

Abstract
Wilms tumor 1 (WT1) has long been implicated in acute myeloid leukemia. It has been described to be both overexpressed and mutated in different forms of acute myeloid leukemia, and overexpression has been reported to play a prognostic role in this disease. However, the precise mechanism through which WT1 may play a role in leukemogenesis has remained elusive. In recent years, new evidence has emerged that points towards a novel role of WT1 mutations in the deregulation of epigenetic programs in leukemic cells through its interaction with TET proteins. Herein we review the current status of the field and its therapeutic and prognostic implications in acute myeloid leukemia.
Introduction
The observations that the Wilms tumor 1 (WT1) protein is expressed in the majority of cases of acute myeloid leukemia (AML), and in addition, is mutated in a proportion of AML cases has led to extensive work over the last several decades to determine the mechanistic role of WT1 in AML. The fact that WT1 may be either overexpressed or mutated has given rise to the concept that it may act as both a tumor suppressor and oncogene, depending on the context. To date, much of the mechanistic work on WT1 has centered on its function as a transcription factor and the role of its many isoforms. However, more recent work stemming from large-scale genomic studies in AML has revealed a new role for WT1 in epigenetic regulation. Indeed, work by several groups has demonstrated that WT1 appears to play a role along with TET proteins in mediating 5-hydroxymethylation of cytosines. These novel observations have given rise to an enhanced understanding of the complexity of this protein and its pathogenic role in leukemogenesis. Herein, we seek to review the major concepts and findings with regard to WT1 in oncogenesis and normal hematopoiesis, as well as any therapeutic strategies that may arise from these observations.
WT structure and function
The WT1 gene was initially identified as an inherited predisposition allele in probands with familial Wilms tumor.1,2 Numerous WT1 missense and nonsense mutations have been described in inherited and sporadic Wilms tumors, a pediatric malignancy affecting the kidneys.3,4 WT1 mutations are identified in approximately 20% of Wilms tumors, and are seen in patients with a familial disposition to these tumors. Wilms tumor is also associated with the Denys-Drash syndrome (DDS), encompassing Wilms tumor, congenital nephrotic syndrome, and XY pseudohermaphroditism, which results from point mutations in WT1.5,6 Mutations in the intron 9 splice site have been identified in patients with Frasier syndrome, a developmental condition that affects kidneys and genitalia.7 In addition, WT1 has been implicated in a number of other malignancies, including desmoplastic small cell tumor, breast cancer, retinoblastoma, and lung carcinoma.8,9 In lung cancer, Wt1 has been shown to play a role in murine lung cancer models driven by Kras mutations where deletion or suppression of Wt1 resulted in senescence of primary murine cells expressing Kras, but had no effect on wild type cells.10 Furthermore, loss of Wt1 led to decreased proliferation and tumor burden in Kras-driven lung carcinoma, thus implicating Wt1 as a necessary component of Kras-driven oncogenesis. Taken together, these observations indicate that a spectrum of mutations in WT1 can lead to both inherited developmental syndromes and cancer predisposition.
The WT1 protein contains an N-terminal transactivation domain and a C-terminus with four zinc-fingers that share DNA binding motifs with EGR11 (Figure 1). Four major WT1 isoforms result from differences in alternative splicing. These isoforms arise through two major splice events. Isoforms resulting through alternative splicing of exon 5, which causes a 17 amino acid insertion between the trans-regulatory domain and the zinc finger domain, have been found more commonly in relapsed AML samples11 (we will refer to the isoform containing this 17 amino acid region as 17+). The second splice event inserts three amino acids –lysine (K), threonine (T), and serine (S)– between exons 9 and 10, resulting in a significant reduction of the DNA binding ability, while enriching RNA binding12,13 (we will refer to the isoform containing the KTS sequence as KTS+). A fifth WT1 isoform produced by an alternative initiation codon within exon 1 lacks the extreme N-terminal repressive domain and augments the protein’s transactivation function.14
Figure 1.

Schematic representation of the WT1 protein. Top: arrows indicate mutations in both AML (red) and those associated with Wilms tumor (green). Bottom: schematic representation of WT1 interactions with other proteins.
