

The pathomechanism of chronic lymphocytic leukemia (CLL) is an interplay of genomic and epigenomic aberrations. Next generation sequencing (NGS) identified mutations in CLL that impact on tumor suppressor and oncogenes like NOTCH1.1 In addition, CLL cells harbor genome-wide aberrant DNA-methylation.2 Of interest, in CLL, DNA-methylation of single CpGs overall correlated only weakly with gene expression. However, no detailed analysis of DNA-methylation on single gene level was performed that would also take larger regions into account.3 In our study, therefore, we profiled gene expression and promoter DNA-methylation in CD19+ cells from 15 CLL patients and B cells from 9 healthy probands (Online Supplementary Table S1) to reveal the impact of aberrant DNA-methylation on transcription. With this approach, we identified the Krüppel-like factor 4 (KLF4) as hypermethylated and down-regulated in CLL. Of note, inhibition of NOTCH signaling induced KLF4 expression. Moreover, overexpression of KLF4 in cell lines modulated components of the B-cell receptor (BCR) signaling pathway.
To identify genes potentially deregulated by promoter DNA-methylation we profiled gene expression and analyzed promoter DNA-methylation using methyl-CpG-immunoprecipitation (MCIp) onto specific custom promoter tiling-arrays that covered −3.8 to +1.8 kb from the transcription start sites (TSS) of all human genes. A total of 1866 genes were differentially expressed in CLL compared to controls (|log2FC|≥1, P≤0.05) and involved genes previously shown to be deregulated or involved in the pathomechanism of CLL (Online Supplementary Figure 1A). Overall, DNA-methylation displayed a broader distribution in CLL compared to normal B cells, which reflects a greater heterogeneity within the patient samples (Online Supplementary Figure 1B), in line with previous results.4 Furthermore, DNA-methylation analysis revealed 2191 gene promoters that showed at least one differentially methylated region (DMR) of at least 500bp with a median |log2FC|≥0.5 (P≤0.05) between CLL and normal B cells. Of these DMRs, 1472 were hypomethylated in CLL and only 719 hypermethylated (Figure 1A). Combining methylation data and expression data, we identified 33 genes to be potentially transcriptionally deregulated by DNA-methylation in CLL with at least one oligonucleotide within the DMR showing a significant negative correlation with gene expression (r≤−0.47 and P≤0.021) (Figure 1B).
Figure 1.
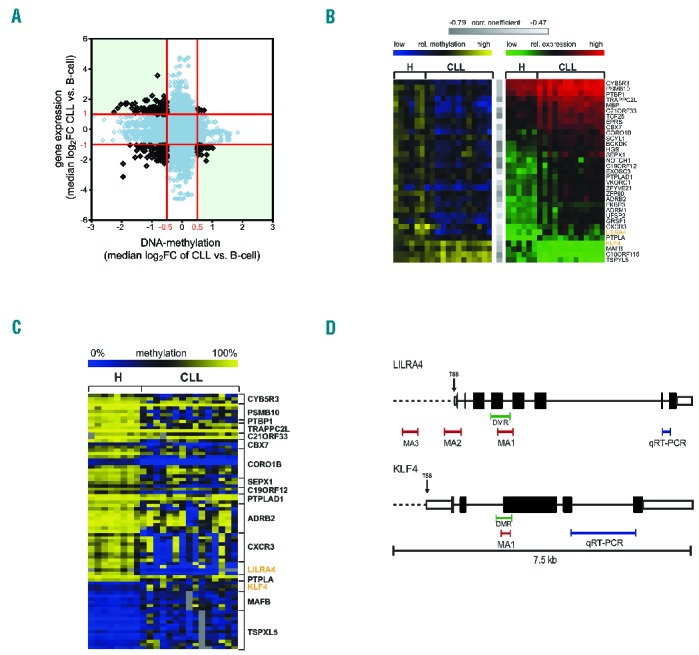
A subset of genes is deregulated by promoter DNA-methylation in chronic lymphocytic leukemia (CLL). (A) Analysis of gene expression and DNA-methylation profiling (MCIp-array) of CD19+ B cells of 9 healthy donors (H) and 15 CLL patient samples. Overall correlation between promoter DNA-methylation and gene expression is depicted. Median log2FC values of CLL versus H for all differentially methylated 500 bp windows (P≤0.05) and the respective differential expression oligonucleotides (P≤0.05) are shown. Red lines indicate cut offs for expression and methylation for further analysis which were |log2FC| > 1 and >0.5, respectively. Genes of interest were identified from top left quadrant (low DNA-methylation and high expression) and bottom right quadrant (high DNA-methylation and low expression). (B) Correlation between DNA methylation and respective gene expression identified 33 genes as potentially deregulated by DNA-methylation. Heatmap in yellow and blue shows DNA-methylation values as log2FC