

Abstract
Bone marrow fibrosis is a central pathological feature and World Health Organization major diagnostic criterion of myelofibrosis. Although bone marrow fibrosis is seen in a variety of malignant and non-malignant disease states, the deposition of reticulin and collagen fibrosis in the bone marrow of patients with myelofibrosis is believed to be mediated by the myelofibrosis hematopoietic stem/progenitor cell, contributing to an impaired microenvironment favoring malignant over normal hematopoiesis. Increased expression of inflammatory cytokines, lysyl oxidase, transforming growth factor-β, impaired megakaryocyte function, and aberrant JAK-STAT signaling have all been implicated in the pathogenesis of bone marrow fibrosis. A number of studies indicate that bone marrow fibrosis is an adverse prognostic variable in myeloproliferative neoplasms. However, modern myelofibrosis prognostication systems utilized in risk-adapted treatment approaches do not include bone marrow fibrosis as a prognostic variable. The specific effect on bone marrow fibrosis of JAK2 inhibition, and other rationally based therapies currently being evaluated in myelofibrosis, has yet to be fully elucidated. Hematopoietic stem cell transplantation remains the only curative therapeutic approach that reliably results in resolution of bone marrow fibrosis in patients with myelofibrosis. Here we review the pathogenesis, biological consequences, and prognostic impact of bone marrow fibrosis. We discuss the rationale of various anti-fibrogenic treatment strategies targeting the clonal hematopoietic stem/progenitor cell, aberrant signaling pathways, fibrogenic cytokines, and the tumor microenvironment.
Introduction
Bone marrow fibrosis (BMF) is characterized by the increased deposition of reticulin fibers and in some cases collagen fibers. The currently accepted methods of evaluating and scoring BMF are primarily dependent on manual grading by the hematopathologist based on the density and type of fibrosis (Table 1).1,2 There are a number of hematologic and non-hematologic disorders that are associated with increased BMF (Table 2).3 Myelofibrosis (MF) refers to the Philadelphia chromosome (BCR-ABL1)-negative myeloproliferative neoplasm (MPN) originating at the level of the multipotent hematopoietic stem cell. MF can present as primary myelofibrosis (PMF), or arise from a pre-existing diagnosis of polycythemia vera or essential thrombocythemia. MF is characterized by variable degrees of cytopenias, a leukoerythroblastic blood picture, and extramedullary hematopoiesis resulting in progressive splenomegaly and debilitating disease-related constitutional symptoms, compromising quality of life.4,5 In addition to increased disease-related morbidity, MF results in early death with the median survival of affected patients being approximately 6 years.6 Causes of early death include leukemic transformation, complications arising from progressive bone marrow failure, portal/pulmonary hypertension, infections, thrombosis and bleeding.7 Pathologically, MF is characterized by thickening and distortion of bony trabeculae, deposition of reticulin and collagen fibers, and megakaryocytic hyperplasia with atypical features.8
Table 1.
European consensus on the grading of bone marrow fibrosis.1

Table 2.
Conditions associated with bone marrow fibrosis.3

The exact pathogenesis of MF is not fully understood. However, better understanding of the role of increased JAK-STAT signaling [either through activating mutations (JAK2V617F, MPL515L/K) within the signaling pathway, or mutations involving CALR], the role of deregulated pro-inflammatory cytokine expression, and the impaired bone marrow microenvironment is transforming the treatment approach for MF. Here we review the pathogenesis of MF and the prognostic impact of BMF, and highlight the potential prospects for anti-fibrogenic strategies that can be utilized in the treatment of MF and other hematologic malignancies.
Pathogenesis
Cytokines
A major biological hallmark of MF is a significant elevation in circulating pro-inflammatory cytokines. The MF inflammatory cytokine signature is believed to be both a consequence of the malignant clone as well an integral modifier of the bone marrow microenvironment, thereby, promoting malignant hematopoiesis.9 Transforming growth factor beta (TGF-β) is a pleiotropic cytokine that potently stimulates fibroblasts to produce extracellular matrix.10–13 It also increases the expression of proteases that inhibit enzymes involved in the degradation of extracellular matrix. Experimental studies have demonstrated that TGF-β1 is important in the development of BMF in animal models.14 Chagraoui et al. compared the pathological changes in irradiated wild-type recipient mice repopulated with thrombopoietin-overexpressing hematopoietic stem cells (HSC) from homozygous TGF-β1−/− or wild-type littermates.15 Mice engrafted with wild-type and TGFβ1−/− cells developed thrombocytosis, leukocytosis, and increased numbers of progenitor cells in the blood and spleen. However, BMF and reticulin deposition in the spleen occurred only in mice reconstituted with wild-type cells. Additionally, osteosclerosis was seen only in mice engrafted with wild-type cells.15
The Gata1low murine model of MF is characterized by normal or mild elevation in TGF-β1 levels. Elevated levels of TGF-β1 have also been documented in patients with MF.16 In order to evaluate the pathological contribution of TGF-β in MF, Zingariello et al. utilized a Gata1low mouse model of MF.17 First, the investigators demonstrated that despite normal or mildly elevated plasma concentrations of total and bioactive TGF-β1 in PMF patients and Gata-1low mice, the TGF-β1 content in megakaryocytes was increased 5- to 10-fold in both. In Gata-1low mice, this increase was accompanied by an increase in TGF-β1, hedgehog, and p53 signaling pathways in the spleen and bone marrow. The mammalian target of rapamycin (mTOR) signaling pathway was found to be increased in the spleen only. The biological consequences of altered gene expression were predicted using the David bioinformatics database and are in concordance with the GATA1low phenotype. Increased TGF-β signaling results in increased levels of osteoblast differentiation in bone marrow but not in the spleen, increased apoptosis, G1 arrest in bone marrow and spleen, reduced ubiquitin-mediated proteolysis in the bone marrow only. Ubiquitin-mediated proteolysis in the bone marrow is important for erythroid maturation. The mTOR signaling pathway is important for erythropoiesis and its selective increase in the spleen may lead to erythroblast maturation and promote extramedullary hematopoiesis. Importantly, the investigators demonstrated that inhibition of TGF-β1 signaling in Gata-1low mice resulted in reduced BMF, neovascularization/osteogenesis and increased hematopoiesis in the bone marrow while a reduction of hematopoiesis in the spleen was seen.17
TGF-β1 also stimulates fibroblasts to produce bone morphogenetic protein-6 in vitro.18 Additionally, the expression of bone morphogenetic protein-4, -2, -5 and -6 genes is increased in GATA-1low mice. Bone morphogenetic proteins are a group of proteins involved in extracellular matrix synthesis, formation, and homeostasis. Although the exact role of bone morphogenetic proteins in MF is not clear, these studies suggest that they have an integral role in the pathogenesis of BMF.18
Advanced MF is not only typified by BMF, but also by osteosclerosis with an increase and thickening of the bone trabeculae. These bone trabeculae are characterized by the absence or sparse presence of osteoclasts. Bock et al. demonstrated that this pathological finding may be related to increased expression of osteoprotegerin, a known inhibitor of osteoclast formation. Levels of osteoprotegerin expression were significantly increased up to 71-fold in bone marrow samples from patients with severe BMF and documented osteosclerosis.19
Studies of cardiac fibrosis in animal models have demonstrated that TGF-β upregulates expression of Lysl oxidase (LOX).20 The LOX family contains genes encoding copper-dependent enzymes that are important for covalent cross-linking of collagen and elastin, ultimately leading to stabilization of extracellular matrix.21 Hypoxia induces LOX expression in different types of cancer cells.22 This increase in expression of LOX is associated with proliferation, invasion, and a worse prognosis.21 In vitro studies and animal models suggest an intricate interplay between LOX, megakaryocytes and the bone marrow matrix.23 LOX, by enhancing the binding of platelet-derived growth factor to its receptor, promotes expansion of the megakaryocyte lineage. Although LOX is not critical for the induction of megakaryocyte ploidy, its expression is increased in low ploidy megakaryocytes and decreased in mature and high ploidy megakaryocytes.23 Eliades et al. showed that LOX expression is increased in megakaryocytes derived from the GATA-1low murine model. Importantly, inhibiting LOX led to a decrease in BMF in these GATA-1low mice.23
Several other studies of MF found increases in various inflammatory cytokines. Tefferi et al. analyzed plasma levels of 30 cytokines/chemokines in 127 patients with PMF, identifying a common cytokine signature. Among the 20 cytokines whose levels were statistically different from those in control samples, interleukin (IL)-8 and IL-2R had the strongest correlation with phenotype and prognosis. Increased plasma levels of these two cytokines were associated with the presence of constitutional symptoms, transfusion need, leukocytosis, and inferior overall and leukemia-free survival.24 IL-8 is a pleiotropic pro-inflammatory cytokine released by numerous cells and has angiogenic, mitogenic and growth factor activity. IL-8 expression has been associated with worse prognosis in numerous solid malignancies. The pathogenic role of IL-8 in MF is not yet fully clear, but it may be involved in leukemic transformation through its growth factor and mitogenic activities.25 An increase in IL-2R levels may be a reflection of either immune activation or an increase in tumor cell burden.24
Lipocalin-2 is a potent inflammatory cytokine that is markedly elevated in the plasma of patients with MPN (particularly MF) and has been shown to preferentially promote proliferation of MF CD34+ cells, induce breaks in double-stranded DNA and cause apoptosis of normal bone marrow cells, as well as enhance stromal cell proliferation through the production of reactive oxygen species.26 A lipocalin-2-induced increase in the expression of the extracellular matrix protein collagen type 1 (COL1A1) by mesenchymal stem cells as well as other disturbances in the bone marrow microenvironment likely contribute to the progression of the malignant HSC.
