

Abstract
Motivation: Chemical cross-linking with mass spectrometry (XL-MS) provides structural information for proteins and protein complexes in the form of crosslinked residue proximity and distance constraints between reactive residues. Utilizing spatial information derived from cross-linked residues can therefore assist with structural modeling of proteins. Selection of computationally derived model structures of proteins remains a major challenge in structural biology. The comparison of site interactions resulting from XL-MS with protein structure contact maps can assist the selection of structural models.
Availability and implementation: XLmap was implemented in R and is freely available at: http://brucelab.gs.washington.edu/software.php.
Contact: jimbruce@uw.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The prediction of protein structures is a major goal of computational structural biology. Accurate prediction of protein structures remains a significant challenge. Chemical cross-linking with mass spectrometry provides residue level distance constraints (DCs) that are useful for refining protein structural models (Hofmann et al., 2015; Rappsilber, 2011). Yet few tools exist for integrating cross-linking data with protein structure prediction. To accommodate this lack of tools, herein we describe the development of XLmap, an algorithm implemented in R, to integrate cross-linked sites originating from cross-link mass spectrometry (XL-MS) data with protein contact maps. A protein structural model can be represented as protein contact map consisting of a binary two-dimensional matrix of the distance between all possible amino acid residue pairs of a three-dimensional protein structure. Euclidean distances between cross-linked residues can be displayed on protein contact maps. The coincidence of cross-linked residues and contact points derived from protein structures can then be determined and quantified, herein as residual scores. We define the sum of all residual scores determined in this way as the CMScore and lower CMScores correspond to better agreement between the cross-linked residue map and the structure-derived protein contact map. These CMScores can then be used to integrate XL-MS experimental data into the process of selection of optimum predicted structural models (Kelley et al., 2015; Yang et al., 2015).
2 Package description
XLmap was implemented in the statistical language R and is provided as an R package in the Supplementary Information. XLmap takes in PDB format protein structural files and cross-linked site information, integrates the data, generates overlaid plots of crosslinked sites on contact maps and assigns a score (CMScore) to each model.
Two files are required as input: (i) a protein structure derived either from model prediction software or from empirical data (i.e. PDB or PDBx/mmCIF file) and (ii) a table of the cross-linked sites in tab delimited text format with the following four columns: protein UniProt accession 1 (‘prot1’), protein UniProt accession 2 (‘prot2’), cross-linked residue 1 (‘mod_pos1’) and cross-linked residue 2 (‘mod_pos2’). Currently, XLmap can automatically extract this data from larger data files such as the outputs from ReACT (Weisbrod et al., 2013) and xQuest (Rinner et al., 2008). Alternatively, a text file containing the above four columns alone can be used as input. An example input file is included in the Supplementary Text and within the package itself (Supplementary Text S4f). The user then specifies a desired maximum allowed contact distance in angstroms (Å) between the alpha carbons of the two cross-linked residues (Cα-Cα). Contact maps are generated through the following command:
>contactmap(DC, UniProt accession)
Images of the contact maps can be exported as ‘.pdf’ files. Additionally, contactmap will output a table that can be used as the input for CMscore.
The overlap of the structural map and the crosslinked array is then calculated as a residual score for the specified DC. By calculating the residual score across multiple DCs and summing these scores, a CMScore is generated using the following formula:
As the overlap between the contact map and cross-linked residue map increases, the CMScore will approach zero. A CMScore and plot are generated through the following command:
>cmscore(DC-range, UniProt accession)
Users will then be prompted for input of a PDB file and tab delimited text file containing cross-link information. Example data in the package includes structures and crosslink information for the Acinetobacter baumannii proteins Ab57_2983 and Oxa23, the yeast protein TbpA (TbpA crosslinks derived from Leitner et al., 2012) and the human protein PPIB.
3 Application and use cases
XLmap generates two features: a protein contact map image with cross-linked sites displayed and a CMScore plot of distance versus residual score, which can be used to gauge the level of agreement between the observed cross-linked sites and the contacting amino acids at various distances.
First, XLmap generates a protein contact map. For example, the 14 cross-linked sites observed by Leitner et al. (2012) in T-complex protein 1 subunit alpha from Saccharomyces cerevisiae (TCPA_YEAST, P12612) are mapped to the crystal structure PDB = 3P9D at DC = 18Å (Fig. 1A).