of all samples present on both, expression- and MCIp arrays (H n=6, CLL n=13). Blue indicates low relative methylation, yellow high methylation. Heatmap in green and red displays expression values for same patients with green indicating low and red high expression of the listed genes. All genes showed significant negative correlation (Pearson correlation) between DNA-methylation and gene expression with correlation coefficient −0.79 ≤ r ≤ −0.47 (indicated by gray bars) and P≤0.21. (C) DNA-methylation analysis was performed with MassARRAY in addition to MCIp in 33 candidate genes to identify genes where both complementary methods show aberrant DNA-methylation. Seventeen of these genes showed aberrant DNA-methylation in at least one CpG unit (CGU; shown in rows) in the promoter (Mann-Whitney U test P≤0.05). Absolute methylation values ranging from 0% to 100% of significantly aberrant CpG units are illustrated in the heat map. Blue color indicates low, yellow high DNA-methylation levels. Positions of MassARRAY amplicons (MA) are indicated in (D). (D) Scheme illustrating the positions of all MassARRAY (MA; red bars) and qRT-PCR (blue bars) amplicons measured for LILRA4 (upper panel) and KLF4 (lower panel). Differentially methylated regions (DMR), identified in the MCIp-array analysis, are marked with green bars. For KLF4 and LILRA4, one amplicon each (MA1) was selected that covers the respective DMR. Two additional amplicons MA2 and MA3 were selected for LILRA4 to represent DMRs previously identified.3 Exons are represented by black boxes, untranslated regions (UTR) by white boxes and the transcription start sites (TSS) are indicated by an arrow.
Out of the 33 genes identified, 17 also displayed aberrant DNA-methylation in the samples using highly quantitative mass spectrometry-based MassARRAY (Online Supplementary Figure 2A).5 Of these genes, the leukocyte immunoglobulin-like receptor subfamily A member 4 (LILRA4) and the Krüppel-like factor 4 (KLF4) were shown to have an impact on ITAM-mediated (immunoreceptor tyrosine-based activation motif) signaling and B-cell activation or B-cell differentiation and may hence play a role in the pathogenesis of CLL (Figure 1D). Furthermore, KLF4 is important for maintaining quiescence of hematopoietic cells, acts as regulator of B-cell numbers and activation-induced B-cell proliferation.6–8
Significant transcriptional upregulation of LILRA4 and downregulation of KLF4 was confirmed in a larger cohort of CD19+ B cells from patients (n≤56) and healthy donors (n≤23) (Figure 2A). Similarly, aberrant DNA-methylation of KLF4 and LILRA4 was validated (Figure 2B and D) and correlated significantly with gene expression (Figure 2C and E). As a tumor suppressor, KLF4 is silenced by promoter methylation also in other B-cell malignancies.9 However, differential methylation could be detected in CLL only in the gene body of KLF4 and not in the upstream sequences reported previously (Figure 1D). Furthermore, the DMR in the KLF4 promoter region identified in our screen could not be detected in recent DNA-methylation analyses of CLL as the 450K arrays used previously do not include this region.3 Analysis of KLF4 promoter DNA-methylation in randomly selected, representative CLL samples from 4 different clinical trials conducted by the German CLL study group (GCLLSG), indicates that KLF4 promoter methylation is an event that occurs at or before Binet stage A and is not influenced by the clinical course (Online Supplementary Figure S2).