Some studies have demonstrated dysregulation in the levels of various other circulating cytokines, although the findings have not been reproducible in all studies.9 This may reflect differences in stage of disease, prior treatments, and technical aspects. However, despite these inconsistencies, cytokines are postulated to play an important role in the initiation, progression, and phenotypic presentation of MF.
Cellular interactions transforming the bone marrow niche
Osteoblastic lineage cells constitute the endosteal niche which is defined anatomically by its close proximity to trabecular or cortical bone. Osteoblastic lineage cells are derived from multipotent stromal cells and are important for the support and maintenance of normal HSC.27,28 Scheper et al. eloquently demonstrated the potential of BCR-ABL+ clonal cells to transform the endosteal bone marrow niche into a “self-reinforcing leukemic niche”. BCR-ABL1 transgenic mice have increased expansion of osteoblastic lineage cells. This increase is associated with an increase in BMF and trabecular thickening. The expansion of osteoblastic lineage cells is driven by an interaction between the MPN HSC and multipotent stromal cells. MPN HSC stimulate production of multipotent stromal cells of osteoblastic lineage through cytokines such as thrombopoietin and CCL3, as well as direct cell contact. Notably, the expansion of osteoblastic lineage cells and the associated BMF are reversible. It was demonstrated that by blocking BCR-ABL expression, the numbers of osteoblastic lineage cells decreased and BMF resolved.28
The expanded osteoblastic lineage cells have increased expression of genes involved in the regulation of extracellular matrix, cell adhesion, and inflammatory responses. These genes include targets of TGF-β1, suggesting increased TGF-β1 signaling in the osteoblastic lineage cells. Importantly the ability of the expanded populations of osteoblastic lineage cells to support normal HSC is compromised, as evidenced by reduced expression of HSC retention factors. Additionally, these cells have increased expression of pathways such as the TGF-β1 pathway, which may promote myeloid neoplastic differentiation. It was demonstrated that a malignant HSC clone can transform the bone marrow niche into a pathological environment preferentially supporting the neoplasm rather than normal HSC.27,28
Genetics
Since the discovery of the Janus kinase 2 (JAK2)V617 mutation in 2005, several other clonal markers have been identified including mutations in MPL and CALR.8,29,30 Understanding genotype-phenotype associations in MPN will likely allow for better prognostication and potentially provide novel targets for therapeutic intervention.
JAK2 is a member of the JAK family kinases (JAK1, JAK3, and TYK2). These cytoplasmic tyrosine kinases are associated with transmembrane class receptors of a number of cytokines (e.g. erythropoietin, thrombopoietin, granulocyte-stimulating factor) involved in cellular growth, differentiation, and survival of hematopoietic and immune cells. JAK2 is important in mediating the signaling pathways of these cytokines.2
JAK2-signal transducers and activators of transcription (STAT) dysregulation plays a central role in the pathogenesis of MPN. JAK2V617F has been identified in approximately 96% of patients with polycythemia vera, 50% of patients with essential thrombocythemia and 60% of patients with PMF. JAK2 signaling is initiated by ligand binding to the cognate cytokine receptor leading to dimerization, which in turn leads to JAK autophosphorylation. The phosphorylated kinases then phosphorylate intracellular receptor tyrosine residues creating a binding side for SH2 domain-containing proteins such as STAT. STAT phosphorylation leads to dimerization and translocation to the nucleus, affecting the transcription of genes important for cell-cycle regulation, apoptosis, and proteasomal degradation.2 Several other signaling pathways are also activated by JAK2 including the PI3K/Akt and mitogen-activated protein kinase (MAPK) signaling pathways. JAK-STAT signaling is negatively regulated by suppressor of cytokine signaling proteins (SOCS), Casitas B-cell lymphoma (CBL) and by protein tyrosine phosphatases (PTP).2
JAK2V617F is constitutively active resulting in chronic activation of the JAK-STAT pathway. This is achieved by a gain of function in the JH1 domain and loss of function in the auto-inhibitory JH2 domain. Furthermore, JAK2V617F escapes negative feedback by SOCS3.2,31 The exact mechanism by which JAK2V617F contributes to the pathogenesis of MF is not fully known. However, as mentioned above, JAK2V617F may promote BMF by transforming the bone marrow microenvironment through various cellular and cytokine-mediated mechanisms.27,28 Alhough Schepers et al. did not observe a defect in megakaryocytopoiesis, earlier studies suggested that the effects of JAK2V617F are in part mediated by its impact on platelet function and structure. Using a JAK2V617F knock-in mouse model of essential thrombocythemia, Hobbs et al. showed that the proportion of JAK2V617F-positive megakaryocytes forming proplatelets is greater than the proportion of control megakaryocytes. Moreover, the JAK2V617F knock-in megakaryocyte has increased fos gene expression. Fos is a regulator of TGF-β signaling.32 Previously Muth et al. studied patients with MPN and reported that proplatelet numbers were higher in patients with essential thrombocythemia and pre-fibrotic MF than in controls.33 Proplatelets were even more abundant in patients with fibrotic MF compared to those with pre-fibrotic MF or essential thrombocythemia.34 Exaggerated production of proplatelets may be associated with increased thrombospondin production. Thrombospondin 1 in PMF is thought to be fibrogenic by activating latent TGF-β1 and inhibiting metalloproteinases.33,35
Calreticulin (CALR) is a multifunctional calcium-binding protein that is located primarily in the endoplasmic reticulum. CALR is important for intracellular calcium homeostasis and intracellular protein chaperoning.36 Recently two groups identified CALR mutations in approximately 20 to 25% of patients with essential thrombocythemia and PMF.29,37 All mutations identified so far are either insertions or deletions in exon 9 of the gene. CALR mutations are mutually exclusive with mutations in both JAK2 and MPL.29,37 Clinically, CALR mutations in PMF are associated with younger age, a higher incidence of thrombocytosis, and lower leukocyte count.36,37 The prognostic impact of CALR is discussed later.
CALR mutations lead to impaired calcium binding and cellular dislocation. This in turn may lead to activation of several pathways including IL-3 and JAK-STAT signaling.30 The functional consequences of CALR mutations have been recently elucidated.38,39 In murine transplant studies, mutant CALR alone was sufficient to induce an MPN phenotype. In vitro, mutant CALR leads to transformation and activation of the JAK-STAT pathway only in MPL-expressing cell lines. Moreover, mutant, but not wild-type, CALR was shown to bind MPL. These findings suggest that mutant CALR is sufficient to induce MPN through an MPL-dependent pathway.