WT1 has been demonstrated to interact with a variety of other proteins. Among these are p53, which WT1 stabilizes and plays a role in preventing apoptosis.15 WT1 also binds to chaperone heat shock protein 90, resulting in WT1 stabilization16 and STAT3, resulting in enhanced cell proliferation of Wilms tumor cells.17 Most recently, WT1 has been demonstrated to interact with the epigenetic modifiers TET2 and TET3 in acute myeloid leukemias. This phenomenon will be discussed in detail later on in this review.18,19
WT1 in normal hematopoiesis
The initial assessment of WT1 expression in normal hematopoiesis was based on observations in normal donors demonstrating that WT1 was expressed in CD34+ bone marrow-derived cells, but not in those lacking CD34 expression. Further analysis demonstrated that WT1 expression occurred in the CD34+ CD38− population.20 Notably, single-cell RT-PCR studies revealed that only a small proportion (1.2%) of bone marrow CD34+ cells actually expressed WT1.21 Further studies demonstrated expression of WT1 in some quiescent primitive progenitors as well as more differentiated populations, while it was undetectable in lineage-committed progenitors.22 In murine hematopoiesis, Wt1 expression has been noted in embryonic murine liver at a time point when the liver is the principle site of hematopoiesis (day 12.5 post conception).23
Attempts to further define the role of WT1 in normal hematopoiesis have been performed by observing the effects of alterations of WT1 levels in hematopoietic cells. Overexpression of WT1 in human umbilical cord derived CD34+ cells –in particular the isoform lacking a KTS sequence– resulted in enhanced differentiation of CD34+ cells in culture conditions, whereas overexpression in CD34+ CD38− cells resulted in a relative increase in this population attributed to quiescence.22 In methylcellulose assays, forced overexpression of WT1 in umbilical cord derived CD34+ cells resulted in reduced myeloid and erythroid colony formation. This observation did not correlate with any differences in cell cycle or viability.24 Finally, overexpression of Wt1 in 32D cl3 cells, an IL-3-dependant myeloid progenitor murine cell line, blocked G-CSF induced differentiation of these cells in culture.25
Several murine studies have been carried out to better characterize the role of Wt1 in normal hematopoiesis in vivo. The complete absence of Wt1 results in embryonic lethality due to failure of renal and gonad development.26 Thus, several alternative approaches, including generating chimeric mice through the injection of normal C57BL6 blastocysts with embryonic stem (ES) cells lacking Wt1, as well as reconstitution of normal mice with hematopoietic cells from embryos lacking Wt1, have been implemented to overcome this limitation. Using these approaches, the investigators demonstrated that while embryonic stem cells lacking Wt1 show a competitive disadvantage in the chimeric context, failing to contribute to mature hematopoiesis, fetal liver cells lacking Wt1 were able to repopulate the hematopoietic system of lethally irradiated recipients, at least for 2 months. In addition, despite Wt1-progenitors displaying multipotent potential on methylcellulose, their colony-forming ability was reduced compared to Wt1+ progenitors.27,28 In a Wt1-GFP transgenic murine model, Wt1 was not found to be expressed in long term-hematopoietic stem cells, and infrequently in multipotent progenitors.29 Deletion of Wt1 in young and adult mice using an inducible system results in the death of the animals within approximately 10 days and results in glomerulosclerosis, atrophy of the pancreas, and diminished extramedullary hematopoiesis.30 Notably, the phenotype of conditional deletion of Wt1 in the adult hematopoietic compartment has not been reported to date.
WT1 in acute myeloid leukemia (AML)
WT1 is overexpressed in both myeloid and lymphoid leukemias. Overexpression has been demonstrated in acute lymphoblastic leukemia (both B cell and T cell) primary patient samples and cell lines.31–33 Likewise, overexpression has been described in both myeloid and lymphoid blast crisis of chronic myeloid leukemia (CML) (but not in chronic phase CML),31 and myelodysplastic syndrome (MDS).34 However, despite this broad range of hematological malignancies harboring overexpression of WT1, this phenomenon has been most extensively studied in acute myeloid leukemia (AML). Indeed, WT1 expression levels have been demonstrated to be higher in both primary human AML samples as well as human leukemia cell lines.32,33,35 Furthermore, recurrent somatic mutations in WT1 have also been described in AML.36,37 The initial observations of WT1 involvement in leukemia have given rise to work examining the potential pathogenic role of WT1 in leukemogenesis. Below, we focus specifically on the clinical implications, pathogenesis and therapeutic strategies with regards to WT1 in AML.
WT1 is overexpressed in the majority of AML patients.35,38 In MDS the expression of WT1 is associated with higher blast counts and an increased risk of progression to AML.34 Several studies have demonstrated that increased levels of WT1 in AML are associated with resistance to therapy, a higher incidence of relapse, and poor overall survival.39,40,41 Furthermore, a failure to reduce WT1 transcript levels to below detectable limits has been associated with a higher incidence of relapse in AML42 (Table 1).
Table 1.
Clinical associations with WT1 overexpression and mutation in AML and MDS.