Figure 2.
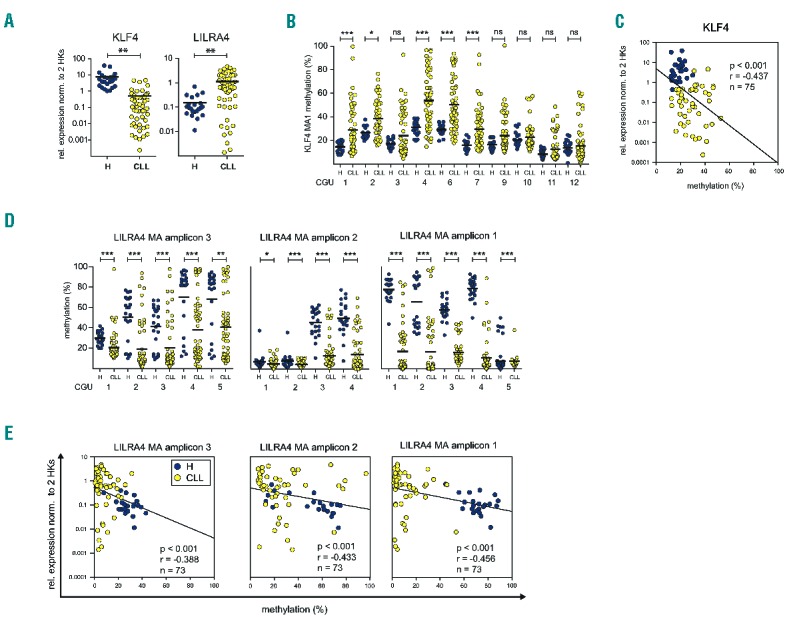
Aberrant DNA-methylation of KLF4 and LILRA4 promoters correlates with their gene expression levels. Expression and methylation of KLF4 and LILRA4 was measured in CD19+ B cells of up to 23 healthy donors and 55 chronic lymphocytic leukemia (CLL) samples. Expression was quantified with qRT-PCR and given values normalized to two housekeeper genes (HPRT and PGK). DNA-methylation was determined with MassARRAY for one intragenic KLF4 amplicon (MA amplicon 1) and 3 amplicons in the LILRA4 promoter region (MA amplicon 1–3). (A) Differential expression of LILRA4 and KLF4 between CLL and healthy donor B cells was confirmed using qRT-PCR in a larger sample cohort (≥73). KLF4 is significantly down-regulated in CLL cells, LILRA4 significantly up-regulated. (B) MassARRAY measurements of KLF4 MA1 confirmed hypermethylation in CLL in 5 out of 10 CpG units (CGUs) that were analyzed. (C) For KLF4 mean methylation values over all CGUs per amplicon show significant negative correlation with gene expression (Spearman rank correlation). (D) LILRA4 MA1, MA2 and MA3 show hypomethylation in all CGUs analyzed in CLL cells. This suggests a larger functional DMR region within the LILRA4 promoter. (E) For LILRA4, mean methylation values over all CGUs per amplicon show significant negative correlation with gene expression (Spearman rank correlation). Mann-Whitney U test was used to test for significance. P-values are indicated by asterisk: *P≤0.05, **P≤0.01, ***P≤0.001. ns: non-significant; n: sample size; r: Spearman rank correlation coefficient; HK: housekeeper; MA: MassARRAY; H: healthy donor. Horizontal lines represent mean.
Also the fact that the aberrant DNA-methylation was found within the gene body rather than upstream of the TSS suggests a more complex mechanism that could include alternative TSS or an enhancer region. The significant correlation of KLF4 expression with its downstream targets (Online Supplementary Figure S3) supports the assumption that the downregulation of KLF4 impacts on the transcriptome of CLL cells. Hence, to understand the functional mechanisms of KLF4 deregulation, we characterized the potential KLF4 downstream target genes and found that NOTCH1 is a possible up-stream effector.
