

Abstract

Leinamycin (LNM) is biosynthesized by a hybrid nonribosomal peptide synthetase (NRPS)–acyltransferase (AT)-less type I polyketide synthase (PKS). Characterization of LnmI revealed ketosynthase (KS)–acyl carrier protein (ACP)–KS domains at the NRPS–PKS interface. Inactivation of the KS domain or ACP domain in vivo abolished LNM production, and the ACP domain can be phosphopantetheinylated in vitro. The LnmI KS–ACP–KS architecture represents a new mechanism for functional crosstalk between NRPS and AT-less type I PKS in the biosynthesis of hybrid peptide–polyketide natural products.
Many clinically important medicines, such as antibiotics (erythromycin and daptomycin), antitumor drugs (bleomycin and epothilone), and immunosuppressants (cyclosporine and rapamycin), are biosynthesized by polyketide synthases (PKSs), nonribosomal peptide synthetases (NRPSs), or hybrid PKS–NRPSs, which have an assembly-line architecture with multifunctional domains organized into modules.1 Polyketide or peptide intermediates are attached to the acyl carrier protein (ACP) or peptidyl carrier protein (PCP) domains in each of the modules, where the polyketide or peptide chain elongation is catalyzed by a β-ketosynthase (KS) domain in PKSs or a condensation (C) domain in NRPSs.2 The remarkably efficient transfer of those peptide or polyketide intermediates between modules is mainly mediated by molecular recognition among the ACP, PCP, KS, or C domains or by the linkers, also known as docking domains, that facilitate communications for modules residing on separate proteins.3
Leinamycin (LNM), a potent antitumor antibiotic produced by Streptomyces atroolivaceus S-140,4 is biosynthesized by a hybrid NRPS–acyltransferase (AT)-less type I PKS megasynthase featuring many unprecedented modular complexities.5 One of them is the presence of two KS domains (KS1 and KS2) in LnmI PKS module 3 at the LNM hybrid NRPS–PKS interface (Figure 1).5b,5g Strikingly, the two KS domains are phylogenetically more similar to KS domains of modular type I PKS than those KS domains found at the hybrid NRPS–PKS interface (Figure S1 and Table S3).1e,6 KS1 is characterized by a mutated catalytic triad (C2090-A2225-H2264), suggesting the lack of a decarboxylation function of a canonical KS domain (C–H–H).7 How this unique domain architecture at the interface of LNM NRPS–PKS megasynthase contributes to LNM biosynthesis, however, is unknown.
Figure 1.

Characterization of the KS–ACP–KS domains at the LnmI NRPS–PKS interface revealing a new mechanism for functional crosstalk between NRPS and PKS in biosynthesis of hybrid peptide–polyketide natural products. Moieties biosynthesized by NRPS, PKS, and other tailoring enzymes are shown in blue, red, and black, respectively. (A) Proposed pathway for LNM biosynthesis featuring a KS–ACP–KS domain architecture at the LnmI NRPS–PKS interface that mediates peptidyl transfer from NRPS module 2 to PKS module 3. (B) Active-site residues of the KS1–ACP1–KS2 domains of LnmI PKS module 3. The ACP1 domain is unusual, featuring two putative phosphopantetheine attachment motifs (highlighted in red), and the S2581 residue is experimentally established to be essential and sufficient for LNM biosynthesis.
We now report that KS1 and KS2 together mediate the transfer of the peptidyl intermediate from LnmI NRPS module 2 to PKS module 3 at the interface of the LNM hybrid NRPS–PKS in LNM biosynthesis (Figure 1). We have further uncovered an additional ACP domain between KS1 and KS2 and established that KS1, KS2, and the newly discovered ACP1 are all essential for LNM biosynthesis. We finally propose that the KS–ACP–KS domain architecture of LnmI PKS module 3 characterized here represents a new hybrid NRPS–PKS interface for hybrid peptide–polyketide biosynthesis.