Somatic mutations of JAK2, MPL, and CALR behave as founding driver mutations responsible for the MPN phenotype. As the disease progresses, the founding malignant clone acquires additional subclonal mutations. There are a number of subclonal mutations which have been reported such as those in ASXL1, EZH2, CBL, IDH1/IDH2, TP53 and SRSF2. These mutations also have prognostic impact as discussed later.30
Prognosticators and predictors beyond the IPSS and DIPSS scores
Risk stratification is at the core of current MF management. The International Prognostic Scoring System (IPSS) and the Dynamic International Prognostic Scoring System (DIPSS) are the two main prognostication schemes used to guide risk-adapted treatment strategies for patients with MF.7 These scores combine patient and MF-specific clinical variables to provide a risk category with an associated median survival. However, these scoring systems do not always reflect the genetic heterogeneity of MF. Several groups have now identified mutational profiles associated with poor prognosis in MF. Vannucchi et al. showed that patients harboring mutations in any one of the following genes, ASXL1, EZH2, SRSF2 or IDH1/IDH2, have a shorter overall survival and a higher risk of their disease transforming into acute myeloid leukemia compared to MF patients who do not have mutations in any of these genes.40,41 The prognostic impact of mutations in CALR versus JAK2 versus MPL mutations versus triple negativity was assessed in a cohort of 254 patients with PMF. Triple negative status and CALR−/ASXL+ patients had the shortest median overall survival of 2.5 years and 2.3 years, respectively.36 Low JAK2V617F allele burden at diagnosis, homozygosity for JAK2V617F, as well as elevated levels of IL-8 and/or IL-2R are also associated with worse overall survival.42,43,24 Other molecular markers have also been associated with poor prognosis; however, they have not yet been validated in large prospective studies.
Impact of bone marrow fibrosis on the prognosis of myeloproliferative neoplasms and other hematologic malignancies
Myelofibrosis
As discussed above, the IPSS and DIPSS are currently the two commonly used scales to assess prognosis in MF and the prognostication may now be further refined through the incorporation of gene mutation profiling. BMF is not at present included in risk stratification schemes for MF or other hematologic malignancies. However, studies suggest a correlation between the grade of BMF and prognosis in MF.44
Several studies have evaluated the prognostic impact of BMF in MF. In most of these studies, high grade BMF was associated with worse outcome.45,46 In a retrospective analysis that included 131 patients with PMF, Lekovic et al. found that BMF grade >1 was associated with shorter overall survival (median: 51 months) compared to grade ≤1 (median: 147 months). Importantly the grade of BMF was an independent risk factor for overall survival when analyzed with IPSS and competing comorbidities.45 In a study that included 196 patients with PMF, higher grade BMF was an independent predictor of poor survival.46 Moreover, higher grade BMF can further refine the prognosis of PMF patients with low and intermediate risk IPSS.46 However, Nazha et al. reported similar rates of overall survival, event-free survival, and disease transformation in MF patients with different grades of BMF. Their study included 512 patients with MF: 1% had MF grade 0, 9% MF grade 1, 33% MF grade 2, and 57% MF grade 3. Higher grade of BMF correlated with clinical manifestations of MF such as lower hemoglobin level, higher percentage of peripheral blood blasts and larger spleen. Although grade of BMF was demonstrated to be correlated with IPSS and DIPSS risk, an impact on overall survival was not evident in this analysis. This may in part be explained by higher numbers of patients with MF grade 2 and MF grade 3 in this cohort.47 In a recent retrospective Italian study of 540 bone marrow specimens from time of PMF diagnosis, higher grade BMF was found to be associated with more constitutional symptoms (P<0.0001), larger splenomegaly (P<0.0001), greater risk of developing anemia (P<0.0001) and thrombocytopenia (P=0.003), as well as with higher risk IPSS score.48 Although there was no association between driver mutation status and grade of BMF, high molecular risk mutations, such as ASXL1 and EZH2, were seen more frequently in PMF patients with higher grade BMF. Importantly, grades 2 and 3 BMF maintained their adverse prognostic values for overall survival [HR 3.9 (1.4–10.8) and HR 4.2 (1.5–12.0), respectively], independently of IPSS score, driver mutation and high-risk molecular status.
Pre-transplant MF grade 3 was associated with worse overall survival in a univariate analysis of patients with MF who underwent hematopoietic stem cell transplantation (HSCT) with reduced intensity conditioning. However, this was not confirmed in multivariate analysis.49 Interestingly, the regression of BMF after HSCT was associated with a better overall survival, as discussed in more detail in the treatment section.44 With the advent of mutation-based prognostication in MF, it remains to be seen whether BMF grade will have an additional prognostic impact.
Essential thrombocythemia/polycythemia vera
Increased reticulin BMF has been reported in approximately 20% of patients with essential thrombocythemia and can also be seen to various degrees in the bone marrow of patients with polycythemia vera.50,51 BMF was associated with an increased risk of transformation to MF or acute myeloid leukemia (HR 1.89, P=0.0359 for essential thrombocythemia; HR 1.71, P=0.0164 for polycythemia vera). However, BMF grade was not associated with worse overall survival.52 These results were replicated in another study by Campbell et al. in which 311 patients with essential thrombocythemia were included. Again, increased reticulin BMF grade at diagnosis was not significantly associated with poorer overall survival. Interestingly, increased grade of reticulin BMF was associated with an increased rate of arterial thrombosis, major hemorrhage and progression to MF.50 The presence of BMF in polycythemia vera has been associated with an increased risk of transformation to MF, but this has not been shown to affect overall survival.51
Chronic myelogenous leukemia
BMF is not specific to MPN and can be seen in many other hematologic malignancies, solid malignancies, and non-malignant conditions, as summarized in Table 2. Patients with chronic myelogenous leukemia have been shown to have increased BMF compared to their normal counterparts53 and, in the pre-imatinib era, an increased degree of BMF prior to starting treatment was associated with a worse outcome.54 Treatment with imatinib was associated with complete reversal of pathological BMF in 73% of treated patients (14/19 patients) in one series.53 The degree of BMF in chronic myelogenous leukemia at baseline in imatinib-treated patients was not shown to be an adverse prognostic factor.55 However, emergence of small foci with abnormal fiber increase or BMF during imatinib treatment is associated with a lower probability of achieving complete cytogenetic response or major molecular response. Furthermore, emergence of abnormal fiber increase can occur in patients who have achieved complete cytogenetic or major molecular response preceding the loss of molecular response. This is believed to be a consequence of the ability of the chronic myelogenous leukemia clone to produce fibrogenic cytokines even when the quantity of the clone is below the threshold of cytogenetic or molecular detection.53,56 Importantly emerging or relapsing abnormal fiber increase was an independent predictor of failure of imatinib treatment. Advanced BMF in chronic myelogenous leukemia has also been associated with an increased risk of developing accelerated phase and blast phase disease.53
Myelodysplastic syndromes
Significant BMF has been reported in approximately 10–20% of patients with myelodysplastic syndromes (MDS). BMF in MDS is associated with profound cytopenias and increased red cell/platelet transfusion dependence.57,58 In a study of 301 patients with MDS, patients with grades 2 and 3 BMF had shorter overall and leukemia-free survival compared to those with grade 0 or 1 BMF. The impact of BMF on overall and leukemia-free survival was seen in WHO subgroups without excess of blasts (overall survival, HR=2.89, P=0.001; leukemia-free survival, HR=2.21, P=0.006) and in refractory anemia with excess blasts type 1 and type 2 (overall survival, HR=2.25, P=0.004; leukemia-free survival, HR=2.03, P=0.01). In MDS patients stratified by the IPSS and WPSS (WHO classification-based Prognostic Scoring System) risk score, grades 2 and 3 BMF maintained their prognostic significance. Furthermore, developing BMF during the course of MDS was shown to be associated with a worse outcome.59 These results held true even when applying the revised-IPSS. Advanced BMF is an adverse risk factor that is not captured by the revised-IPSS.58 In this study, BMF grade 2–3 was an independent variable that negatively influenced overall and leukemia-free survival in MDS patients treated with a non-transplant modality. Moderate/severe BMF was identified in 17% of cases of therapy-related MDS, but was not associated with an increased risk of leukemic transformation or inferior outcome.57 This may in part be explained by the intrinsic high-risk features of therapy-related MDS resulting from genetic and epigenetic alterations rather than histopathomorphological aspects. The presence of severe BMF was associated with reduced overall survival even in MDS patients who underwent allogeneic HSCT.58,60 It has also been shown that in MDS patients treated with azacitdine, grade 3 BMF is associated with a poor response to treatment.61
The potential prognostic impact of BMF extends beyond myeloid malignancies. Among patients with chronic lymphocytic leukemia, those with high grade BMF (grades 2 and 3) had a worse 5-year overall survival rate than those with grade 0–1 BMF (51.9% versus 86.92 %, respectively). Fluorescence in situ hybridization analysis was performed in only a few patients in this study and none was tested for P53 status. It, therefore, remains unknown whether BMF has true prognostic significance independent of modern prognostic features.62
Grading of bone marrow fibrosis
Increased reticulin and collagen deposition in the bone marrow is the hallmark of MF. Reticulin and collagen are connective tissue fibers that provide the structural framework of bone marrow stroma. BMF is routinely assessed and graded in core biopsies from patients with a known or suspected MPN, and is a major diagnostic criterion in the 2008 WHO classification system of MPN. Reticulin is detectable by silver staining methods (Figure 1) while collagen deposition is detected by trichrome staining (Figure 2).