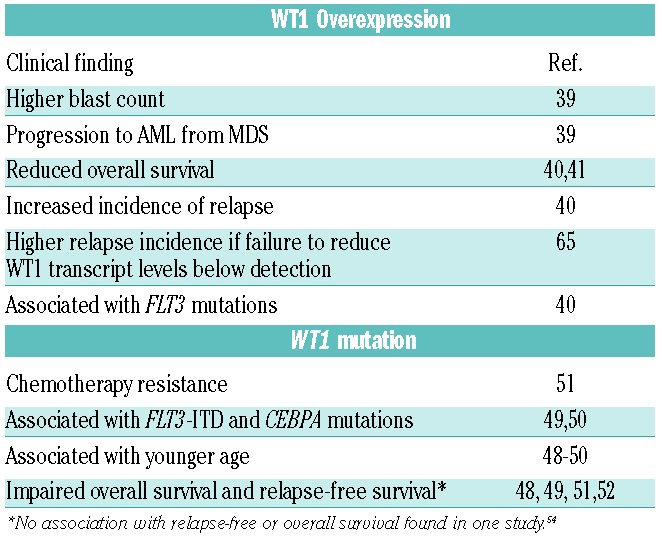
Significant strides in understanding the role of WT1 overexpression in AML have been made by studies in human leukemia cell lines and murine models. Notably, downregulation of WT1 expression in the K562 cell line, as well as a proportion of tested primary AML and CML cases (chronic phase and blast crisis) resulted in inhibition of cell growth.43 Further studies have demonstrated that downregulation of the 17+ isoform specifically, but not the 17- isoform, was able to induce apoptosis in leukemia cell lines K562, HL-60, and Kasumi-1. Conversely, the expression of the 17+ isoform led to a decrease in the expression of proapoptotic Bak.44
In order to further define the role of WT1 overexpression in leukemogenesis, a transgenic murine model of WT1 overexpression in the hematopoietic system was established by Nishida and colleagues.45 Bone marrow from transgenic mice overexpressing WT1 was transduced with a construct expressing the AML1-ETO fusion product, which by itself is insufficient to produce AML. By contrast, in cooperation with WT1 overexpression AML1-ETO led to rapid onset leukemia, with a median time to leukemia development of 50 days. To further characterize WT1’s contribution to leukemogenesis, Hosen and colleagues generated a knock-in reporter WT1-Green Fluorescent Protein (WT1GFP/+) murine model that allowed them to track WT1 expression in normal and leukemic bone marrow cells.29 Using this model they characterized the expression of WT1 in an AML1-ETO+TEL-PDGRFB leukemia model. Recipient mice transplanted with bone marrow transduced with AML1-ETO+TEL-PDGRFB developed a rapid leukemia and displayed higher expression of WT1 in the lineage negative, Sca+,cKit+ (LSK) fraction compared to normal WT1GFP/+ mice. The same observation was made in WT1GFP/+ mice transduced with BCR-ABL. WT1 was found to be expressed in a subset of LSK cells capable of transplanting the disease phenotype into recipient animals; however, a fraction of GFP negative LSK cells was also able to transplant the leukemia phenotype, indicating that WT1 was not required for the propagation of leukemia. Also of note, enforced expression of WT1 in hematopoietic stem cells did not result in proliferation or expansion of these cells or their downstream progeny, nor did this overexpression result in a differentiation block in these cells.
Thus, the precise role of WT1 overexpression in leukemogenesis remains elusive. The disparate results in these models may be due to cooperating oncogenes or other factors. Further evaluation in models using other cooperating genomic alterations, based on the current understanding of the spectrum and co-occurrence of mutational events in human AML may be necessary, as it is possible that the pathogenic role of WT1 is context-specific.
WT1 mutations in AML
Recurrent somatic mutations in WT1 appear to occur in approximately 6–15% of de novo AML.46,47 WT1 mutations associate with younger age,47–49 and the presence of FLT3-ITD mutations48,49 and CEBPA mutations.49 The clinical impact of WT1 in AML has been assessed in several studies, with conflicting results. Multivariate analysis of 470 de novo AML patients (excluding Acute Promyelocytic Leukemia) demonstrated that WT1 mutations were associated with worsened overall survival (OS) and relapse-free survival (RFS) both in the entire cohort as well as when the analysis was restricted to normal karyotype AML patients.47 These results were supported by other studies of adult AML patients.48,50,51 By contrast, a study including patients from three German-Austrian AML study protocols demonstrated no association with RFS or OS.49 A possible explanation for this difference in findings may reside in the fact that patients in the German-Austrian trials received higher doses of cytarabine during consolidation (18–54 g/m2 vs. 6–25 g/m2) than those in the other studies. In addition to the impact of WT1 mutation on OS and RFS, the presence of WT1 mutations has also been associated with resistance to induction chemotherapy50 (Table 1).
The functional effects of WT1 mutations have been assessed in various studies. Several different WT1 mutations have been described in AML, which occur primarily in exons 1, 7 and 9 (Figure 1). These include base substitutions, deletions, and insertions. Significantly, however, the vast majority of mutations result in the creation of stop codons and reading frame shifts, resulting in loss-of-function and expression of a truncated protein lacking the zinc-finger domain.52,37 Additionally, more recent data indicate that WT1 mutant transcripts with frameshift mutations are subject to nonsense-mediated RNA decay without expression of the truncated protein.53,35 Thus, these WT1 mutations may result in the loss of DNA binding ability due to loss of the zinc-finger domain, or result in loss of expression of the WT1 protein altogether.