It was shown that the activity of KLF4 can either be transcriptionally induced or blocked by NOTCH1 in different malignant tissues.10,11 As NOTCH1 is constitutively activated and frequently mutated in CLL,1 we investigated their functional relationship. Despite the upregulation of NOTCH1 in CLL cells (P≤0.01), we did not detect a correlation of NOTCH1 and KLF4 mRNAs in our sample cohort (Online Supplementary Figure S4A). To investigate whether NOTCH1 activity, but not expression, correlated with KLF4 induction, we inhibited NOTCH signaling with γ-Secretase-inhibitor (GSI-I). Upon inhibition of γ-Secretase, cleavage of the NOTCH1 intra-cellular domain (NICD) does not take place and NOTCH1 signaling is abrogated.12
Six different leukemia and lymphoblastoid (LCL) cell lines as well as frozen peripheral blood mononuclear cells (PBMCs) from 8 CLL patients were treated with GSI-I or DMSO as solvent control.12 Treatment with GSI-I resulted in reduction of full-length NOTCH1 protein (NP) and the NICD domain (Figure 3A) as well as a dose-dependent reduction in viability in cell lines and patient samples after 24 h (Online Supplementary Figure 4B). Patient samples showed strong inhibition of NOTCH1 upon GSI-I treatment with almost no NICD left. The reduction of NP, which was also reported by others,12 could be the result of a feedback mechanism on transcriptional level for example via the activation of KLF4, which was shown to inhibit NOTCH1 transcription.13
Figure 3.
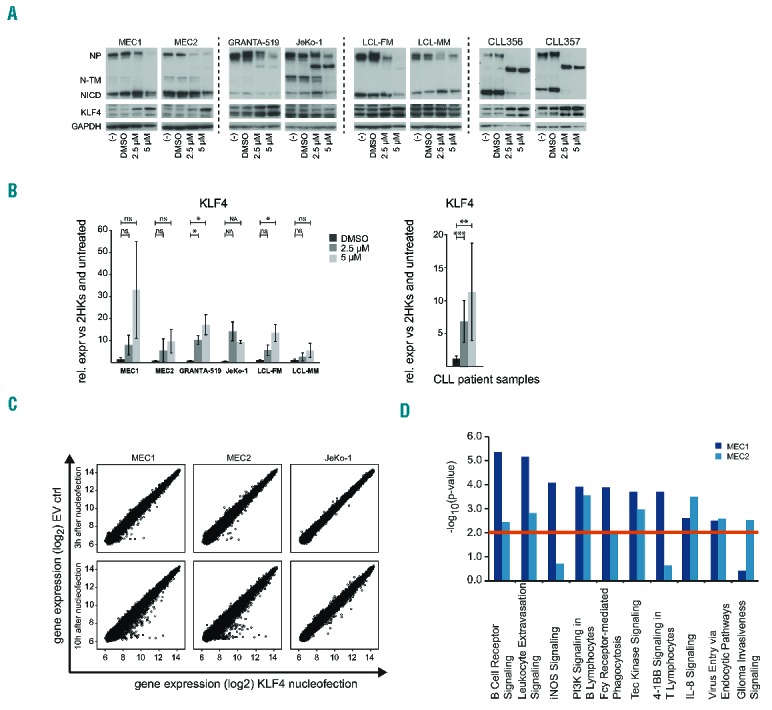
KLF4 is induced by inhibition of NOTCH and modulates BCR signaling components. (A and B) Cell lines MEC1, MEC2, GRANTA-519, JeKo-1, LCL-FM, LCL-MM and PBMCs of 8 chronic lymphocytic leukemia (CLL) patients (CD19+ cells ≥91.6%) were treated with 2.5 μM and 5 μM γ-Secretase inhibitor (GSI-I) or 0.002% DMSO (“−“) as carrier control for 24 h. Three independent experiments were conducted per cell line. (A) Western blot detecting NOTCH1 and KLF4 in cell line samples (left panel) and CLL patient samples (right panel) after 24 h GSI-I treatment. Exemplary blots for one experiment each per cell line and 2 chronic lymphocytic lymphoma (CLL) patients are shown. GAPDH was used as loading control. Treatment with GSI-I shows inhibition of NOTCH1 activation by reduced presence of NP and NICD and increased expression of KLF4. NP: NOTCH1 full-length protein; N-TM: NOTCH1 transmembrane protein; NICD: NOTCH1 intracellular domain. (B) qRT-PCR measurement of KLF4 expression in cell lines (left) and primary CLL samples (right) is shown. Represented is the relative gene expression normalized to 2 housekeeper genes and untreated cells. KLF4 expression is induced in CLL patient samples and to some degree in cell lines (paired t-test). Error bars represent standard deviation, P-values are indicated by asterisk: *P≤0.05, **P≤0.01, ***P≤0.001. ns: non-significant; NA: not available (only 2 out of 3 experiments could be analyzed). (C) Overexpression of KLF4 in MEC1, MEC2 and JeKo-1 cells compared to empty vector control (EV) 3 h, 10 h and 24 h after nucleofection. Expression profiling was conducted with samples from 3 h and 10 