Figure 1.
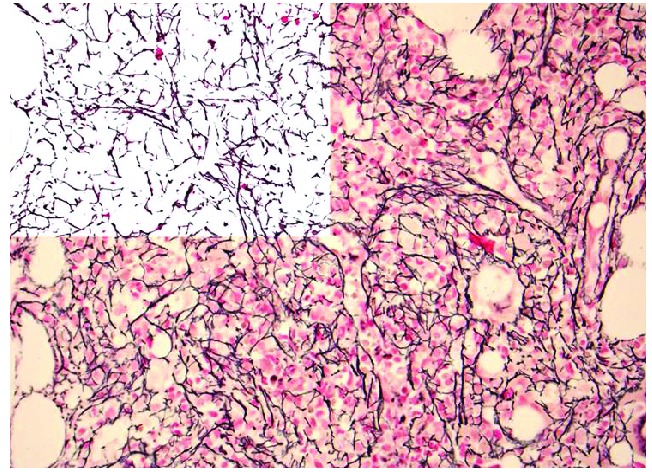
Photomicrograph showing reticulin (silver) stain in a specimen from a patient with myelofibrosis. Note the diffuse and dense increase in reticulin with extensive intersections which would be graded as 2+ myelofibrosis. The left upper corner is a mark up image of post-image analysis processing with the reticulin fibers being de-convoluted from background tissue. The area occupied by the fibers, as well as branching points, could be objectively quantified using computer assisted image analysis (original magnification, 400X).
Figure 2.
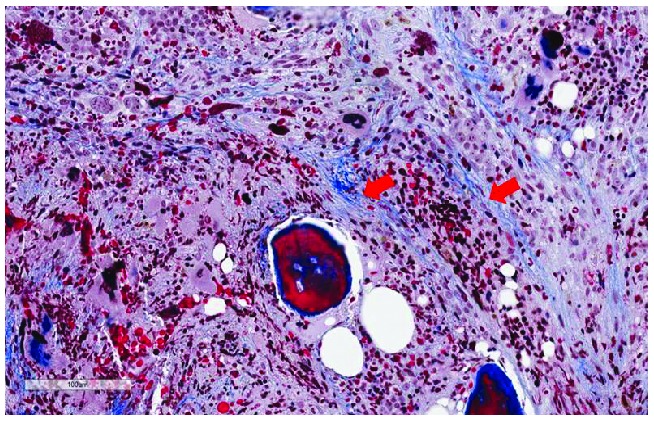
Trichrome stain showing deposits of collagen fibers. The fibers are arranged in bundles that are marked with red arrows (original magnification 400X).
Several reporting systems have been proposed to quantify reticulin and collagen deposition in the bone marrow. The most frequently used grading systems are based on reticulin grade scored using the Bauermeister system from 1971 (0–4 scale) and the recently revised European consensus system (0–3 scale) (Table 1 and Figure 1). All accepted traditional grading systems are dependent on manual grading of reticulin by a histopathologist. These traditional grading systems are semi-quantitative, and suffer major limitations related to subjectivity.63 This problem is further confounded by the heterogeneity of fibrosis within a given sample, variability of pre-analytical processing, staining inconsistency and subjective assessment from lack of a positive staining internal standard, and inadequate guidance to disregard lymphoid nodules, vessels as well as fibers framing adipocytes. Reports that address these limitations are scarce to non-existent.
Whole slide imaging with computer-assisted image analysis has emerged recently as a more objective approach to provide numerical assessment of both fibrosis and osteosclerosis, offering a more precise way of following patients over time.63 The most significant advantages of this approach are its contribution to standardizing assessment of fibrosis among histopathologists and hematologists and that it makes it possible to assess disease progression or to serially assess the impact of a therapy on marrow histology as a surrogate endpoint of efficacy of therapy.
The microscopic assessment of BMF by skilled hematopathologists remains the standard of care in the diagnosis MPN. However, computer-assisted image analysis is being increasingly utilized in clinical trials for objective assessment of BMF as an adjunct to semi-quantitative grading. Computer-assisted image analysis correlates well with morphology but was more sensitive than histopathology for detecting BMF level changes in MF patients undergoing treatment.64
Recently, a stereology-based computer-assisted image analysis method using systematic uniform random sampling and line counting to calculate length density of the reticulin network as well as to measure heterogeneity across the bone marrow sample was developed by Salama et al.65 Computer-based stereology proved to be more reproducible than manual scoring at predicting a therapeutic actionable cut-point. This novel stereology-based method is fast and can be easily implemented in the clinical laboratory with a high level of reproducibility. A major advantage of this approach is that it can provide a measure of heterogeneity of the BMF.
Another promising stain-free modality that employs two photon excitation and second harmonic generation phenomena has great potential as an application for quantification of BMF where pre-analytical variability related to stain limitation can be eliminated. Accurate scoring of BMF remains a major area of academic interest and an unmet clinical need.
Treatment strategies for bone marrow fibrosis
Most of the data regarding treatments targeting BMF has been garnered from MF. However, because of a potential overlap in the pathogenesis of BMF among different disease entities, some treatment strategies can be used in diseases other than MF. Historically, BMF has been considered a secondary or reactive process driven by the clonal malignancy and not produced by the malignant cell population itself. In an early, small study, the incidence of cytogenetic abnormalities in fibroblasts from patients with MF was comparable to that in patients with non-malignant causes of anemias. Importantly, the karyotypic abnormalities seen in fibroblasts were often different from those seen in the hematopoietic cells. This finding supports the hypothesis that the fibroblast population in MF is the result of a reactive phenomenon rather than a malignant outgrowth.66 However, there are evolving data to suggest clonal involvement of fibroblasts in PMF as evidenced by the detection of JAK2V617F and other chromosomal abnormalities in fibroblasts that are concordant with the MPN cell population.67
In general, the goals of MF-directed treatment can be divided into two broad categories: clonal eradication and treatments targeting various signaling pathways and mediators implicated in BMF. As discussed in the section on pathogenesis, eradicating the BCR-ABL1+ clonal cells in animal models of chronic myelogenous leukemia was associated with reversal of pathological BMF.28 In PMF, allogeneic HSCT is the only therapeutic intervention with curative potential, effectively eradicating the malignant MPN HSC clone, leading to reversal of BMF and MPN-associated histomorphological features of the bone marrow. In a study by Rondelli et al., 21 patients with intermediate/high risk MF underwent reduced intensity conditioning-HSCT. Nineteen had grade 3 or 4 BMF at baseline. After transplantation, all evaluable patients had BMF grade 0 – 2 for at least 12 months of follow up.68 Importantly, regression of BMF at day +100 after HSCT in patients with MF is associated with improved survival, independently of IPSS score at the time of transplantation, as demonstrated by Kroger et al.44 This conclusion was reached in a study that included 57 patients with PMF or MF evolving from polycythemia vera or essential thrombocythemia who underwent reduced intensity conditioning-HSCT. Bone marrow histology was available for 35 patients at days +30 and +100, while 13 patients only had a day +30 bone marrow biopsy, and nine patients only had a day +100 bone marrow biopsy. Patients with grade 2/3 BMF at day +100 had a higher rate of dependence on red blood cell and platelet transfusions compared to patients who had grade 0/1 BMF at day +100. Importantly, patients with grade 0/1 BMF at day +100 after HSCT had a 5-year overall survival rate of 96% compared to 57% in those with persistent grade 2/3 BMF (P=0.04). This survival advantage was attributed to a lower risk of treatment-related mortality and a decrease in relapse rate. Additionally, patients with a reduction of BMF by 2 or 3 grades at day +100 after HSCT had a trend towards a better overall survival rate than that of patients with unchanged BMF (95% versus 71%, respectively; P=0.19). The regression of BMF achieved in patients after reduced intensity conditioning-HSCT did not correlate with JAK2V617F allele burden or with IPSS risk group at the time of transplantation.44 This study highlights the prognostic significance of BMF in MF not accounted for by the current prognostic scoring systems. It also raises the potential of improving outcomes of HSCT by combining anti-fibro-genic strategies prior to or even after the transplant.