We first examined the role of KS1 and KS2 of LnmI PKS module 3 in LNM biosynthesis by site-specifically mutating the active-site residues C2090 of KS1 and C2824 and H2959 of KS2 to Ala in vivo [Figure S4 and the Supporting Information (SI)]. Thus, the lnmI KS1–KS2 locus was first replaced with an apramycin-resistant aac(3)IV gene cassette in S. atroolivaceus S-140 to generate the mutant strain SB3035. Plasmids pBS3118 (C2090A), pBS3119 (C2824A), and pBS3120 (H2959A) (Table S1) containing the designed point mutations were constructed in vitro following standard site-directed mutagenesis protocols (see the SI). Each of the mutant constructs was then introduced into the SB3035 strain by conjugation, first screening for the single-crossover mutants resistant to both apramycin and thiostrepton followed by serial transfer to isolate the double-crossover mutant strains sensitive to both antibiotics, to afford the mutant strains SB3036 (C2090A), SB3037 (H2959A), and SB3038 (C2824A) (Table S2).5b−5d,5f The genotypes of mutant strains SB3035, SB3036, SB3037, and SB3038 were confirmed by Southern analysis (Figure S4). The mutant strains SB3036, SB3037, and SB3038 were fermented under the standard conditions for LNM production, with the S. atroolivaceus S-140 wild-type strain as a control.5 Fermentation cultures were subjected to HPLC and electron-spray ionization mass spectrometry (ESI-MS) analysis.5 LNM production was completely abolished in all three mutant strains SB3036, SB3037, and SB3038 (Figure 2A, panels I–III).
Figure 2.

Investigation of the KS1–ACP1–KS2 domain of LnmI PKS module 3 at the LnmI NRPS–PKS interface for LNM biosynthesis by site-directed mutagenesis in vivo. (A) HPLC analysis of LNM production from KS1 or KS2 mutants as well as the complementation strains: I, SB3036 (C2090A); II, SB3037 (H2959A); III, SB3038 (C2824A); IV, SB3045; V, SB3046; VI, SB3047; VII, S. atroolivaceus S-140 wild-type; VIII, LNM standard (●). (B) HPLC analysis of LNM production from the ACP1 mutants: I, SB3039; II, SB3040; III, SB3041; IV, SB3042; V, SB3043; VI, SB3044; VII, S. atroolivaceus S-140 wild-type; VIII, LNM standard (●).
We next constructed an lnmI expression plasmid to complement the three mutant strains of KS1 and KS2 in trans (see the SI). Thus, a 14,756 bp DNA fragment containing the intact lnmI gene5b was placed under the control of the constitutively expressed ErmE* promoter and subcloned into an integrative plasmid pSET152 to afford pBS3121 (Table S1). Introduction of pBS3121 into SB3036, SB3037, and SB3038 by conjugation, followed by selection of exconjugants resistant to apramycin, afforded the complementation strains SB3045, SB3046, and SB3047, respectively (Table S2). These strains were fermented under the standard LNM production conditions, with the S. atroolivaceus wild-type strain as a control,5 and the fermentation cultures were similarly analyzed by HPLC and ESI-MS.5 LNM production was restored in all three complementation strains (Figure 2A, panels IV–VI), unambiguously establishing that both KS1 and KS2 are essential for LNM biosynthesis. Taken together, these results establish that both KS1 and KS2 are involved in LNM biosynthesis, most likely catalyzing the peptidyl intermediate transfer (KS1) and elongation (KS2) at the LnmI hybrid NRPS–PKS interface from NRPS module 2 to PKS module 3 (Figure 1).
Inspired by the finding that both KS1 and KS2 are essential for LNM biosynthesis, we re-examined LnmI PKS module 3 closely and uncovered an additional ACP domain (i.e., ACP1) between KS1 and KS2, which was confirmed to be essential for LNM biosynthesis (Figure 1). Although ACP1 is indistinguishable phylogenetically from ACPs of canonical modular type I PKSs (Figure S3), ACP1 is very unusual, featuring two putative phosphopantetheine (P-pant) attachment motifs [2578GLSSR2582] and [2597GVSST2601] that are separated by 18 amino acids and differ from the highly conserved signature motif [G(X)DSL] found in canonical ACPs (where X is any amino acid and S is the P-pant attachment site) (Figure 1B).7
The role of ACP1 in LNM biosynthesis was first studied through site-directed mutagenesis of the putative P-pant attachment sites in vivo (Figure S4 and the SI). Plasmids containing the designed point mutations in ACP1 were constructed by following standard protocols, in which one or both P-pant attachment sites were site-directly mutated to the conserved ACP motif [DS], inactivated by mutating both Ser into Ala [AA], or kept unchanged [SS], affording plasmids pBS3130 (D2580S2581-D2599S2600), pBS3132 (A2580A2581-D2599S2600), pBS3135 (A2580A2581-A2599A2600), pBS3136 (A2580A2581-S2599S2600), pBS3137 (S2580S2581-A2599A2600), and pBS3138 (D2580S2581-A2599A2600) (in which the mutated amino acid residues are shown in italics; Table S1). They were then introduced into SB3035 individually by conjugation. The desired double-crossover mutants were similarly isolated via the intermediacy of the corresponding single-crossover mutants by selecting for apramycin- and thiostrepton-resistant or -sensitive phenotypes, respectively, affording the six ACP1 mutant strains SB3039 (D2580S2581-D2599S2600), SB3040 (A2580A2581-D2599S2600), SB3041 (A2580A2581-A2599A2600), SB3042 (A2580A2581-S2599S2600), SB3043 (S2580S2581-A2599A2600), and SB3044 (D2580S2581-A2599A2600) (Table S2).5b−5d,5f The genotypes of these mutant strains were confirmed by Southern analysis (Figure S4).
