopamine concentrations would be minimal, resulting in reduced DAT function and slower dopamine clearance from the extracellular space. It is exceedingly difficult, if not impossible, to measure cytosolic dopamine content in vivo, making direct measures of such a mechanism unfeasible in this model system. However, evidence from mice with either increased DAT (DAT-Tg) or decreased VMAT2 (VMAT2-deficient) expression shows that differential transporter activity contributes to the accumulation of toxic compounds within the cytosol (eg, MPTP metabolites) and the accumulation of dopamine metabolites (Caudle et al., 2007; Gainetdinov et al., 1998; Lohr et al., 2014; Masoud et al., 2015; Takahashi et al., 1997). These studies provide in vivo evidence of the accumulation and clearance of cytosolic dopamine following manipulation of transporter levels.
The second option is also based on the DAT's ability to reverse transport and cause an efflux of dopamine when cytosolic dopamine concentrations become high. The best known example of this is when amphetamines are applied to dopamine neurons (Sulzer et al., 2005). Amphetamines deplete vesicular stores, elevating cytosolic dopamine concentrations, and this high concentration gradient (high cytosolic dopamine, low extracellular dopamine) causes DAT to reverse transport directions, dumping dopamine into the extracellular space (Pifl et al., 1995). Similarly, wild-type mice that are unable to store excess dopamine may have high cytosolic dopamine levels in this paradigm, resulting in DAT-mediated dopamine efflux. In voltammetry, this would be recorded as a slower recovery of the signal to baseline and an increase in tau and half-width. In contrast, VMAT2-HI mice would have lower cytosolic dopamine levels because of their ability to store dopamine, resulting in less DAT-mediated dopamine efflux. This reduced efflux would be recorded as a faster recovery of the signal and decreased tau and half-width. There has been previous evidence that increases in cytosolic dopamine levels will increase DAT-mediated efflux. MPP+, the metabolite of MPTP, displaces dopamine into the cytosol and induces a stimulation-independent efflux of dopamine, much like an amphetamine-like compound (Choi et al., 2015). Additionally, it is known that compounds that inhibit VMAT2, and presumably elevate cytosolic dopamine, can also reduce DAT function (Meyer et al., 2011).
Finally, it is possible that vesicular function modifies DAT-mediated synaptic dopamine uptake by a direct interaction between DAT and the synaptic vesicle. The DAT has been shown to interact with known vesicle associated proteins, including syntaxin (Lee et al., 2004) and the synaptic vesicle protein synaptogyrin-3 (Egaña et al., 2009). Synaptogyrin-3 has been speculated to tether synaptic vesicles to the DAT based on evidence showing a biochemical complex involving DAT, synaptogyrin-3, and VMAT2, and a reserpine-sensitive effect of synaptogyrin-3 on DAT function (Egaña et al., 2009). Additionally, it has recently been shown that a variety of DAT-inhibiting compounds (cocaine, nomifensine and the “bath salt” component, methylene-dioxypyrovalerone) are capable of augmenting vesicular release through unknown mechanisms, perhaps via calcium-dependent interactions with vesicular proteins (Hoffman et al., 2016). Based on these results, it is possible that the dopamine shuttled into the presynaptic terminal via DAT may be directly or more efficiently packaged into synaptic vesicle pools near the transporter. Finally, future studies with VMAT2-HI mice should also consider the redistribution and posttranslational modification of the DAT because these changes are known to modify transporter function and have yet to be examined in this paradigm (German et al., 2015).
Benefits of Increased VMAT2 Function
In this study, we demonstrated that vesicular storage acts on a continuum with a large range of amplitudes of neurochemical output. For the first time, we are able to assess genetic changes to vesicular function using mice congenic for the C57BL/6 background. As expected, VMAT2 level opposes dopaminergic toxicity following MPTP exposure and enhances dopamine release both with and without a lesion. Interestingly, it also appears that vesicular neurotransmitter storage modifies the handling of an exogenous flood of dopamine by L-DOPA treatment. Although no compound is currently available to enhance vesicular filling, a positive allosteric modulator of VMAT2 or pharmacological manipulation of another vesicular protein responsible for altering vesicular storage would be of interest in the context of disease. In a disorder like Parkinson’s disease, which is defined by vulnerability to insult, decreased dopamine output, and dysfunctional handling of L-DOPA, enhanced vesicular filling could preserve and improve the output of dopaminergic circuitry.