Apart from HSCT, none of the conventional therapies utilized in the treatment of MF is considered curative. However, there are several published reports describing reversibility of BMF with interferon-α (IFN-α) based therapy. Early data from patients with MPN did not show significant changes in grade of reticulin BMF with the use of recombinant IFN-α.69 However, in a subsequent, small, prospective study, 17 patients with early stage MF, without grade 3/4 BMF, were treated with IFN-α (n=14) or pegylated IFN-α (n=3) and four of the patients had documented improvements in bone marrow reticulin and collagen fibrosis. Two of these patients had complete resolution of BMF and megakaryocytic atypia after 1–4 years of treatment. The median duration of bone marrow responses was 1.9 years.70 A large retrospective analysis by Ianotto et al. suggests that IFN-α2a can be clinically effective in patients with MF who do not have massive splenomegaly (spleen size <6 cm), marked leukopenia or thrombocytopenia, and BMF <grade 3.71 However, there were no data regarding the effect of IFN-α treatment on BMF in this study. Evidence of reduction in grade of BMF with IFN-α therapy in patients with MF is mostly restricted to case reports.72 Collectively, these reports suggest IFN-α therapy may have the most beneficial impact on BMF early on in the disease course. However, the clinical significance of this finding and the impact on overall survival of reducing/eliminating BMF with IFN-α-based therapy remain uncertain.
Impact of ruxolitinib on bone marrow fibrosis
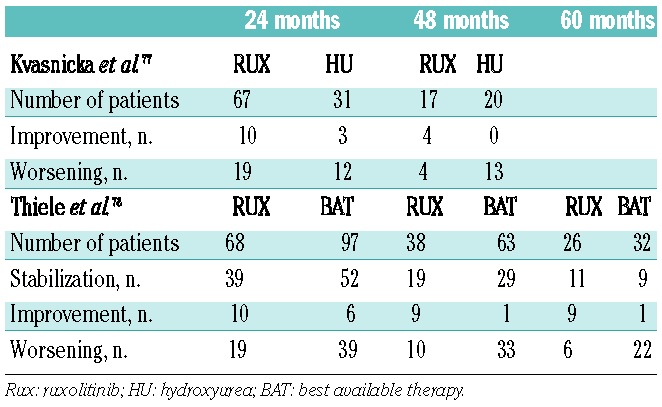
With the introduction of selective JAK2 inhibitors into clinical investigation over the last 10 years, initial expectation of clonal suppression as measured by elimination of molecular and karyotypic abnormalities was dampened by the observations of persistent clonal hematopoiesis and unaltered MPN bone marrow pathological features. None of the approximately dozen oral JAK2 tyrosine kinase inhibitors evaluated in the preclinical and clinical MF setting has recapitulated the success of the BCR-ABL1 inhibitors in chronic myelogenous leukemia. An analysis of results from bone marrow biopsy specimens obtained from MF patients enrolled in the pivotal phase III COMFORT studies at 6 and 12 months of therapy with ruxolitinib failed to show improvements in histopathological abnormalities and did not confirm reductions in grade of BMF.73,74 However, several case reports and retrospective analyses suggest that longer treatment duration with ruxolitinib may have a modest impact on BMF in a subset of patients. In several published case reports, resolution of BMF was noted after 17 and 48 weeks of ruxolitinib treatment.75,76 Recently, reports of long-term follow-up of bone marrow responses in MF patients treated in the phase I/II trial of ruxolitinib have suggested modest responses in BMF in selected cases. The changes in grade of BMF at 24 and 48 months of ruxolitinib treatment were analyzed and compared to those in a similar, matched cohort of MF patients treated with hydroxyurea (Table 3). At 24 months, BMF had stabilized in 57% and 52% of the ruxolitinib and hydroxurea treatment groups, respectively and 15% and 10%, respectively, had improvements in grade of BMF. At 48 months, stabilization was achieved in 53% of ruxolitinib-treated patients versus 35% of the hydroxyurea-treated patients. Improvements in BMF grade were achieved in 24% of the ruxolitinib group, but in none of those treated with hydroxyurea.77 These results were further validated in a larger cohort with a longer follow-up. In this study, changes in the grade of BMF were determined at 24, 48 and 60 months of ruxolitinib treatment and compared with those in a group of MF patients treated with “best available therapy”. The majority of patients treated with “best available therapy” received hydroxyurea or various sequential therapies, or were being observed; only a few patients were treated with IFN-α. As summarized in Table 3, compared to this control cohort, a higher percentage of patients in the ruxolitinib group had stabilization or improvement of BMF. Ruxolitinib-treated patients who achieved stabilization or improvement in grade of BMF at 24 months had a reduced relative risk of death in this analysis.78
Table 3.
Ruxolitinib therapy and effect on bone marrow fibrosis.
Targeting fibrogenic cytokines
Pirfenidone is an anti-fibrotic agent that inhibits fibrogenic cytokines including platelet-derived growth factor, tumor necrosis factor-α and TGF-β and was studied in 28 patients with MF. None of the patients had improvement in BMF or osteosclerosis. Additionally, there were no significant clinical benefits seen in terms of improvement in anemia or reduction in splenomegaly.79
Targeting fibrogenic cytokine expression either alone or in combination with a JAK2 inhibitor is an active area of research in MF. As previously mentioned, the TGF-β signaling pathway plays a central role in the pathogenesis of BMF in MF. Monoclonal antibodies antagonizing TGF-β are being evaluated in various fibrotic conditions including pulmonary fibrosis, glomerulosclerosis, and MF. GC1008 (fresolimumab, Genzyme) is a human IgG4 monoclonal antibody capable of neutralizing TGF-β isoforms 1, 2, and 3. The relative tolerability of GC1008 was demonstrated in a phase I trial in advanced melanoma and renal cell cancer.80 In an aborted phase I trial, three MF patients were treated with GC1008 and changes in bone marrow pathology were assessed after 6 and 12 months.14 There were no observed changes in bone marrow reticulin or collagen fibrosis after six or 12 cycles of treatment and no appreciable decrease in spleen size after six cycles of treatment. Interestingly, two treated patients obtained responses in anemia that were durable, including transfusion independence for over 16 months in one patient (unpublished observation, JM). Of interest, plasma levels of TGF-β were significantly higher in the three patients than in normal controls and after treatment with GC1008 were undetectable in two evaluable patients. Further clinical evaluation of TGF-β inhibition alone or in combination with other agents will be necessary for a complete assessment of this targeted therapeutic approach.14 The Myeloproliferative Disorder Research Consortium (MPD-RC) will be evaluating the oral selective TGF-β receptor 1 kinase inhibitor, galunisertib (LY2157299, Lilly), in patients with MF and at least MF grade 2 BMF.
Figure 3.
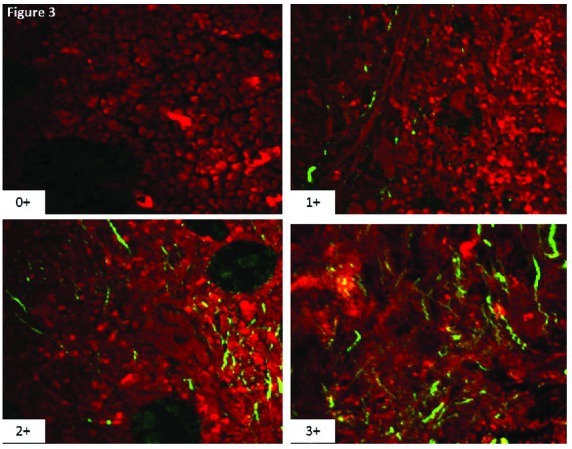
Four images from unstained bone marrow biopsies obtained from patients with myeloproliferative neoplasms with a spectrum of marrow fibrosis ranging from 0–3 according to the revised European consensus system for grading bone marrow fibrosis. The red is a pseudo color that highlights all tissue elements in the bone marrow according to two-photon excitation. The technology utilizes the second harmonic generation phenomenon to highlight fibrillar collagen with high specificity as highlighted by the green fluorescent colored structures.