The fact that WT1 is overexpressed in many patients with AML, and that overexpression of WT1 can contribute to driving leukemogenesis in a murine model, yet it is also mutated –with presumed loss-of-function– in a significant proportion of AML cases presented a paradox as to the role of this protein in AML. Microarray profiling has identified a gene expression signature characterizing WT1-mutant disease, including overexpression of GTSF1, CD96, MLL, PML, and MACC1, and down-regulation of SNRPN, SNURF, FCHO2, MTX3, INSR, and IRS2. In total, the WT1-mutant gene expression signature included 74 upregulated and 40 downregulated genes. When analyzed from a functional point of view, this signature included genes involved in gene regulation, cell proliferation and metabolic homeostasis.54 Beyond this, however; little was known regarding the mechanism by which WT1 mutations might contribute to leukemogenesis. Recent large-scale genetic studies in AML have begun to shed light on this problem. Mutational analysis of a large cohort of AML cases who were treated on the ECOG 1900 clinical trial55,56 revealed that TET2 and IDH1/2 mutations are mutually exclusive in AML, and that TET2/IDH mutant AML is characterized by a common DNA hypermethylation phenotype.57 TET2 is a member of the TET protein family, a group of three iron(II)/αKG-dependent dioxygenases that catalyze the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC).58,59 AMLs carrying mutations in IDH1 or IDH2 mutant produce the oncometabolite 2-hydroxyglutarate, which inhibits TET2 activity.60 Thus, both TET2 loss-of-function mutations and IDH1/2 mutations result in inhibition of the DNA demethylation pathways with accumulation of 5-mC and decrease of 5-hmC, thus providing a convergent mechanism for TET2 and IDH1/2 mutations in AML57 (Figure 2). Interestingly, analysis of the ECOG 1900 cohort also demonstrated that WT1 mutations are strongly anticorrelated with TET2 and IDH1/2 mutations – a finding confirmed in the TCGA AML dataset–, leading to the hypothesis that WT1 mutations may also correspond to a subgroup of AMLs characterized by low levels of 5-hmC, and further, that WT1 may have a previously unrecognized role in this epigenetic pathway. Examination of primary AML samples from the ECOG 1900 cohort using promoter DNA methylation microarrays demonstrated a hypermethylation signature when compared to AMLs that were wild type for all known epigenetic modifier mutations. This hypermethylation signature very strongly overlapped with the TET2 hypermethylation signature. In turn, the TET2 and WT1 hypermethylated loci showed a near complete overlap with the IDH1/2 hypermethylation signature, demonstrating a site-specific overlap of the epigenetic abnormalities in all three types of AMLs. Furthermore, analysis of both global 5-hmC levels in primary AML patient samples as well as genome-wide distribution of 5-hmC by next-generation sequencing demonstrated that WT1-mutant AMLs presented a global reduction in 5-hmC levels comparable to that seen in TET2 and IDH1/2 mutant AMLs when compared with AMLs wild type for WT1/TET2/IDH1/2.18
Figure 2.

The effects of mutations in mediators of the 5-hmC pathway. (A) pathway under normal conditions; (B) IDH mutations lead to production of 2-hydroxyglutarate which inhibits TET2 function; (C) TET2 mutations disrupt interaction with WT1; (D) WT1 mutations fail to properly direct TET2 to its target sites, either by disruption of the interaction itself or by failing to bind to DNA.
The observations that WT1-mutant AMLs are characterized by similar epigenetic alterations as found in TET2 and IDH1/2 mutant AMLs raised the question as to whether WT1 plays a direct role in DNA hydroxymethylation. Knockdown of Wt1 in murine mesonephron cells (which overexpress Wt1) resulted in a significant reduction in 5-hmC levels, while overexpression of WT1 in 32D cells (which do not express endogenous Wt1) led to increases in 5-hmC, thus demonstrating a dynamic response of 5-hmC to WT1 levels. In vitro characterization of the effects of Wt1 downregulation in murine bone marrow cells demonstrated that such downregulation led to increased ckit expression and an increase in the LSK stem/progenitor population, similar to prior observations with loss of Tet2 in vivo and in vitro.61–63 These observations raised the question as to the mechanism by which WT1 contributes to alteration in 5-hmC levels. Analysis of primary patient samples and hematopoietic cell lines demonstrated that the presence or absence of WT1 mutations had no bearing on TET1/TET2/TET3 expression levels, or on 2-hydroxyglutarate levels. WT1 was subsequently found to bind to TET2 in co-immunoprecipitation assays in transfected cell lines, as well as in AML cell lines. Furthermore, WT1 overexpression was able to attenuate the biochemical and immunophenotypic alterations observed in Tet2 knockout murine hematopoietic cells. However, the ability of WT1 to alter 5-hmC levels was noted to be attenuated by exposure to a cell-permeable form of 2-HG, which argued that WT1 likely mediates its effect via TET enzymes, and that in the absence of Tet2, WT1 might exert its effects through other TET enzymes. Subsequent experiments demonstrated that WT1 also binds to TET3, but not TET1.18 Work by Wang et al.19 confirmed and extended these findings. These authors likewise demonstrated that WT1 mutations are exclusive of TET2 and IDH1/2 mutations in AML using a meta-analysis of six studies, and also demonstrated that WT1 indeed binds to TET2. Moreover, overexpression of a catalytically active form of TET2 in cell lines resulted in the activation of known WT1-target genes; this activation required the presence of WT1. Further analysis using chromatin immunoprecipitation and PCR demonstrated that TET2 binds to transcription start sites and CpG islands of WT1 target genes, and that overexpression of both WT1 and TET2 led to increases in 5-hmC levels near the transcription start site of WT1-target genes. Finally these authors demonstrated that a proportion of TET2 mutations commonly found in AML result in the loss of the ability of WT1 to bind to TET2 (Figure 2).