FUNDING
This work was supported by The National Institutes of Health (Grants R01ES023839, P30ES019776, F31NS084739, F31DA037652, F31NS089242, P50NS071669, and T32ES012870) and The Lewis Dickey Memorial Fund.
REFERENCES
- Alter S. P., Stout K. A., Lohr K. M., Taylor T. N., Shepherd K. R., Wang M., Guillot T. S., Miller G. W. (2016). Reduced vesicular monoamine transport disrupts serotonin signaling but does not cause serotonergic degeneration. Exp. Neurol. 275(Pt 1), 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascherio A., Chen H., Weisskopf M. G., O’Reilly E., McCullough M. L., Calle E. E., Schwarzschild M. A., Thun M. J. (2006). Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol. 60, 197–203. [DOI] [PubMed] [Google Scholar]
- Bemis J. C., Seegal R. F. (2004). PCB-induced inhibition of the vesicular monoamine transporter predicts reductions in synaptosomal dopamine content. Toxicol. Sci. 80, 288–295. [DOI] [PubMed] [Google Scholar]
- Brighina L., Riva C., Bertola F., Saracchi E., Fermi S., Goldwurm S., Ferrarese C. (2013). Analysis of vesicular monoamine transporter 2 polymorphisms in Parkinson’s disease. Neurobiol. Aging 34, 1712.e9–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M., Carlsson T., Muñoz A., Kirik D., Björklund A. (2008). Serotonin-dopamine interaction in the induction and maintenance of L-DOPA-induced dyskinesias. Prog. Brain Res. 172, 465–478. [DOI] [PubMed] [Google Scholar]
- Caudle W. M., Guillot T. S., Lazo C., Miller G. W. (2012). Parkinson’s disease and the environment: Beyond pesticides. Neurotoxicology 33, 585. [DOI] [PubMed] [Google Scholar]
- Caudle W. M., Richardson J. R., Wang M. Z., Taylor T. N., Guillot T. S., McCormack A. L., Colebrooke R. E., Di Monte D. a., Emson P. C., Miller G. W. (2007). Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci M. A. (2014). Presynaptic mechanisms of L-DOPA-induced dyskinesia: the findings, the debate, and the therapeutic implications. Front. Neurol. 5, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry F. A., Boulland J. L., Jenstad M., Bredahl M. K. L., Edwards R. H. (2008). Pharmacology of neurotransmitter transport into secretory vesicles. Handb. Exp. Pharmacol. 184, 77–106. [DOI] [PubMed] [Google Scholar]
- Cheshire P., Ayton S., Bertram K. L., Ling H., Li A., McLean C., Halliday G. M., O’Sullivan S. S., Revesz T., Finkelstein D. I., et al. (2015). Serotonergic markers in Parkinson’s disease and levodopa-induced dyskinesias. Mov. Disord. 30(6), 796–804. [DOI] [PubMed] [Google Scholar]
- Choi S. J., Panhelainen A., Schmitz Y., Larsen K. E., Kanter E., Wu M., Sulzer D., Mosharov E. V. (2015). Changes in neuronal dopamine homeostasis following 1-Methyl-4-phenylpyridinium (MPP+) exposure. J. Biol. Chem. 290, 6799–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colliver T. L., Pyott S. J., Achalabun M., Ewing A. G. (2000). VMAT-mediated changes in quantal size and vesicular volume. J. Neurosci. 20, 5276–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden J. R., Graybiel A. M. (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egaña L. A., Cuevas R. A., Baust T. B., Parra L. A., Leak R. K., Hochendoner S., Peña K., Quiroz M., Hong W. C., Dorostkar M. M., et al. (2009). Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-3. J. Neurosci. 29, 4592–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson J. D., Eiden L. E., Hoffman B. J. (1992). Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc. Natl. Acad. Sci. 89, 10993–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon E. A., Pothos E. N., Sun B. C., Killeen N., Sulzer D., Edwards R. H. (1997). Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron 19, 1271–1283. [DOI] [PubMed] [Google Scholar]
- Fumagalli F., Gainetdinov R. R., Wang Y. M., Valenzano K. J., Miller G. W., Caron M. G. (1999). Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J. Neurosci. 19, 2424–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov R. R., Fumaga F., Wang Y., Jones S. R., Levey A. I., Miller G. W., Caron M. G. (1998). Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J. Neurochem. 70, 1973–1978. [DOI] [PubMed] [Google Scholar]