Pentraxin (PTX) family proteins include C-reactive protein (CRP; PTX1), serum amyloid P (SAP; PTX2), and pentraxin-3 (PTX3).81 PTX2 is a circulating plasma acute phase response protein made by the liver which localizes to the site of injury and affects monocyte differentiation and function in the removal of damaged tissue. Physiologically, PTX2 may act as a powerful antifibrotic agent because of its ability to inhibit human derived fibrocyte differentiation.82 Injections of PTX2 reduce fibrosis significantly in several animal models (pulmonary fibrosis, ischemic cardiac fibrosis, renal fibrosis).83–88 On the other hand, PTX3 promotes human and murine fibrocyte differentiation.81 PRM-151 is a recombinant PTX2 being actively investigated in the treatment of various fibrotic diseases including MF (NCT01981850).89 In a phase II study that included 26 patients with MF, PRM-151 (alone or in combination with ruxolitinib) was associated with symptom improvement and/or BMF reduction in 43% of the treated patients. Specifically, a ≥1 grade reduction of BMF was seen in 35% of patients at 24 weeks of treatment. The therapy was well tolerated without significant infusion reactions or grade 3/4 treatment-associated adverse events.90,91 Longer term follow-up of 13 patients treated for at least 72 weeks with PRM-151 continued to demonstrate reductions of ≥1 grade of BMF in nearly 70% of patients, and improvements in spleen size, anemia and thrombocytopenia, as well as symptom burden.92 The second stage of this trial is now evaluating PRM-151 monotherapy in patients who are intolerant of, refractory to or ineligible for ruxolitinib therapy. The relative pre-treatment levels of PTX2 and PTX3 in the bone marrow may serve as a predictor of response to PRM-151.
As mentioned above, LOX levels are increased in GATA-1low mice and LOX inhibition led to a decrease in BMF in these animal models. Bone marrow samples from patients with PMF overexpress all LOX genes. Furthermore, serum LOX levels were significantly higher in PMF patients than in a control group.93 Simtuzumab is a humanized monoclonal antibody that binds to and inhibits LOXL2. A phase II trial of simtuzumab monotherapy (stage 1) and combination therapy with ruxolitinib (stage 2) in patients with intermediate-1 or higher risk MF has completed recruitment and the results were recently presented (NCT01369498).94 A total of 24 patients were treated in the stage 1 part of the study and 30 in the stage 2 part, for a minimum median duration of 22 weeks, without a clear signal of response in terms of reduction of BMF. Larger studies are required to determine whether serum levels of LOX can be used to refine diagnosis and prognosis, and to determine the predictive potential of response to LOX-directed therapy in patients with MF.
In the Gata1low mouse model of MF, the animals have increased expression of the hedgehog pathway in both bone marrow and spleen.17 An increase in the expression of hedgehog target genes has also been observed in granulocytes isolated from MPN patients.95 The exact role of the hedgehog pathway in MF and its contribution to BMF are not fully understood. However, preclinical and clinical data suggest that hedgehog pathway inhibitors have therapeutic activity in MF. Combining sonidegib (LDE225, Novartis), a hedgehog inhibitor, with ruxolitinib in a murine transplant model of essential thrombocythemia/MF resulted in a reduction of mutant allele burden in the bone marrow and a significant reduction in BMF compared to that achieved by ruxolitinib alone.
Sonidegib, saridegib, and PF-04449913 are currently under clinical investigation, alone or in combination with other agents, in MF. Seven patients with MF were included in a phase I study investigating PF-04449913 in myeloid malignancies. Two of these patients achieved durable clinical responses (>50% reduction in spleen size) and one patient had a significant reduction in BMF. The therapy was relatively well tolerated and is currently being investigated as a second-line agent in MF (NCT02226172).96,97 Sonidegib in combination with ruxolitinib was tested in a multicenter, phase Ib/II study: the maximum tolerated dose was not reached during the dose escalation phase, but myelosuppression and elevation of creatinine kinase levels were reported.98 A total of 27 MF patients were treated at the recommended phase II dose of 400 mg once daily with ruxolitinib 20 mg twice daily. Although the combination was relatively well tolerated and resulted in approximately 50% of patients achieving a ≥35% decrease in spleen volume, only two treated patients had at least a one grade reduction in BMF.99
Disrupting the myelofibrosis bone marrow microenvironment
As discussed above, modulation of the bone marrow microenvironment by the neoplastic hematopoietic clone plays an integral role in the pathogenesis of myeloid malignancies. Disrupting the tumor-microenvironment interaction is a potential therapeutic strategy in MPN.28 The effects of disrupting the tumor microenvironment by targeting Eph receptor tyrosine kinases are currently being studied in both solid and hematologic malignancies.100,101 EphA3 is important in cell positioning during fetal development and is not expressed in normal adult tissues. However, EphA3 expression has been demonstrated in various hematologic and solid tumors.88,89 In solid malignancies, EphA3 is preferentially expressed in tumor stroma, vasculature, and bone marrow-derived mesenchymal stem cells.101 Data from Vail et al. suggest that Eph is not ligated in stromal cells derived from the solid tumor so it is kinase-dormant. In solid tumors, kinase-dormant Eph lead to cell-cell adhesion, invasion and tumor maintenance. Targeting Epha3 by ChIIIA4 led to EphA3 kinase activation, cell contraction and apoptosis of solid tumor-resident multipotent stromal cells. In turn, this led to disruption of the integrity of tumor stromal architecture, microvasculature, and ultimately to inhibition of tumor growth. Epha3 is expressed in blood and mone marrow leukemic HSC as well as the stromal compartment.102
Humaneered® IIIA4 (KB004, Kalobios) is currently being evaluated in the treatment of hematologic malignancies including MF (NCT01211691). Early data from a phase I/II trial of 58 patients (the majority with acute myeloid leukemia) suggest a well-tolerable side effect profile with infections (41.4%), febrile neutropenia (20.7%), and infusion-related reactions (13.8%) being the most frequent side effects.103 Efficacy data are also encouraging. The potential impact of KB004 on BMF in responding patients is of interest. In a single patient with acute myeloid leukemia, a sustained complete remission was observed for over 1 year and this clinical response was also accompanied by a decrease in reticulin and collagen BMF. In another patient with MF, clinical improvement (decrease in spleen size, transfusion independence) was achieved and accompanied by a significant improvement in BMF.100 The anti-fibrotic mechanism of action of KB004 is unknown.
Conclusion
BMF is seen in many hematologic and non-hematologic conditions and is a prominent pathologic feature of MF. The extent to which BMF contributes to a disorganized bone marrow microenvironment and promotes disease progression rather than serving as a biomarker reflecting disease activity remains incompletely understood. BMF is a culminating effect of a complex interplay between MPN cells and supporting stromal cells through the interaction of various inflammatory cytokines such as TGF-β. Increased reticulin and collagen fibrosis may have a prognostic significance that is not accounted for by current prognostic scoring systems. Better understanding of the molecular and cellular mechanisms governing MF will potentially lead to more effective therapies targeting the MPN HSC. Anti-fibrotic strategies are currently being evaluated in a number of therapeutic trials aimed at disrupting the malignant bone marrow niche and promoting normal over malignant hematopoiesis. In the future, validating the prognostic impact of BMF, identifying soluble surrogates to allow for easier monitoring of changes in BMF, and combining anti-fibrogenic strategies with currently available treatments, including HSCT, will likely result in improved clinical outcomes.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/6/660
References
- 1.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128–1132. [PubMed] [Google Scholar]
- 2.Meyer SC, Levine RL. Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin Cancer Res. 2014;20(8):2051–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuter DJ, Bain B, Mufti G, Bagg A, Hasserjian RP. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007;139(3):351–362. [DOI] [PubMed] [Google Scholar]
- 4.Reilly JT, McMullin MF, Beer PA, et al. Guideline for the diagnosis and management of myelofibrosis. Br J Haematol. 2012;158(4):453–471. [DOI] [PubMed] [Google Scholar]