Collectively, these studies have identified a previously unrecognized role for WT1 as an epigenetic modifier and place WT1 mutations in the mutational class defined by alterations in TET2 function in AML. Much work remains to fully describe the mechanism of leukemogenesis in WT1 mutant AMLs. For example, the precise subsets of genes that are regulated by the TET2/WT1 interaction and which become deregulated by a mutation in either remain an area of investigation. Toward this end, work by Sinha et al.64 also identified an association between WT1 mutations and DNA CpG hypermethylation using an algorithm based on Boolean implications on the TCGA AML patient data set. In particular, hypermethylation of polycomb repressive complex 2 (PRC2) target genes marked by H3K27 trimethylation, in both embryonic stem cells and the leukemia cell lines was observed. Gene expression analysis further revealed overexpression of EZH2, the catalytic subunit of the PRC2 complex, in WT1-mutant AMLs compared to normal karyotype AMLs. Taken together these two findings imply that silencing of polycomb target genes may be a necessary event in WT1-mediated leukemogenesis. Further delineation of the pathways involved in WT1 mutant mediated leukemogenesis is under way. In addition, no murine models of leukemogenesis that are driven by Wt1 mutations have yet been published. Such studies will likely give rise to a more precise understanding of the mechanism through which WT1 mutations mediate leukemogenesis.
Therapeutic implications
The observation that failure to reduce high levels of WT1 transcript after therapy was associated with a higher incidence of relapse in AML42 spurred efforts to utilize WT1 as a biomarker of minimal residual disease (MRD) in AML. Earlier efforts to use WT1 were met with mixed results as some investigators utilized primers targeting the 3′ region, which is where many WT1 mutations cluster.65 To address these discrepancies, Cilloni et al. performed an evaluation of nine published and other real-time quantitative polymerase chain reaction assays for WT1 quantification.66 The authors noted marked variation between assays. Using the best-performing assay to assess WT1 levels in diagnostic and follow-up samples they demonstrated that the kinetics of WT1 transcript reduction following induction was prognostically significant in terms of prediction of relapse.67 Patients in whom WT1 expression is not normalized following consolidation were at particular risk of relapse. By contrast, pretreatment WT1 levels were not found to be predictive of outcome. Of additional note, WT1 levels were not significantly modulated upon marrow regeneration following chemotherapy. Moreover, correcting for background WT1 expression in control peripheral blood and bone marrow, the degree of WT1 expression was not sufficiently robust to be used as a marker for sequential MRD analysis. Thus, the optimal utilization of WT1 in disease response assessment remains to be determined.
The involvement of both WT1 overexpression and WT1 mutations in AML has given rise to both clinical and preclinical therapeutic strategies. Perhaps the most prominent strategy in this regard has been the attempt to use peptide vaccines against WT1, given its overexpression in AML. Numerous studies have been carried out to date using a variety of different vaccination strategies (HLA-restricted versus non HLA-restricted peptides, for example). Clinically meaningful responses have been reported in several trials in both AML and MDS patients, with associated increases in WT1-specific T-cell frequencies (Table 2).68 Given the fact that WT1 is normally expressed in several tissues and plays a role in hematopoiesis, concern had been raised about the potential for WT1-vaccine therapies to elicit autoimmune reactions. However, this has not been reported thus far.69 This approach has thus demonstrated clinical activity, but still requires further large-scale evaluation. Another approach has been the development of a monoclonal antibody that recognizes a peptide fragment of WT1 complexed with HLA-A0201. This antibody demonstrated efficacy in a NOD/SCID mouse xenotransplanted with human leukemias.70
Table 2.
Overview of Select Trials of WT1 vaccines in AML and MDS patients.
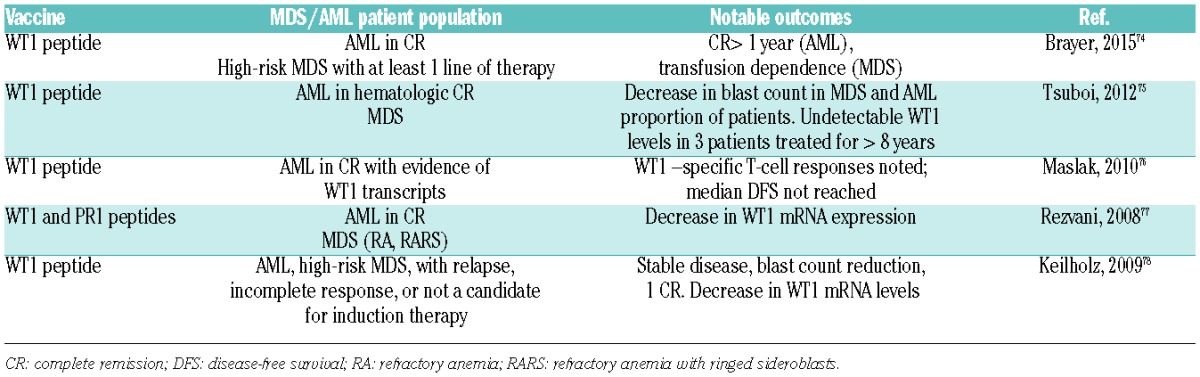
Given the epigenetic alterations catalogued in WT1 mutant (as well as TET2 and IDH mutant AMLs), epigenetic-targeted therapy has been explored as a potential mechanism to deal with this subgroup of leukemias. Indeed retrospective data has demonstrated an increased response rate with the hypomethylating agent azacitidine in MDS and AML patients with TET2 mutations compared with those without TET2 mutations. However, this increased response rate did not translate to improved overall survival of TET2-mutant cases.71,72 Finally, AML patients with IDH1/2 mutations were noted to have a higher response rate compared to those without mutations (71% vs. 23%, P=0.01) in a retrospective analysis of 42 patients receiving either decitabine or azacitidine. However, conclusions from this study are limited by the fact that it only included 42 patients, 7 of whom had mutations in IDH1/2, and larger studies have failed to detect this association.72,73 Thus, while there is evidence of a possible increased response to hypomethylating agents in patients bearing mutations in the DNA hydroxymethylation pathway, this still requires further exploration. Whether this holds true for WT1 mutations has not been established yet, and prospective evaluations of these findings are required. Finally, as previously discussed, WT1 mutations are associated with higher expression levels of EZH2 and appear to impact PRC2 target genes. In vitro genetic downregulation of the PRC2 member EZH2 in a WT1 mutant AML cell line, as well as pharmacologic inhibition of this enzyme in WT1 mutant cell lines or primary AML specimens resulted in increased differentiation of these cells.64 Thus, there are several treatment strategies, both clinical and preclinical, that warrant further testing both for WT1 overexpressing and WT1 mutant AML cases.