h after transfection. Scatter plots of gene expression values (log2 values of signal intensities) for empty vector control (y-axis) and KLF4 overexpression (x-axis) are displayed for all 3 cell lines. Plots show changes in gene expression patterns after 10 h in all 3 cell lines. (D) Pathway analysis of top deregulated genes (|log2FC|≥0.38) in MEC1 (dark blue bars) and MEC2 (light blue) 10 h after nucleofection. The combination of the 7 top deregulated canonical pathways for MEC1 and MEC2 is shown. Cut off for significance was P≤0.01 (red line).
Importantly, after GSI-I treatment, we detected a significant upregulation of KLF4 mRNA and protein levels in primary CLL samples, and to a lesser extent also in cell lines (Figure 3A and B). This indicates an impact of NOTCH1 signaling on KLF4 expression in CLL. As DNA-demethylation is usually linked to DNA-replication or DNA-repair mechanisms, it is likely that regulation of KLF4 by NOTCH1 occurs via an independent mechanism in addition to DNA-methylation. Yet, as inhibition of γ-Secretase does not only target NOTCH1, but also additional pathways, the exact mechanism will need further clarification.
To investigate what downstream targets are affected by KLF4 deregulation, we performed expression profiling of the CLL cell lines MEC1, MEC2 and the mantle cell lymphoma (MCL) cell line JeKo-1 after transient overexpression of KLF4. Despite visible overexpression of KLF4 after 3 h (Online Supplementary Figure S5A), no significant changes were detected in the majority of genes at this time point. In contrast, changes in expression profiles of MEC1, MEC2, and to a lesser degree in JeKo-1, were detectable after 10 h (Figure 3C).
In order to identify which pathways are affected by KLF4, we analyzed genes that were deregulated 10 h after transfection with a cut off of |log2FC≥0.38| that included a higher number of genes [n(MEC1)=1218, n(MEC2)=1245, n(JeKo-1)=705] (Online Supplementary Table S2). We aimed to identify relevant pathways that are affected by deregulation of a larger number of genes, albeit at low levels on single gene level. In MEC1 and MEC2 cells, KLF4 targeted genes were involved in hematopoiesis, hematologic diseases and system development (Online Supplementary Table S3). Furthermore, pathways deregulated both in MEC1 and MEC2 were BCR signaling, genes involved in leukocyte extravasation, iNOS-(nitric oxide synthase) and PI3K-signaling, which are important for almost all B-cell stages (Figure 3D and Online Supplementary Tables S3 and S4). Hence, KLF4 deregulation likely increases the activation state of CLL cells, which is beneficial for survival, proliferation and homeostasis.14 Of the genes involved in BCR signaling, a significant proportion became deregulated after KFL4 overexpression (in MEC1 25 of 67; P=4.34e−06) (Online Supplementary Figure 5B), suggesting an impact of KLF4 inactivation on the BCR-signaling pathway that is central to the pathomechanism of CLL. While KLF4 might not directly bind to promoters of affected genes, the modulation of BCR signaling components by KLF4 is of interest in CLL, be it direct primary or secondary effects.