The six ACP1 mutant strains were next fermented under the standard conditions for LNM production, with the S. atroolivaceus S-140 wild-type as a control, and the fermentation cultures were subjected to HPLC and ESI-MS analysis.5b−5d,5f The strains with mutations at S2580 and S2581, as exemplified by SB3040 (A2580A2581-D2599S2600), SB3041 (A2580A2581-A2599A2600), and SB3042 (A2580A2581-S2599S2600), completely lost LNM production (Figure 2B, panels II–IV), while the strains with mutations at S2599 and S2600, as exemplified by SB3043 (S2580S2581-A2599A2600) and SB3044 (D2580S2581-A2599A2600), retained LNM production (Figure 2B, panels V and VI). Taken together, these findings unambiguously establish that S2580 and S2581, but not S2599 and S2600, are essential for LNM biosynthesis (Figure 1B). The fact that both SB3039 (D2580S2581-D2599S2600) and SB3044 (D2580S2581-A2599A2600) (Figure 2B, panels I and VI) still produced LNM unambiguously pinned down the S2581 residue that is essential and sufficient for LNM biosynthesis (Figure 1B).
We finally confirmed S2581 as the P-pant attachment site of ACP1 by phosphopantetheinylating selected recombinant ACP1 proteins in vitro using CoA in the presence of Sfp, a known promiscuous phosphopantetheinyl transferase from Bacillus subtilis(8) (see the SI). Expression plasmids pBS3139 for the wild-type ACP1 domain (S2580S2581-S2599S2600) and pBS3143, pBS3141, pBS3142, and pBS3140 for four mutant variants, ACP1 (D2580S2581–S2599S2600), ACP1 (S2580S2581-A2599A2600), ACP1 (A2580A2581-D2599S2600), and ACP1 (D2580S2581-A2599A2600), were constructed (Table S1) and introduced into Escherichia coli BL21 (DE3). The wild-type and mutant variants of ACP1 were overproduced in their apo forms and purified to near homogeneity (Figure 3). In vitro phosphopantetheinylation of apo-ACP1 and its variants using CoA and Sfp was carried out by following literature procedure, with TcmM, a known ACP, as a control.6 The formation of phosphopantetheinylated products (i.e., holo-ACPs) was monitored by ESI-MS analysis.5a,5c,5e
Figure 3.
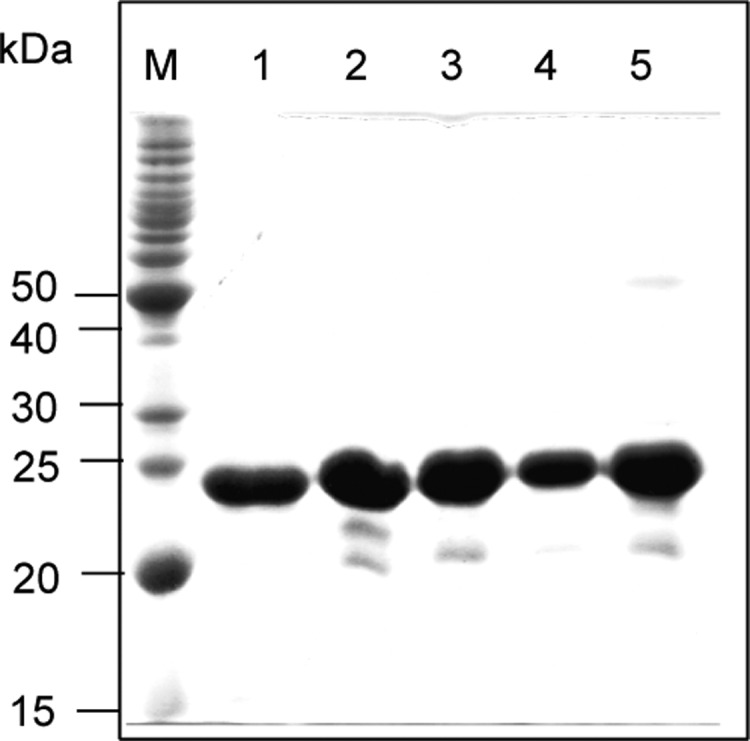
Production in E. coli and purified wild-type ACP1 and its mutant variants upon SDS-PAGE analysis. Lanes: M, MW standards; 1, ACP1 (S2580S2581–S2599S2600); 2, ACP1 (D2580S2581–S2599S2600); 3, ACP1 (D2580S2581-A2599A2600); 4, ACP1 (S2580S2581-A2599A2600); 5, ACP1 (A2580A2581-D2599S2600).
