

Though rare, inherited forms of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are being recognized with increasing frequency. Germline mutations in well-characterized genes such as RUNX1, CEBPA, GATA2 and ETV6 have been found to predispose to MDS/AML.1–4 Families with mutations in these genes display an autosomal dominant mode of inheritance and a variable latency period until disease onset. The mutations are heterozygous, most commonly producing a loss of function allele and subsequent haploinsufficiency of gene dosage, though gain of function mutations have also been reported.5 Progression to hematologic malignancy is thought to require the acquisition of somatic mutations in bone marrow stem or progenitor cells, often in the same gene that contains the germline mutation. Furthermore, all of these genes have been found to be recurrently mutated in sporadic disease, and investigation into their function has provided an insight into normal hematopoiesis as well as the pathogenesis of hematologic malignancies. Recently, heterozygous germline mutations in the DEAD/H-box helicase gene DDX41 were shown to predispose to MDS/AML.6,7 The most frequent DDX41 mutation identified was a frameshift duplication causing a premature stop codon (p.D140Gfs*2); however, missense and splice variants were also described. These families displayed the characteristics of other MDS/AML syndromes, including an autosomal dominant inheritance pattern and a long latency period.
The gene for DDX41 encodes an RNA helicase protein thought to function in RNA splicing; however, its role in hematopoiesis and leukemogenesis remains unknown. In addition, the prevalence and penetrance of DDX41 mutations, as well as the prognosis of individuals that develop disease, are unclear. Crucial to the investigation of this recently discovered MDS/AML predisposition syndrome is the identification of additional families with germline DDX41 mutations. Characterization of the pathological inherited variant is important for diagnosis and for genetic counseling of patients and their families. This is especially true for asymptomatic carriers, a frequent occurrence given the long latency period associated with this disease. Here, we report a Caucasian family with inherited MDS/AML with two germline mutations in the DDX41 gene. The proband is a 66-year old male who presented with cytopenias and was diagnosed with acute erythroid leukemia (erythroid/myeloid subtype) on bone marrow biopsy. Family history revealed a mother who had died of AML at 86 years of age and 2 brothers diagnosed with MDS at 59 and 70 years of age, as well as 2 additional brothers with no evidence of hematologic malignancy. All 3 brothers with MDS or AML were treated with chemotherapy followed by allogeneic bone marrow transplant and are currently in complete remission (Table 1).
Table 1.
Summary of clinical and genetic characteristics.
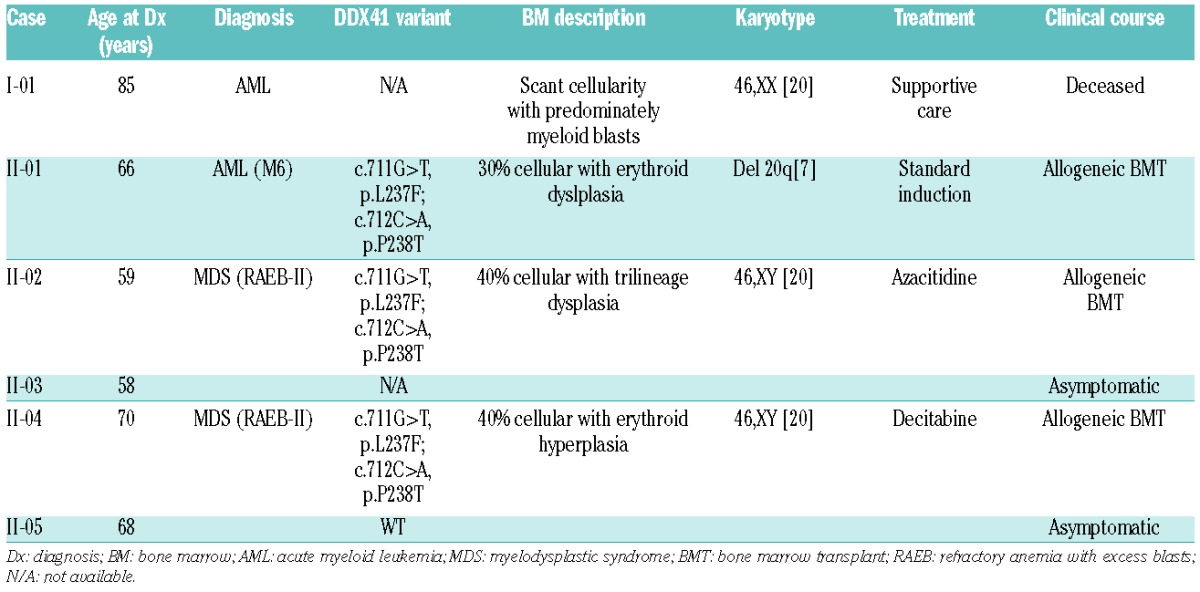
The pedigree for this family exhibits an autosomal dominant inheritance pattern with a long latency period, consistent with other known familial MDS/AML syndromes (Figure 1A). To identify the inherited causal variant of the phenotype in this family, exome sequencing of the proband (II-1) and his affected siblings (II-2 and II-4) was performed on germline DNA using the Illumina HiSeq2500 platform. Using the PhenoDB variant analysis tool,8 we selected heterozygous functional variants with a minor allele frequency (MAF) of less than 0.01 in the 1000 Genome Project and in the Exome Variant Server shared by the 3 affected brothers. This analysis produced a list of 59 candidate single nucleotide variants (SNVs) (Online Supplementary Table S1). These were ranked by SIFT and Polyphen-2 scores,9,10 generating 18 SNVs as likely to be pathological inherited variants. Of these, two consecutive SNVs in the DDX41 gene (c.711G>T, p.L237F and c.712C>A, p.P238T) were identified and confirmed by Sanger sequencing (Figure 1B). We then performed Sanger sequencing on a sample available from one unaffected brother (II-5), which did not possess either of the variants (Figure 1B); thus, these DDX41 variants co-segregate with the disease in this family. While no other SNVs in genes known to predispose to MDS or AML were identified in our analysis, it is important to note that exome sequencing may fail to identify all possible genetic abnormalities, including copy number variants and non-canonical splice variants. To determine whether the DDX41 variants were located on the same allele (cis or trans), PCR products generated from targeted sequencing of genomic DNA of the proband were cloned and re-sequenced. Out of nine clones, three displayed the wild-type (WT) allele while six displayed both variants, consistent with a cis configuration (Online Supplementary Figure S1A).