- 5.Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. [DOI] [PubMed] [Google Scholar]
- 6.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. [DOI] [PubMed] [Google Scholar]
- 7.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. [DOI] [PubMed] [Google Scholar]
- 8.Tabarroki A, Tiu RV. Molecular genetics of myelofibrosis and its associated disease phenotypes. Transl Med UniSa. 2014;8:53–64. [PMC free article] [PubMed] [Google Scholar]
- 9.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24(2):133–145. [DOI] [PubMed] [Google Scholar]
- 10.Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83(12):4167–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura A, Katoh O, Hyodo H, Kuramoto A. Transforming growth factor-beta regulates growth as well as collagen and fibronectin synthesis of human marrow fibroblasts. Br J Haematol. 1989;72(4):486–491. [DOI] [PubMed] [Google Scholar]
- 12.Massagué J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. [DOI] [PubMed] [Google Scholar]
- 13.Ignotz RA, Endo T, Massagué J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem. 1987;262(14):6443–6446. [PubMed] [Google Scholar]
- 14.Mascarenhas J, Li T, Sandy L, et al. Anti-transforming growth factor beta (TGF-β) therapy in patients with myelofibrosis. Leuk Lymphoma. 2014;55(2):450–452. [DOI] [PubMed] [Google Scholar]
- 15.Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood. 2002;100(10):3495–3503. [DOI] [PubMed] [Google Scholar]
- 16.Chou JM, Li CY, Tefferi A. Bone marrow immunohistochemical studies of angiogenic cytokines and their receptors in myelofibrosis with myeloid metaplasia. Leuk Res. 2003;27(6):499–504. [DOI] [PubMed] [Google Scholar]
- 17.Zingariello M, Martelli F, Ciaffoni F, et al. Characterization of the TGF-β1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood. 2013;121(17):3345–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bock O, Höftmann J, Theophile K, et al. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am J Pathol. 2008;172(4):951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bock O, Loch G, Schade U, et al. Osteosclerosis in advanced chronic idiopathic myelofibrosis is associated with endothelial overexpression of osteoprotegerin. Br J Haematol. 2005;130(1):76–82. [DOI] [PubMed] [Google Scholar]
- 20.Voloshenyuk TG, Landesman ES, Khoutorova E, Hart AD, Gardner JD. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine. 2011;55(1):90–97. [DOI] [PubMed] [Google Scholar]
- 21.Nishioka T, Eustace A, West C. Lysyl oxidase: from basic science to future cancer treatment. Cell Struct Funct. 2012;37(1): 75–80. [DOI] [PubMed] [Google Scholar]
- 22.Schietke R, Warnecke C, Wacker I, et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem. 2010;285(9):6658–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliades A, Papadantonakis N, Bhupatiraju A, et al. Control of megakaryocyte expansion and bone marrow fibrosis by lysyl oxidase. J Biol Chem. 2011;286(31):27630–27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10): 1356–1363. [DOI] [PubMed] [Google Scholar]
- 25.Zarogoulidis P, Katsikogianni F, Tsiouda T, Sakkas A, Katsikogiannis N, Zarogoulidis K. Interleukin-8 and interleukin-17 for cancer. Cancer Invest. 2014;32(5):197–205. [DOI] [PubMed] [Google Scholar]
- 26.Lu M, Xia L, Liu YC, et al. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood. 2015;126(8):972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullally A, Ebert BL. Sinister symbiosis: pathological hematopoietic-stromal interactions in CML. Cell Stem Cell. 2013;13(3): 257–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014;123(24):3714–3719. [DOI] [PubMed] [Google Scholar]
- 31.Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol. 2012;19(8):754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hobbs CM, Manning H, Bennett C, et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood. 2013;122(23):3787–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muth M, Büsche G, Bock O, Hussein KH, H K. Aberrant proplatelet formation in chronic myeloproliferative neoplasms. Leuk Res. 2010;34(11):1424–1429. [DOI] [PubMed] [Google Scholar]
- 34.Kreipe H, Büsche G, Bock O, Hussein K. Myelofibrosis: molecular and cell biological aspects. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muth M, Engelhardt BM, Kröger N, et al. Thrombospondin-1 (TSP-1) in primary myelofibrosis (PMF) - a megakaryocyte-derived biomarker which largely discriminates PMF from essential thrombocythemia. Ann Hematol. 2010;90(1):33–40.36. [DOI] [PubMed] [Google Scholar]
- 36.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28(7):1472–1477. [DOI] [PubMed] [Google Scholar]
- 37.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 38.Elf S, Abdelfattah N, Chen E, et al. Physical interaction between mutant calreticulin and the thrombopoietin receptor is required for hematopoietic transformation. ASH. 2015;57th Annual Meeting and Exposition (LBA-4). [Google Scholar]
- 39.Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2015. December 14 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 40.Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–1810. [DOI] [PubMed] [Google Scholar]
- 41.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. [DOI] [PubMed] [Google Scholar]
- 42.Guglielmelli P, Barosi G, Specchia G, et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood. 2009;114(8):1477–1483. [DOI] [PubMed] [Google Scholar]
- 43.Barosi G, Poletto V, Massa M, et al. JAK2 V617F genotype is a strong determinant of blast transformation in primary myelofibrosis. PLoS One. 2013;8(3):e59791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kröger N, Zabelina T, Alchalby H, et al. Dynamic of bone marrow fibrosis regression predicts survival after allogeneic stem cell transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2014;20(6):812–815. [DOI] [PubMed] [Google Scholar]
- 45.Lekovic D, Gotic M, Perunicic-Jovanovic M, et al. Contribution of comorbidities and grade of bone marrow fibrosis to the prognosis of survival in patients with primary myelofibrosis. Med Oncol. 2014;31(3):869. [DOI] [PubMed] [Google Scholar]
- 46.Gianelli U, Vener C, Bossi A, et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod Pathol. 2012;25(9):1193–1202. [DOI] [PubMed] [Google Scholar]
- 47.Nazha A, Estrov Z, Cortes J, Bueso-Ramos CE, Kantarjian H, Verstovsek S. Prognostic implications and clinical characteristics associated with bone marrow fibrosis in patients with myelofibrosis. Leuk Lymphoma. 2013;54(11):2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guglielmelli P, Rotunno G, Pacilli A, et al. Prognostic impact of bone marrow fibrosis in primary myelofibrosis: a study of Agimm Group on 540 patients. ASH. 2015;57th Annual Meeting and Exposition:634. [DOI] [PubMed] [Google Scholar]
- 49.Alchalby H, Yunus DR, Zabelina T, et al. Risk models predicting survival after reduced-intensity transplantation for myelofibrosis. Br J Haematol. 2012;157(1): 75–85. [DOI] [PubMed] [Google Scholar]
- 50.Campbell PJ, Bareford D, Erber WN, et al. Reticulin accumulation in essential thrombocythemia: prognostic significance and relationship to therapy. J Clin Oncol. 2009;27(18):2991–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbui T, Thiele J, Passamonti F, et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood. 2012;119(10):2239–2241. [DOI] [PubMed] [Google Scholar]
- 52.Abdulkarim K, Ridell B, Johansson P, Kutti J, Safai-Kutti S, Andréasson B. The impact of peripheral blood values and bone marrow findings on prognosis for patients with essential thrombocythemia and polycythemia vera. Eur J Haematol. 2011;86(2): 148–155. [DOI] [PubMed] [Google Scholar]
- 53.Buesche G, Ganser A, Schlegelberger B, et al. Marrow fibrosis and its relevance during imatinib treatment of chronic myeloid leukemia. Leukemia. 2007;21(12):2420–2427. [DOI] [PubMed] [Google Scholar]
- 54.Kvasnicka HM, Thiele J, Schmitt-Graeff A, et al. Bone marrow features improve prognostic efficiency in multivariate risk classification of chronic-phase Ph(1+) chronic myelogenous leukemia: a multicenter trial. J Clin Oncol. 2001;19(12):2994–3009. [DOI] [PubMed] [Google Scholar]
- 55.Kantarjian HM, Bueso-Ramos CE, Talpaz M, et al. The degree of bone marrow fibrosis in chronic myelogenous leukemia is not a prognostic factor with imatinib mesylate therapy. Leuk Lymphoma. 2005;46(7):993–997. [DOI] [PubMed] [Google Scholar]
- 56.Kimura A, Katoh O, Hyodo H, Kuramoto A, Satow Y. Platelet derived growth factor expression, myelofibrosis and chronic myelogenous leukemia. Leuk Lymphoma. 1995;18(3–4):237–242. [DOI] [PubMed] [Google Scholar]
- 57.Fu B, Ok CY, Goswami M, et al. The clinical importance of moderate/severe bone marrow fibrosis in patients with therapy-related myelodysplastic syndromes. Ann Hematol. 2013;92(10):1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu B, Jaso JM, Sargent RL, et al. Bone marrow fibrosis in patients with primary myelodysplastic syndromes has prognostic value using current therapies and new risk stratification systems. Mod Pathol. 2014;27(5):681–689. [DOI] [PubMed] [Google Scholar]