Under the assay conditions, Sfp catalyzed rapid phosphopantetheinylation of apo-TcmM, quantitatively converting apo-TcmM to holo-TcmM in 15 min (Table 1, entry 6), while omitting Sfp or CoA in the assays led to no formation of detectable amounts of holo-TcmM.8b Compared with apo-TcmM, apo-ACP1 and its mutant variants are relatively poor substrates for Sfp, with only 12–33% conversion to the corresponding holo-ACPs even after prolonged incubation for 120 min (entries 1–4). This is not surprising considering the atypical P-pant attachment motif (Figure 1B). Among the four possible Ser residues, i.e., (S2580S2581) and (S2599S2600) within the two putative P-pant attachment motifs of ACP1, [2578GLSSR2582] and [2597GVSST2601], only one phosphopantetheinylation was observed, as evidenced by the observed 340 molecular mass increase in the resultant holo-ACP1 products. In agreement with the in vivo studies, the fact that ACP1 (D2580S2581-A2599A2600), but not ACP1 (A2580A2581-D2599S2600), was specifically phosphopantetheinylated in vitro conclusively established S2581 as the site of P-pant attachment (Figure 1B). Taken together, these results show that ACP1 is a novel ACP with an atypical P-pant attachment motif [GLSSR] that plays an essential role in mediating peptidyl transfer at the hybrid NRPS–PKS interface for hybrid peptide–polyketide biosynthesis (Figure 1).
Table 1. In Vitro Phosphopantetheinylation of apo-ACP1 and Its Variants by Sfp upon ESI-MS Analysis.
Studies of intermediate channeling in modular PKSs, NRPSs, and hybrid PKS–NRPSs continue to reveal new mechanistic details of these remarkable megasynthases.2,3 In this study, we discovered that the KS1–ACP1–KS2 domain of LnmI PKS module 3 at the LnmI NRPS–PKS interface is required for LNM biosynthesis. Site-directed mutagenesis of the critical residues of the active sites of the KS1, ACP1, and KS2 domains in vivo all abolished LNM production (e.g., SB3036 and SB3038, Figure 2A, panels I and III, and SB3042, Figure 2B, IV), while mutation of residues outside the ACP1 active sites still produced LNM (e.g., SB3043 and SB3044, Figure 2B, panels V and VI). In addition, both in vivo mutagenesis studies and in vitro phosphopantetheinylation assays of ACP1 definitely established S2581 as the P-pant attachment site. On the basis of these results, we now propose that (i) the PCP-tethered peptidyl intermediate of NRPS module 2 is first transferred to ACP1 of PKS module 3 to yield the ACP1-tethered peptidyl intermediate, with KS1 acting as the acyl transfer agent to catalyze this process, and (ii) KS2 then catalyzes decarboxylative condensation between peptidyl-S-ACP1 and malonyl-S-ACP2, completing the elongation of a peptidyl intermediate with a polyketide extender (Figure 1A). While it has been speculated previously on the basis of bioinformatics analysis,5,9 the current study, to our knowledge, represents the first KS–ACP–KS domain architecture characterized experimentally at the NRPS–PKS interface responsible for hybrid peptide–polyketide biosynthesis.