Of note, upon KLF4 overexpression, the expression of NOTCH1 was induced in MEC1 and MEC2 (log2FC=1.08/1.25) and to some degree in JeKo-1 (log2C=0.31), suggesting a regulatory feedback interaction of these two genes (Online Supplementary Table S1).
In summary, we screened all human promoters for aberrant DNA-methylation in CLL patients in comparison to CD19+ B cells of healthy donors. We identified a subset of 17 genes whose aberrant promoter DNA-methylation in CLL cells correlates significantly with gene expression levels, including LILRA4 and KLF4. As DNA-methylation is a stable long-term marker for gene expression, we focused on the upstream and downstream mechanisms of KLF4 deregulation. Subsequent functional analyses, including inhibition of γ-Secretase, uncovered NOTCH1 as a potential repressor of KLF4 in leukemia and lymphoblastoid cell lines, as well as in primary CLL cells. Pathway analysis showed that genes deregulated after KLF4 overexpression in MEC1 and MEC2 are involved in iNOS-, BCR- and PI3K-signaling. CLL cells are known to be dependent on BCR signaling, especially in the lymph node microenvironment, where stimulation of the BCR is thought to induce tumor proliferation.15 Furthermore, upon B-cell activation, KLF4 was shown to be down-regulated.7 The repression of KLF4 in CLL might, therefore, support the highly activated state of the CLL cells and exert a supportive effect on the survival and expansion of the cells.14
Acknowledgments
We would like to acknowledge Sibylle Ohl and Karin Müller for excellent technical support, Silke Brüderlein for kindly providing cell lines and Marc Seifert, Martina Seiffert and Claudia Scholl for helpful discussions. We would also like to thank the patients for generous donation of primary material and the German CLL study group (GCLLSG) for collection and characterization of samples and enabling their molecular analysis. We thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Whole-Genome Expression Beadchips and related services. Detailed description of methods and primers is given in Online Supplementary Materials and Online Supplementary Table S5.
Footnotes
Funding: this work was supported by the Else Kröner-Fresenius-Stiftung (2012_A146), the Virtual Helmholtz Institute (VH-VI-404), the DFG (SFB1074 projects B1/B2), the BMBF-Network “CancerEpiSys” (0316049C) and the Deutsche Jose Carreras Leukaemie Stiftung (DJCLS R 11/01).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garding A, Bhattacharya N, Claus R, et al. Epigenetic upregulation of lncRNAs at 13q14.3 in leukemia is linked to the In Cis downregulation of a gene cluster that targets NF-kB. PLoS Genet. 2013;9(4):e1003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–1242. [DOI] [PubMed] [Google Scholar]
- 4.Oakes CC, Claus R, Gu L, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4(3):348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005; 102(44):15785–15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao W, Bover L, Cho M, et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med. 2009;206(7):1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Good K, Tangye S. Decreased expression of Krüppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. PNAS. 2007;104(33):13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaewsongkram J, Yang Y, Golech S, Katz J, Kaestner KH, Weng N-P. Krüppel-like factor 4 regulates B cell number and activation-induced B cell proliferation. J Immunol. 2007;179(7):4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan H, Xie L, Leithäuser F, et al. KLF4 is a tumor suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood. 2010;116(9):1469–1478. [DOI] [PubMed] [Google Scholar]
- 10.Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15(1):50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H-F, Huang C-H, Liu C-J, et al. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun. 2014;5:4697. [DOI] [PubMed] [Google Scholar]
- 12.Rosati E, Sabatini R, De Falco F, et al. γ-Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and notch down-regulation. Int J Cancer. 2013;132(8):1940–1953. [DOI] [PubMed] [Google Scholar]
- 13.Lambertini C, Pantano S, Dotto GP. Differential control of Notch1 gene transcription by Klf4 and Sp3 transcription factors in normal versus cancer-derived keratinocytes. PLoS One. 2010;5(4):e10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damle R, Calissano C, Chiorazzi N. Chronic lymphocytic leukaemia: a disease of activated monoclonal B cells. Best Pract Res Clin. 2010;23(1):33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]