Figure 1.
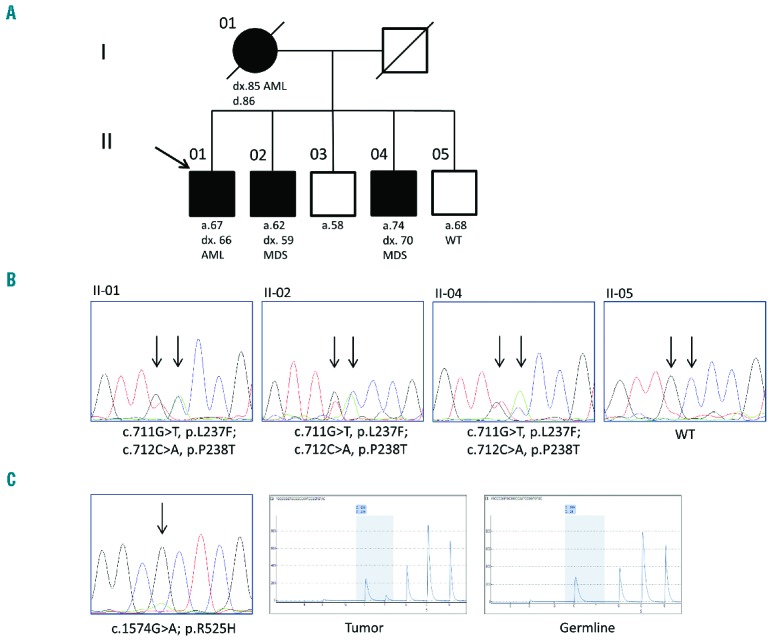
Genetic analysis of family with inherited myelodysplastic syndrome/acute myeloid leukemia. (A) Family pedigree is consistent with an autosomal dominant mode of inheritance. Filled circles/boxes denote affected family members, while slashes denote deceased individuals. A black arrow denotes the proband. (B) Representative chromatograms from Sanger sequencing of family members. The affected nucleotides are indicated with black arrows. (C) Analysis of the tumor sample of subject II-1 shows evidence of a p.R525H mutation both by Sanger (right panel) and pyrosequencing (center panel). Pyrosequencing of genomic DNA shows only the reference allele (left panel). Note that the sequencing primer for pyrosequencing aligns to the reverse complement sequence.
The two variants identified in this family are located in exon 8 of the DDX41 gene, corresponding to the DEAD/ATP binding domain of the DDX41 protein; the amino acids affected by these SNVs are highly conserved across species and across the DDX family of proteins (Figure 2). The amino acid residues are in a conserved motif that includes the ATP binding site.11 Neither variant is found in the Exome Variant Server [NHLBI Exome Sequencing Project (EVS)12] or the Catalogue Of Somatic Mutations In Cancer (COSMIC).13 However, a rare c.712C>T, p.P238S SNV (rs376093707) with an MAF of 0.0077% is noted in the EVS. The Exome Aggregation Consortium (ExAC) identifies both c.711G>T, p.L237F and c.712C>A, p.P238T in 1/120,916 alleles, indicating both SNVs are extremely rare.14 They are characterized as probably damaging (Polyphen) and deleterious (SIFT), further supporting their role as the inherited mutations predisposing to disease in this family.
Figure 2.