Similar KS–ACP–KS domain architecture at the NRPS–PKS interface, in fact, is present in other hybrid peptide–polyketide biosynthetic machineries, e.g., chivosazol, rhizoxin, rhizopodin, and calyculin A (Table S3 and Figure S1).9 The first KSs all feature the mutated catalytic triad [C-A-H], as exemplified by ChiD-KS10,9a RhiB-KS2,9b RizD-KS11,9c and CalC-KS7,9d consistent with their proposed role as acyltransferases to catalyze peptidyl transfer only (Table S3 and Figure S2). These findings therefore support the proposal that the KS–ACP–KS domain architecture at the NRPS–PKS interface, as exemplified by LnmI, represents a new general mechanism for functional crosstalk between NRPS and PKS in biosynthesis of hybrid peptide and polyketide natural products (Figure 1).3,9 The removal of the decarboxylation function from KSs by mutation of its first active-site His has been observed previously.10 These KSs are functionally equivalent to acyltransferases but catalyze acyl or peptidyl transfer between ACPs or PCPs, engineering of which would increase our toolbox of acyltransferases for combinatorial biosynthesis. Taken together, our findings should inspire new strategies to engineer hybrid peptide–polyketide biosynthetic machinery for natural product structural diversity.
Acknowledgments
We thank Kyowa Hakko Kogyo Co. Ltd. (Tokyo, Japan) for the wild-type S. atroolivaceus S-140 strain and the Analytical Instrumentation Center of the School of Pharmacy, UW-Madison, for support in obtaining ESI-MS data. This work was supported in part by NIH Grant CA106150.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b02033.
Methods and additional data (PDF)
Author Present Address
⊥ Y.H.: Xiangya International Academy of Translational Medicine, Central South University, Changsha, Hunan 410013, China.
Author Present Address
# G.-L.T.: State Key Laboratory of Bioorganic and Natural Product Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, China.
The authors declare no competing financial interest.
Supplementary Material
References
- a Cane D. E.; Walsh C. T.; Khosla C. Science 1998, 282, 63–68. 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]; b Hertweck C. Angew. Chem., Int. Ed. 2009, 48, 4688–4716. 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]; c Butler M. S.; Robertson A. A.; Cooper M. A. Nat. Prod. Rep. 2014, 31, 1612–1661. 10.1039/C4NP00064A. [DOI] [PubMed] [Google Scholar]; d Medema M.; Fischbach M. A. Nat. Chem. Biol. 2015, 11, 639–648. 10.1038/nchembio.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Helfrich E. J. N.; Piel J. Nat. Prod. Rep. 2016, 33, 231–316. 10.1039/C5NP00125K. [DOI] [PubMed] [Google Scholar]; f Smanski M. J.; Zhou H.; Claesen J.; Shen B.; Fischbach M. A.; Voigt C. A. Nat. Rev. Microbiol. 2016, 14, 135–149. 10.1038/nrmicro.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shen B. Top. Curr. Chem. 2000, 209, 1–51. 10.1007/3-540-48146-X_1. [DOI] [Google Scholar]; b Staunton J.; Weissman K. J. Nat. Prod. Rep. 2001, 18, 380–416. 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]; c Finking R.; Marahiel M. A. Annu. Rev. Microbiol. 2004, 58, 453–488. 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]; d Fischbach M. A.; Walsh C. T. Chem. Rev. 2006, 106, 3468–3496. 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; e Walsh C. T. Nat. Prod. Rep. 2016, 33, 127–135. 10.1039/C5NP00035A. [DOI] [PubMed] [Google Scholar]
- a Gokhale R. S.; Tsuji S. Y.; Cane D. E.; Khosla C. Science 1999, 284, 482–485. 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]; b Du L.; Sanchez C.; Shen B. Metab. Eng. 2001, 3, 78–95. 10.1006/mben.2000.0171. [DOI] [PubMed] [Google Scholar]; c Tsuji S. Y.; Cane D. E.; Khosla C. Biochemistry 2001, 40, 2326–2331. 10.1021/bi002463n. [DOI] [PubMed] [Google Scholar]; d Jiang H.; Zirkle R.; Metz J. G.; Braun L.; Richter L.; Van Lanen S. G.; Shen B. J. Am. Chem. Soc. 2008, 130, 6336–6337. 10.1021/ja801911t. [DOI] [PubMed] [Google Scholar]; e Richter C. D.; Nietlispach D.; Broadhurst R. W.; Weissman K. J. Nat. Chem. Biol. 2008, 4, 75–81. 10.1038/nchembio.2007.61. [DOI] [PubMed] [Google Scholar]; f Jenke-Kodama H.; Dittmann E. Nat. Prod. Rep. 2009, 26, 874–883. 10.1039/b810283j. [DOI] [PubMed] [Google Scholar]