Conclusions
WT1 appears to be an important factor in both normal development and oncogenesis. However, the precise role of this protein has been difficult to pinpoint, owing to its complexity, both in terms of protein isoforms as well as the fact that it may participate in oncogenesis by way of either overexpression or loss-of-function mutations. This is a particular challenge in AML, where both scenarios are found. Recent work on WT1 mutations in AML have shed new light on the function of this protein, as there is a clear role for WT1 in epigenetic modifications. Thus far, this has only been studied in the context of AML and in the context of WT1 mutations. It remains to be determined what epigenetic role WT1 plays in normal development and other WT1-associated cancers. Likewise, whether the overexpression of WT1 in AML does or doesn’t lead to the perturbation of the epigenome on a genome-wide manner still remains to be explored. In addition, it is likely that the effects of mutations or overexpression are in part context dependent, and that cooperating genomic alterations modulate and are modulated by WT1’s effects on gene expression. As such, new answers revealed by recent work have given rise to new questions. Importantly, this recent work has given vital clues and brought us one step closer to unraveling this enigma wrapped in a mystery.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/6/672
References
- 1.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60(3):509–520. [DOI] [PubMed] [Google Scholar]
- 2.Gessler M, Poustka A, Cavenee W, et al. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature. 1990;343(6260): 774–778. [DOI] [PubMed] [Google Scholar]
- 3.Huff V. Wilms tumor genetics. Am J Med Genet. 1998;79(4):260–267. [DOI] [PubMed] [Google Scholar]
- 4.Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47(6):461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Little M, Holmes G, Bickmore W, et al. DNA binding capacity of the WT1 protein is abolished by Denys-Drash syndrome WT1 point mutations. Hum Mol Genet. 1995;4(3):351–358. [DOI] [PubMed] [Google Scholar]
- 6.Little MH, Williamson KA, Mannens M, et al. Evidence that WT1 mutations in Denys-Drash syndrome patients may act in a dominant-negative fashion. Hum Mol genet. 1993;2(3):259–264. [DOI] [PubMed] [Google Scholar]
- 7.Klamt B, Koziell A, Poulat F, et al. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum Mol genet. 1998;7(4):709–714. [DOI] [PubMed] [Google Scholar]
- 8.Yang L, Han Y, Saiz FS, Minden MD. A tumor suppressor and oncogene: the WT1 story (vol 21, pg 868, 2007). Leukemia. 2007;21(5):1603–1603. [DOI] [PubMed] [Google Scholar]
- 9.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997;9(3):209–225. [DOI] [PubMed] [Google Scholar]
- 10.Vicent S, Chen R, Sayles LC, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120(11): 3940–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu W, Hu S, Chen Z, et al. High expression of WT1 gene in acute myeloid leukemias with more predominant WT1+17AA isoforms at relapse. Leuk Res. 2010;34(1):46–49. [DOI] [PubMed] [Google Scholar]
- 12.Bor YC, Swartz J, Morrison A, et al. The Wilms’ tumor 1 (WT1) gene (+KTS isoform) functions with a CTE to enhance translation from an unspliced RNA with a retained intron. Genes Dev. 2006;20(12): 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison AA, Venables JP, Dellaire G, Ladomery MR. The Wilms tumour suppressor protein WT1 (+KTS isoform) binds alpha-actinin 1 mRNA via its zinc-finger domain. Biochem Cell Biol. 2006;84(5):789–798. [DOI] [PubMed] [Google Scholar]
- 14.Hossain A, Nixon M, Kuo MT, Saunders GF. N-terminally truncated WT1 protein with oncogenic properties overexpressed in leukemia. J Biol Chem. 2006;281(38): 28122–28130. [DOI] [PubMed] [Google Scholar]
- 15.Maheswaran S, Park S, Bernard A, et al. Physical and functional interaction between WT1 and p53 proteins. Proc Natl Acad Sci USA. 1993;90(11):5100–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bansal H, Bansal S, Rao M, et al. Heat shock protein 90 regulates the expression of Wilms tumor 1 protein in myeloid leukemias. Blood. 2010;116(22):4591–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rong Y, Cheng L, Ning H, et al. Wilms’ tumor 1 and signal transducers and activators of transcription 3 synergistically promote cell proliferation: a possible mechanism in sporadic Wilms’ tumor. Cancer Res. 2006;66(16):8049–8057. [DOI] [PubMed] [Google Scholar]
- 18.Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9(5):1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell 2015;57(4):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird PN, Simmons PJ. Expression of the Wilms’ tumor gene (WT1) in normal hemopoiesis. Exp Hematol. 1997;25(4):312–320. [PubMed] [Google Scholar]