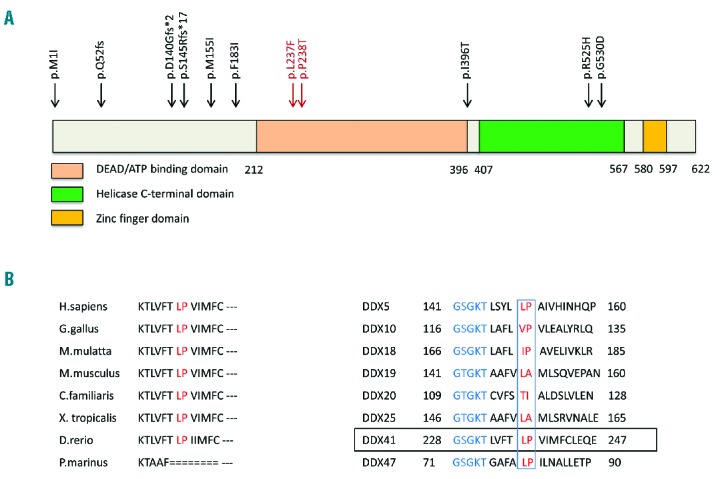
Structure and alignment of the DDX41 protein. (A) Schematic diagram of the DDX41 protein indicating sites of known germline mutations. The variants described in this report are shown in red. Known functional domains are indicated. (B and C) Protein sequence of DDX41 flanking the Leu237Phe and Pro238Thr variants (red) and alignment to the corresponding regions of various species and the DEAD-box family members. The ATP-binding site (blue) is located close to the variants (red) among the eight homologs shown.
Approximately 50% of individuals with a germline DDX41 mutation were reported to have an additional acquired somatic DDX41 mutation in their tumor DNA.7 The most frequently identified mutation is c.1574G>A, p.R525H, which corresponds to a highly conserved region across the DDX family in the helicase domain thought to contribute to nucleotide co-ordination.11 We sequenced the entire DDX41 coding region in the tumor sample of the proband via Sanger sequencing and found evidence of a somatic mutation at this site. This was confirmed by pyrosequencing which showed an adenosine at this position in approximately 20% of the amplicons (Figure 1C). No other mutations were identified; however, the presence of a small subclone containing a different DDX41 mutation cannot be ruled out given the allele frequency detection limit of Sanger sequencing. While the pathophysiology of leukemogenesis in DDX41-driven disease is still not known, Polprasert et al. showed that knockdown of DDX41 mRNA leads to enhanced proliferation, possibly due to defects in RNA splicing. The most common DDX41 mutation identified in families is an N-terminal frameshift, consistent with a loss of function defect. The presence of a ‘second hit’ in the tumor sample of the proband in this study also supports a loss of function model. To determine whether the variants reported here result in a loss of function, we over-expressed both the WT and mutant forms of DDX41 in 293T cells using an EGFP expression vector. Approximately 30%–40% of transfected cells expressed WT DDX41-GFP protein, which localized to the nucleus. Conversely, expression of mutant DDX41-GFP was ab-sent or barely detectable in the vast majority of transfected cells (Online Supplementary Figure S1B). In cells that did express mutant DDX41-GFP, localization was pre-dominately nuclear, though partial cytoplasmic expression was evident in some cells, indicating a possible defect in nuclear localization. Western blot corroborated these results, revealing absent mutant DDX41-GFP expression in transfected cells (Online Supplementary Figure S1C). Additional studies of DDX41 function in hematopoiesis will provide critical insight into its role in disease predisposition and may help in the treatment of MDS/AML.
In summary, we have identified two previously unreported consecutive mutations in the DDX41 gene that predispose to MDS/AML. All 3 affected individuals found to possess these mutations are currently in complete remission following bone marrow transplant. The clinical and genetic investigation of the phenotype in this family as well as others with inherited hematologic malignancies is crucial to provide appropriate counseling and treatment to patients and families, and to raise awareness of these diseases.
Acknowledgments
The authors would like to thank the patients and family members for participation in this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Sanger and pyrosequencing was conducted at the Genetic Resources Core Facility, Johns Hopkins Institute of Genetic Medicine, Baltimore, MD, USA
Footnotes
Funding: this work was supported by NHLBI K12HL087169 (EMB) and by NHGRI 1U54HG006542 to the Baylor-Hopkins Center for Mendelian Genomics.
The online version of this letter has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403–2407. [DOI] [PubMed] [Google Scholar]
- 4.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. [DOI] [PubMed] [Google Scholar]
- 5.Godley LA. Inherited predisposition to acute myeloid leukemia. Semin Hematol. 2014;51(4):306–321. [DOI] [PubMed] [Google Scholar]
- 6.Lewinsohn M, Brown AL, Weinel LM, et al. Novel germline DDX41 mutations define families with a lower age of MDS/AML onset, and lymphoid malignancies. Blood. 2016;127(8):1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polprasert C, Schulze I, Sekeres MA, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27(5):658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobreira N, Schiettecatte F, Boehm C, Valle D, Hamosh A. New tools for mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum Mutat. 2015;36(4):425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schutz P, Karlberg T, van den Berg S, et al. Comparative structural analysis of human DEAD-box RNA helicases. PLoS One. 2010;5(9):e12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, USA. Available from: http://evs.gs.washington.edu/EVS/Last accessed Nov 2015.
- 13.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lek M, Karczewski K, Minikel E, et al. Exome aggregation consortium. Analysis of protein-coding genetic variation in 60,706 humans. BioRxiv October 30 2015. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]