- 59.Della Porta MG, Malcovati L, Boveri E, et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol. 2009;27(5):754–762. [DOI] [PubMed] [Google Scholar]
- 60.Kröger N, Zabelina T, van Biezen A, et al. Allogeneic stem cell transplantation for myelodysplastic syndromes with bone marrow fibrosis. Haematologica. 2011;96(2): 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanna A, Gozzini A, Donnini I, et al. Influence of mild bone marrow fibrosis on response of int-2/high risk MDS patients to 5-azacitidine. Leuk Res. 2011;35:S126–S127. [Google Scholar]
- 62.Tadmor T, Shvidel L, Aviv A, et al. Significance of bone marrow reticulin fibrosis in chronic lymphocytic leukemia at diagnosis: a study of 176 patients with prognostic implications. Cancer. 2013;119(10):1853–1859. [DOI] [PubMed] [Google Scholar]
- 63.Teman CJ, Wilson AR, Perkins SL, Hickman K, Prchal JT, Salama ME. Quantification of fibrosis and osteosclerosis in myeloproliferative neoplasms: a computer-assisted image study. Leuk Res. 2010;34(7):871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pozdnyakova O, Hasserjian R, Salama M, et al. Bone marrow fibrosis by WHO Grade and quantitative image analysis is reduced by PRM-151 in patients with myelofibrosis and associated with improved bone marrow morphology and increased platelet counts. Haematologica. 2015;100(s1):677.25661441 [Google Scholar]
- 65.Salama ME, Hagendorn E, Perkins SL, et al. Stereology and computer-based image analysis quantifies heterogeneity and improves reproducibility for grading reticulin In: Bone Marrow. Histopathology in Pharmaceutical Product Development. 1st edition. Eberhard DA, Potts SJ, Young GD, eds. Springer Publishing, NY, USA: 2014. [Google Scholar]
- 66.Wang JC, Lang HD, Lichter S, Weinstein M, Benn P. Cytogenetic studies of bone marrow fibroblasts cultured from patients with myelofibrosis and myeloid metaplasia. Br J Haematol. 1992;80(2):184–188. [DOI] [PubMed] [Google Scholar]
- 67.Mascarenhas J. Rationale for combination therapy in myelofibrosis. Best Pract Res Clin Haematol. 2014;27(2):197–208. [DOI] [PubMed] [Google Scholar]
- 68.Rondelli D, Barosi G, Bacigalupo A, et al. Allogeneic hematopoietic stem-cell transplantation with reduced-intensity conditioning in intermediate- or high-risk patients with myelofibrosis with myeloid metaplasia. Blood. 2005;105(10):4115–4119. [DOI] [PubMed] [Google Scholar]
- 69.Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107(3):451–458. [DOI] [PubMed] [Google Scholar]
- 70.Silver RT, Vandris K, Goldman JJ. Recombinant interferon-α may retard progression of early primary myelofibrosis: a preliminary report. Blood. 2011;117(24): 6669–6672. [DOI] [PubMed] [Google Scholar]
- 71.Ianotto JC, Boyer-Perrard F, Gyan E, et al. Efficacy and safety of pegylated-interferon α-2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol. 2013;162(6):783–791. [DOI] [PubMed] [Google Scholar]
- 72.Hebballi S, Akiki S, Bareford D. Resolution of post-polycythaemic myelofibrosis with a combination of thalidomide and interferon. J Clin Pathol. 2012;65(8):762–763. [DOI] [PubMed] [Google Scholar]
- 73.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. March 1 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 75.Wilkins BS, Radia D, Woodley C, Farhi SE, Keohane C, Harrison CN. Resolution of bone marrow fibrosis in a patient receiving JAK1/JAK2 inhibitor treatment with ruxolitinib. Haematologica. 2013;98(12):1872–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Molica M, Serrao A, Saracino R, et al. Disappearance of fibrosis in secondary myelofibrosis after ruxolitinib treatment: new endpoint to achieve? Ann Hematol. 2014;93(11):1951–1952. [DOI] [PubMed] [Google Scholar]
- 77.Kvasnicka HM, Thiele J, Bueso-Ramos C, et al. Exploratory analysis of the effect of ruxoli, tinib on bone marrow morphology in patients with myelofibrosis. J Clin Oncol. 2013;31(Suppl): [abstract 703]. [Google Scholar]
- 78.Thiele J, Bueso-Ramos CE, Sun W, et al. Effects of five-years of ruxolitinib therapy on bone marrow morphology in patients with myelofibrosis and comparison with best available therapy. Blood. 2013;122(21):4055 [abstract]. [Google Scholar]
- 79.Mesa RA, Tefferi A, Elliott MA, et al. A phase II trial of pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a novel anti-fibrosing agent, in myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;114(1):111–113. [DOI] [PubMed] [Google Scholar]
- 80.Morris JC, Shapiro GI, Tan AR, et al. Phase I/II study of GC1008: a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). J Clin Oncol. 2008;26(15S):9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pilling D, Cox N, Vakil V, Verbeek JS, Gomer RH. The long pentraxin PTX3 promotes fibrocyte differentiation. PLoS One. 2015;10(3):e0119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crawford JR, Pilling D, Gomer RH. FcγRI mediates serum amyloid P inhibition of fibrocyte differentiation. J Leukoc Biol. 2012;92(4):699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haudek SB, Xia Y, Huebener P, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103(48):18284–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pilling D, Roife D, Wang M, et al. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol. 2007;179(6):4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Castaño AP, Lin SL, Surowy T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1(5):5ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maharjan AS, Roife D, Brazill D, Gomer RH. Serum amyloid P inhibits granulocyte adhesion. Fibrogenesis Tissue Repair. 2013;6(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murray LA, Rosada R, Moreira AP, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One. 2010;5(3):e9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dillingh MR, van den Blink B, Moerland M, et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm Pharmacol Ther. 2013;26(6):672–676. [DOI] [PubMed] [Google Scholar]
- 89.Duffield JS, Lupher MLJ. PRM-151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect. 2010;23(5):305–315. [DOI] [PubMed] [Google Scholar]
- 90.Verstovsek S, Mesa RA, Foltz LM, et al. Phase 2 trial of PRM-151, an antifibrotic agent, in patients with myelofibrosis: Stage 1 results. J Clin Oncol. 2014;32:5s (abstr 7114). [Google Scholar]
- 91.Verstovsek S, Pozdnyakova O, Hasserjian R, et al. Outcomes in myelofibrosis patients completing 24 and 36 weeks of treatment with PRM-151. Clin Lymphoma Myeloma Leuk. 2015;15:S58–S59. [DOI] [PubMed] [Google Scholar]
- 92.Verstovsek S, Mesa RA, Foltz LM, et al. PRM-151 in myelofibrosis: durable efficacy and safety at 72 weeks. ASH. 2015;57th Annual Meeting and Exposition. [Google Scholar]
- 93.Tadmor T, Bejar J, Attias D, et al. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am J Hematol. 2013;88(5):355–358. [DOI] [PubMed] [Google Scholar]
- 94.Verstovsek S, Savona MR, Mesa RA, et al. A phase 2 study to evaluate the efficacy and safety of simtuzumab in adult subjects with primary, post polycythemia vera (PV) or post essential thrombocythemia (ET) myelofibrosis. ClinicalTrials.gov. 2015; NCT01369498. [Google Scholar]
- 95.Bhagwat N, Keller MD, Rampal R, et al. Improved efficacy of combination of JAK2 and hedgehog inhibitors in myelofibrosis. Blood. 2013;122:s666. [Google Scholar]
- 96.Martinelli G, Oehler VG, Papayannidis C, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015;2(8):e339–e346. [DOI] [PubMed] [Google Scholar]
- 97.Tibes R, Mesa RA. Targeting hedgehog sig-naling in myelofibrosis and other hematologic malignancies. J Hematol Oncol. 2014;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gupta V, Koschmieder S, Harrison CN, et al. Phase 1b dose-escalation study of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Blood. 2014;124(21):712. [Google Scholar]
- 99.Gupta V, Harrison CN, Hasselbalch H, et al. Myeloproliferative ssyndromes: clinical: combination therapy in MPN. ASH. 2015;57th Annual Meeting and Exposition:634. [Google Scholar]
- 100.Lancet J, Wei AH, Durrant ST, et al. A phase I study of KB004, a novel non-fucosylated humaneered® antibody, targeted against the receptor tyrosine kinase EphA3, in advanced hematologic malignancies. ASH. 2013:3838 [abstract]. [Google Scholar]
- 101.Vail ME, Murone C, Tan A, et al. Targeting EphA3 inhibits cancer growth by disrupting the tumor stromal microenvironment. Cancer Res. 2014;74(16):4470–4481. [DOI] [PubMed] [Google Scholar]
- 102.Slape CI. EphA3 is expressed on leukemia stem cells, and eph/ephrin signalling features in the remodelling of the leukemia stem cell niche. Blood. 2014;124(21):3756. [Google Scholar]
- 103.Swords RT, Wei AH, Durrant S, et al. KB004, a novel non-fucosylated humaneered® antibody, targeting EphA3, is active and well tolerated in a phase I/II study of advanced hematologic malignancies. Blood. 2014;124(21):3756. [Google Scholar]