- 21.Hosen N, Sonoda Y, Oji Y, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Naematol. 2002;116(2):409–420. [DOI] [PubMed] [Google Scholar]
- 22.Ellisen LW, Carlesso N, Cheng T, Scadden DT, Haber DA. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20(8): 1897–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraizer GC, Patmasiriwat P, Zhang X, Saunders GF. Expression of the tumor suppressor gene WT1 in both human and mouse bone marrow. Blood. 1995;86(12): 4704–4706. [PubMed] [Google Scholar]
- 24.Svedberg H, Richter J, Gullberg U. Forced expression of the Wilms tumor 1 (WT1) gene inhibits proliferation of human hematopoietic CD34(+) progenitor cells. Leukemia. 2001;15(12):1914–1922. [DOI] [PubMed] [Google Scholar]
- 25.Inoue K, Tamaki H, Ogawa H, et al. Wilms’ tumor gene (WT1) competes with differentiation-inducing signal in hematopoietic progenitor cells. Blood. 1998;91(8):2969–2976. [PubMed] [Google Scholar]
- 26.Kreidberg JA, Sariola H, Loring JM, et al. Wt-1 Is Required for Early Kidney Development. Cell. 1993;74(4):679–691. [DOI] [PubMed] [Google Scholar]
- 27.Alberta JA, Springett GM, Rayburn H, et al. Role of the WT1 tumor suppressor in murine hematopoiesis. Blood. 2003;101(7): 2570–2574. [DOI] [PubMed] [Google Scholar]
- 28.King-Underwood L, Little S, Baker M, et al. Wt1 is not essential for hematopoiesis in the mouse. Leuk Res. 2005;29(7):803–812. [DOI] [PubMed] [Google Scholar]
- 29.Hosen N, Shirakata T, Nishida S, et al. The Wilms’ tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia. 2007;21(8): 1783–1791. [DOI] [PubMed] [Google Scholar]
- 30.Chau YY, Brownstein D, Mjoseng H, et al. Acute Multiple Organ Failure in Adult Mice Deleted for the Developmental Regulator Wt1. Plos Genet. 2011;7(12):e1002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyagi T, Ahuja H, Kubota T, et al. Expression of the candidate Wilm’s tumor gene, WT1, in human leukemia cells. Leukemia. 1993;7(7):970–977. [PubMed] [Google Scholar]
- 32.Miwa H, Beran M, Saunders GF. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia. 1992;6(5):405–409. [PubMed] [Google Scholar]
- 33.Menssen HD, Renkl HJ, Rodeck U, et al. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia. 1995;9(6):1060–1067. [PubMed] [Google Scholar]
- 34.Tamaki H, Ogawa H, Ohyashiki K, et al. The Wilms’ tumor gene WT1 is a good marker for diagnosis of disease progression of myelodysplastic syndromes. Leukemia. 1999;13(3):393–399. [DOI] [PubMed] [Google Scholar]
- 35.Miyagi T, Ahuja H, Kubota T, et al. Expression of the Candidate Wilms-Tumor Gene, Wt1, in Human Leukemia-Cells. Leukemia. 1993;7(7):970–977. [PubMed] [Google Scholar]
- 36.King-Underwood L, Pritchard-Jones K. Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood. 1998;91(8): 2961–2968. [PubMed] [Google Scholar]
- 37.KingUnderwood L, Renshaw J, PritchardJones K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood. 1996;87(6):2171–2179. [PubMed] [Google Scholar]
- 38.Miwa H, Beran M, Saunders GF. Expression of the Wilms-Tumor Gene (Wt1) in Human Leukemias. Leukemia. 1992;6(5):405–409. [PubMed] [Google Scholar]
- 39.Barragan E, Cervera J, Bolufer P, et al. Prognostic implications of Wilms’ tumor gene (WT1) expression in patients with de novo acute myeloid leukemia. Haematologica. 2004;89(8):926–933. [PubMed] [Google Scholar]
- 40.Bergmann L, Miething C, Maurer U, et al. High levels of Wilms’ tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood. 1997;90(3):1217–1225. [PubMed] [Google Scholar]
- 41.Karakas T, Miething CC, Maurer U, et al. The coexpression of the apoptosis-related genes bcl-2 and wt1 in predicting survival in adult acute myeloid leukemia. Leukemia. 2002;16(5):846–854. [DOI] [PubMed] [Google Scholar]
- 42.Cilloni D, Renneville A, Hermitte F, et al. Real-Time Quantitative Polymerase Chain Reaction Detection of Minimal Residual Disease by Standardized WT1 Assay to Enhance Risk Stratification in Acute Myeloid Leukemia: A European LeukemiaNet Study. J Clin Oncol. 2009;27(31):5195–5201. [DOI] [PubMed] [Google Scholar]
- 43.Yamagami T, Sugiyama H, Inoue K, et al. Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: Implications for the involvement of WT1 in leukemogenesis. Blood. 1996;87(7):2878–2884. [PubMed] [Google Scholar]
- 44.Ito K, Oji Y, Tatsumi N, et al. Antiapoptotic function of 17AA(+)WT1 (Wilms’ tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. 2006;25(30):4217–4229. [DOI] [PubMed] [Google Scholar]
- 45.Nishida S, Hosen N, Shirakata T, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107(8):3303–3312. [DOI] [PubMed] [Google Scholar]