- German C. L., Baladi M. G., McFadden L. M., Hanson G. R., Fleckenstein A. E. (2015). Regulation of the dopamine and vesicular monoamine transporters: Pharmacological targets and implications for disease. Pharmacol. Rev. 67, 1005–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt C. E., Wahner A. D., White D. J., Ruiz-linares A. (2006). Gain-of-function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Hum. Mol. Gen. 15, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot T. S., Miller G. W. (2009). Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol. 2, 149–170. [DOI] [PubMed] [Google Scholar]
- Guillot T. S., Shepherd K. R., Richardson J. R., Wang M. Z., Li Y., Emson P. C., Miller G. W. (2008). Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J. Neurochem. 106, 2205–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher-Martin J. M., Gearing M., Steenland K., Levey A. I., Miller G. W., Pennell K. D. (2012). Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicology 33, 1298–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A. F., Spivak C. E., Lupica C. R. (2016). Enhanced dopamine release by dopamine transport inhibitors described by a restricted diffusion model and fast-scan cyclic voltammetry. ACS Chem. Neurosci. 7, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. (2008). Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 9, 665–677. [DOI] [PubMed] [Google Scholar]
- Kile B. M., Walsh P. L., McElligott Z. A., Bucher E. S., Guillot T. S., Salahpour A., Caron M. G., Wightman R. M. (2012). Optimizing the temporal resolution of fast-scan cyclic voltammetry. ACS Chem. Neurosci. 3, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H., Kim M. Y., Kim D. H., Lee Y. S. (2004). Syntaxin 1A and receptor for activated C kinase interact with the N-terminal region of human dopamine transporter. Neurochem. Res. 29, 1405–1409. [DOI] [PubMed] [Google Scholar]
- Liu Y., Peter D., Roghani A., Schuldiner S., Privé G. G., Eisenberg D., Brecha N., Edwards R. H. (1992a). A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70, 539–551. [DOI] [PubMed] [Google Scholar]
- Liu Y., Roghani A., Edwards R. H. (1992b). Gene transfer of a reserpine-sensitive mechanism of resistance to N-methyl-4-phenylpyridinium. Proc. Natl. Acad. Sci. 89, 9074–9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr K. M., Bernstein A. I., Stout K. A., Dunn A. R., Lazo C. R., Alter S. P., Wang M., Li Y., Fan X., Hess E. J., et al. (2014). Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. 111, 9977–9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr K. M., Stout K. A., Dunn A. R., Wang M., Salahpour A., Guillot T. S., Miller G. W. (2015). Increased vesicular monoamine transporter 2 (VMAT2; Slc18a2) protects against methamphetamine toxicity. ACS Chem. Neurosci. 6, 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariussen E., Fonnum F. (2001). The effect of polychlorinated biphenyls on the high affinity uptake of the neurotransmitters, dopamine, serotonin, glutamate and GABA, into rat brain synaptosomes. Toxicology 159, 11–21. [DOI] [PubMed] [Google Scholar]
- Masoud S. T., Vecchio L. M., Bergeron Y., Hossain M. M., Nguyen L. T., Bermejo M. K., Kile B., Sotnikova T. D., Siesser W. B., Gainetdinov R. R., et al. (2015). Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol. Dis. 74, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. C., Horton D. B., Neugebauer N. M., Wooters T. E., Nickell J. R., Dwoskin L. P., Bardo M. T. (2011). Tetrabenazine inhibition of monoamine uptake and methamphetamine behavioral effects: Locomotor activity, drug discrimination and self-administration. Neuropharmacology 61, 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G., Lohr K., Chen M., Hoffman C., McDaniel M., Stout K., Dunn A., Wang M. (2016). Data from: Vesicular monoamine transporter 2 (VMAT2) level regulates MPTP vulnerability and clearance of excess dopamine in mouse striatal terminals. Dryad Digit. Repos. doi: http://dx.doi.org/10.5061/dryad.d689j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooslehner K. A., Chan P. M., Xu W., Liu L., Smadja C., Humby T., Allen N. D., Wilkinson L. S., Emson P. C. (2001). Mice with very low expression of the vesicular monoamine transporter 2 gene survive into adulthood: Potential mouse model for parkinsonism. Mol. Cell. Biol. 21, 5321–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov E. V., Borgkvist A., Sulzer D. (2014). Presynaptic effects of levodopa and their possible role in dyskinesia. Mov. Disord. 30(1), 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifl C., Drobny H., Reither H., Hornykiewicz O., Singer E. A. (1995). Mechanism of the dopamine-releasing actions of amphetamine and cocaine: Plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol. Pharmacol. 47, 368–373. [PubMed] [Google Scholar]