- a Hara M.; Asano K.; Kawamoto I.; Takiguchi T.; Katsumata S.; Takahashi K.-I.; Nakano H. J. Antibiot. 1989, 42, 1768–1774. 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]; b Hara M.; Saitoh Y.; Nakano H. Biochemistry 1990, 29, 5676–5681. 10.1021/bi00476a005. [DOI] [PubMed] [Google Scholar]; c Huang S.-X.; Yun B.-S.; Ma M.; Basu H. S.; Church D. R.; Ingenhorst G.; Huang Y.; Yang D.; Lohman J. R.; Tang G.-Li; Ju J.; Liu T.; Wilding G.; Shen B. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8278–8283. 10.1073/pnas.1506761112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cheng Y.; Tang G.; Shen B. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3149–3154. 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tang G.; Cheng Y.; Shen B. Chem. Biol. 2004, 11, 33–45. 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]; c Tang G.; Cheng Y.; Shen B. J. Nat. Prod. 2006, 69, 387–393. 10.1021/np050467t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tang G.; Cheng Y.; Shen B. J. Biol. Chem. 2007, 282, 20273–20282. 10.1074/jbc.M702814200. [DOI] [PubMed] [Google Scholar]; e Liu T.; Huang Y.; Shen B. J. Am. Chem. Soc. 2009, 131, 6900–6901. 10.1021/ja9012134. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Huang Y.; Huang S.-Y.; Ju J.; Tang G.; Liu T.; Shen B. Org. Lett. 2011, 13, 498–501. 10.1021/ol102838y. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ma M.; Lohman J. R.; Liu T.; Shen B. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10359–10364. 10.1073/pnas.1508437112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman J. R.; Ma M.; Osipiuk J.; Nocek B.; Kim Y.; Chang C.; Cuff M.; Mack J.; Bigelow L.; Li H.; Endres M.; Babnigg G.; Joachimiak A.; Phillips G. N. Jr.; Shen B. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12693–12698. 10.1073/pnas.1515460112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay A. T. Nat. Prod. Rep. 2012, 29, 1050–1053. 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- a Lambalot R. H.; Gehring A. M.; Flugel R. S.; Zuber P.; LaCelle M.; Marahiel M. A.; Reid R.; Khosla C.; Walsh C. T. Chem. Biol. 1996, 3, 923–936. 10.1016/S1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]; b Sanchez C.; Du L.; Edwards D. J.; Toney M. D.; Shen B. Chem. Biol. 2001, 8, 725–738. 10.1016/S1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- a Perlova O.; Gerth K.; Kaiser O.; Hans A.; Muller R. J. Biotechnol. 2006, 121, 174–191. 10.1016/j.jbiotec.2005.10.011. [DOI] [PubMed] [Google Scholar]; b Partida-Martinez L. P.; Hertweck C. ChemBioChem 2007, 8, 41–45. 10.1002/cbic.200600393. [DOI] [PubMed] [Google Scholar]; c Pistorius D.; Müller R. ChemBioChem 2012, 13, 416–426. 10.1002/cbic.201100575. [DOI] [PubMed] [Google Scholar]; d Wakimoto T.; Egami Y.; Nakashima Y.; Wakimoto Y.; Mori T.; Awakawa T.; Ito T.; Kenmoku H.; Asakawa Y.; Piel J.; Abe I. Nat. Chem. Biol. 2014, 10, 648–655. 10.1038/nchembio.1573. [DOI] [PubMed] [Google Scholar]
- a Kwon H.-J.; Smith W. C.; Scharon A. J.; Hwang S. H.; Kurth M. J.; Shen B. Science 2002, 297, 1327–1330. 10.1126/science.1073175. [DOI] [PubMed] [Google Scholar]; b El-Sayed A. K.; Hothersall J.; Cooper S. M.; Stephens E.; Simpson T. J.; Thomas C. M. Chem. Biol. 2003, 10, 419–430. 10.1016/S1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]; c Simunovic V.; Zapp J.; Rachid S.; Krug D.; Meiser P.; Müller R. ChemBioChem 2006, 7, 1206–1220. 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]; d Robbins T.; Kapilivsky J.; Cane D. E.; Khosla C. Biochemistry 2016, 55, 4476–4484. 10.1021/acs.biochem.6b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.