- 46.King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood. 1996;87(6):2171–2179. [PubMed] [Google Scholar]
- 47.Hou HA, Huang TC, Lin LI, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010;115(25):5222–5231. [DOI] [PubMed] [Google Scholar]
- 48.Renneville A, Boissel N, Zurawski V, et al. Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer. 2009;115(16):3719–3727. [DOI] [PubMed] [Google Scholar]
- 49.Gaidzik VI, Schlenk RF, Moschny S, et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood. 2009; 113(19):4505–4511. [DOI] [PubMed] [Google Scholar]
- 50.Virappane P, Gale R, Hills R, et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol. 2008;26(33):5429–5435. [DOI] [PubMed] [Google Scholar]
- 51.Paschka P, Marcucci G, Ruppert AS, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2008;26(28):4595–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pritchardjones K, Renshaw J, Kingunderwood L. The Wilms-Tumor (Wt1) Gene Is Mutated in a Secondary Leukemia in a Wagr Patient. Hum Mol Genet. 1994;3(9):1633–1637. [DOI] [PubMed] [Google Scholar]
- 53.Abbas S, Erpelinck-Verschueren CAJ, Goudswaard CS, Lowenberg B, Valk PJM. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia. 2010;24(3):660–663. [DOI] [PubMed] [Google Scholar]
- 54.Becker H, Marcucci G, Maharry K, et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116(5):788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell. 2010;18(6):553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310): 1129–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tahiliani M, Koh KP, Shen YH, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science. 2009;324(5929):930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ward PS, Patel J, Wise DR, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting alpha-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell. 2011;20(1):11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quivoron C, Couronne L, Della Valle V, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell. 2011;20(1):25–38. [DOI] [PubMed] [Google Scholar]
- 63.Li Z, Cai XQ, Cai CL, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinha S, Thomas D, Yu L, et al. Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood. 2015;125(2):316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grimwade D, Vyas P, Freeman S. Assessment of minimal residual disease in acute myeloid leukemia. Curr Opin Oncol. 2010;22(6):656–663. [DOI] [PubMed] [Google Scholar]
- 66.Cilloni D, Renneville A, Hermitte F, et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European LeukemiaNet study. J Clin Oncol. 2009;27(31):5195–5201. [DOI] [PubMed] [Google Scholar]
- 67.Van Dijk JP, Knops GH, Van De Locht LT, et al. Abnormal WT1 expression in the CD34-negative compartment in myelodysplastic bone marrow. Br J Haematol. 2002;118(4): 1027–1033. [DOI] [PubMed] [Google Scholar]
- 68.Di Stasi A, Jimenez AM, Minagawa K, Al-Obaidi M, Rezvani K. Review of the Results of WT1 Peptide Vaccination Strategies for Myelodysplastic Syndromes and Acute Myeloid Leukemia from Nine Different Studies. Front Immunol. 2015;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Driessche A, Berneman ZN, Van Tendeloo VF. Active specific immunotherapy targeting the Wilms’ tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: lessons from early clinical trials. Oncologist. 2012;17(2):250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dao T, Yan S, Veomett N, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5(176):176ra133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–1152. [DOI] [PubMed] [Google Scholar]
- 72.Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124(17):2705–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emadi A, Faramand R, Carter-Cooper B, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90(5):E77–79. [DOI] [PubMed] [Google Scholar]
- 74.Brayer J, Lancet JE, Powers J, et al. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am J Hematol. 2015;90(7):602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsuboi A, Oka Y, Kyo T, et al. Long-term WT1 peptide vaccination for patients with acute myeloid leukemia with minimal residual disease. Leukemia. 2012;26(6): 1410–1413. [DOI] [PubMed] [Google Scholar]
- 76.Maslak PG, Dao T, Krug LM, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116(2):171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rezvani K, Yong AS, Mielke S, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111(1): 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Keilholz U, Letsch A, Busse A, et al. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113(26):6541–6548. [DOI] [PubMed] [Google Scholar]