- Pifl C., Rajput A., Reither H., Blesa J., Cavada C., Obeso J. A., Rajput A. H., Hornykiewicz O. (2014). Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 34, 8210–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothos E. N. (2002). Regulation of dopamine quantal size in midbrain and hippocampal neurons. Brain Behav. Res. 130, 203–207. [DOI] [PubMed] [Google Scholar]
- Pothos E. N., Larsen K. E., Krantz D. E., Liu Y., Haycock J. W., Setlik W., Gershon M. D., Edwards R. H., Sulzer D. (2000). Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J. Neurosci. 20, 7297–7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadarshi A., Khuder S. A., Schaub E. A., Priyadarshi S. S. (2001). Environmental risk factors and Parkinson’s disease: A metaanalysis. Environ. Res. 86, 122–127. [DOI] [PubMed] [Google Scholar]
- Richardson J. R., Miller G. W. (2004). Acute exposure to aroclor 1016 or 1260 differentially affects dopamine transporter and vesicular monoamine transporter 2 levels. Toxicol. Lett. 148, 29–40. [DOI] [PubMed] [Google Scholar]
- Rilstone J., Alkhater R., Minassian B. (2013). Brain dopamine-serotonin vesicular transport disease and its treatment. N. Engl. J. Med. 368, 543–550. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos J., Facca A., Basit A., Song S. (1998). Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures. Exp. Neurol. 150, 263–271. [DOI] [PubMed] [Google Scholar]
- Specht C. G., Schoepfer R. (2001). Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal R. G., Hogan K. A., Liang C. L., German D. C., Sonsalla P. K. (2000). In vitro studies of striatal vesicles containing the vesicular monoamine transporter (VMAT2): Rat versus mouse differences in sequestration of 1-methyl-4-phenylpyridinium. J. Pharmacol. Exp. Ther. 293, 329–335. [PubMed] [Google Scholar]
- Steenland K., Wesseling C., Román N., Quirós I., Juncos J. L. (2013). Occupational pesticide exposure and screening tests for neurodegenerative disease among an elderly population in Costa Rica. Environ. Res. 120, 96–101. [DOI] [PubMed] [Google Scholar]
- Sulzer D., Pothos E. N. (2000). Regulation of quantal size by presynaptic mechanisms. Rev. Neurosci. 11, 159–212. [DOI] [PubMed] [Google Scholar]
- Sulzer D., Sonders M. S., Poulsen N. W., Galli A. (2005). Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 75, 406–433. [DOI] [PubMed] [Google Scholar]
- Sulzer D., Zecca L. (1999). Intraneuronal dopamine-quinone synthesis: A review. Neurotox. Res. 1, 181–195. [DOI] [PubMed] [Google Scholar]
- Takahashi N., Miner L. L., Sora I., Ujike H., Revay R. S., Kostic V., Jackson-Lewis V., Przedborski S., Uhl G. R. (1997). VMAT2 knockout mice: Heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc. Natl. Acad. Sci. 94, 9938–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T. N., Alter S. P., Wang M., Goldstein D. S., Miller G. W. (2014). Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology 76, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T. N., Caudle W. M., Miller G. W. (2011). VMAT2-deficient mice display nigral and extranigral pathology and motor and nonmotor symptoms of Parkinson’s disease. Parkinsons Dis. 2011, 124165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T. N., Caudle W. M., Shepherd K. R., Noorian A., Jackson C. R., Iuvone P. M., Weinshenker D., Greene J. G., Miller G. W. (2009). Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 29, 8103–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. M., Gainetdinov R. R., Fumagalli F., Xu F., Jones S. R., Bock C. B., Miller G. W., Wightman R. M., Caron M. G. (1997). Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron 19, 1285–1296. [DOI] [PubMed] [Google Scholar]