>contactmap(18,’P12612’)
Fig. 1.
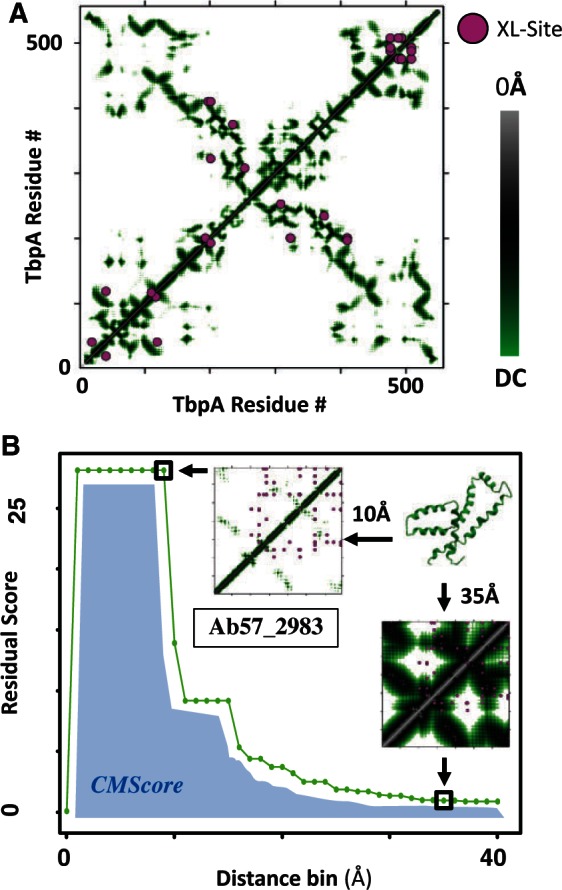
XLmap crosslink map for TbpA (A) and CMScore generation for Ab57_2983 Phyre2 de novo model (B)
Second, XLmap generates a CMScore for a given protein structure or model based on the number of crosslinked sites that overlap with the generated contact map across a range of DCs (Fig. 1B). CMScores can then be used to determine how well sites of crosslinking overlap across multiple structural models. This is exemplified for the top three de novo structure predictions of Ab57_2983 from both Phyre2 and iTASSER (6 total models). The model with the best match to XL-MS data was returned as the second highest scoring iTASSER model (Supplementary Fig. S1).
Finally, we validated the use of CMScores to differentiate between a published crystal structure and a decoy structure derived from the same amino acid sequence. We show that for the human PPIB protein (Uniprot: P23284), CMScore can accurately differentiate the true (PDB: 1CYN) and decoy structure based on the coincidence of structure contact sites and crosslinked residue positions within the protein (Supplementary Fig. S2).
4 Conclusions
XLmap is an R package, which integrates cross-linked site information with protein contact maps. XLmap evaluates the overlap between the cross-linking information with the protein contact map over a specified distance range resulting in a CM score. In silico prediction of protein structures by common algorithms such as Phyre2 and iTASSER can generate multiple putative models for one protein sequence (Kelley et al., 2015; Yang et al., 2015). When comparing multiple models of the same protein using the package described here, the lowest CMScore corresponds with the model with the highest incidence of overlap between crosslinking data and structural models at the lowest Cα-Cα distance. Therefore, CMScores can be used to assess predicted structural data based on protein crosslinking. Importantly, XLmap is not restricted by either the model prediction software (e.g. Phyre2 and iTasser) or XL-MS platform (e.g. crosslinker or MS instrumentation/searching) used to generate input data. In summary, XLmap provides a new tool to utilize experimentally derived cross-linking information in protein structural analysis.
Supplementary Material
Acknowledgement
The authors thank Jimmy Eng at the University of Washington Proteomics core for technical assistance.
Funding
This study was supported by National Institutes of Health grants: U19-AI107775-01, R01-AI101307-02, R01-HL110879-04, R01-GM086688-06 and R01-GM097112-04.
Conflict of Interest: none declared.
References
- Hofmann T., et al. (2015) Protein structure prediction guided by crosslinking restraints—a systematic evaluation of the impact of the crosslinking spacer length. Methods, 89, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., et al. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc., 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A., et al. (2012) The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure, 20, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J. (2011) The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol., 173, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O., et al. (2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods, 5, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod C.R., et al. (2013) In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J. Proteome Res., 12, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., et al. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods, 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.