

ABSTRACT
The transcription factor VP30 of the non-segmented RNA negative strand Ebola virus balances viral transcription and replication. Here, we comprehensively studied RNA binding by VP30. Using a novel VP30:RNA electrophoretic mobility shift assay, we tested truncated variants of 2 potential natural RNA substrates of VP30 - the genomic Ebola viral 3′-leader region and its complementary antigenomic counterpart (each ∼155 nt in length) - and a series of other non-viral RNAs. Based on oligonucleotide interference, the major VP30 binding region on the genomic 3′-leader substrate was assigned to the internal expanded single-stranded region (∼ nt 125–80). Best binding to VP30 was obtained with ssRNAs of optimally ∼ 40 nt and mixed base composition; underrepresentation of purines or pyrimidines was tolerated, but homopolymeric sequences impaired binding. A stem-loop structure, particularly at the 3′-end or positioned internally, supports stable binding to VP30. In contrast, dsRNA or RNAs exposing large internal loops flanked by entirely helical arms on both sides are not bound. Introduction of a 5´-Cap(0) structure impaired VP30 binding. Also, ssDNAs bind substantially weaker than isosequential ssRNAs and heparin competes with RNA for binding to VP30, indicating that ribose 2′-hydroxyls and electrostatic contacts of the phosphate groups contribute to the formation of VP30:RNA complexes. Our results indicate a rather relaxed RNA binding specificity of filoviral VP30, which largely differs from that of the functionally related transcription factor of the Paramyxoviridae which binds to ssRNAs as short as 13 nt with a preference for oligo(A) sequences.
KEYWORDS: Ebola virus (EBOV), electrophoretic/gel mobility shift assay, RNA binding specificity, transcription factor, VP30
Introduction
Non-segmented, negative-sense RNA viruses of the family Filoviridae1 include Ebola virus (EBOV), Marburg virus (MARV) and the recently discovered species Lloviu (LLOV) cuevavirus.2 Filoviruses can cause severe fever in humans and non-human primates with fatality rates of up to 90% in humans.3 The recent EBOV outbreak in West Africa caused more than 29,000 cases and claimed more than 11,000 lives. To date neither prophylactic vaccines nor antiviral drugs are available for human use.
The (−) RNA genome of EBOV, which has a length of about 19 kb, consists of 7 genes flanked by 2 regulatory regions, the 3′-leader and the 5′-trailer region.4 The genomic 3′-leader contains the bipartite replication promoter as well as the transcription start site for the first gene, while the 5′-trailer encodes the complementary sequence of the replication promoter utilized by the viral RNA-dependent RNA polymerase to synthesize (−) RNA copies of (+) RNA antigenomes.5,6,7 The RNA genome is encapsidated by the nucleoprotein NP which forms the helical nucleocapsid together with the viral proteins (VP) 24, VP30 and VP35 and the RNA-dependent RNA polymerase L. While L and VP35 are essential for replication and transcription, VP30 is only essential for transcription but dispensable for replication.7 Phosphorylation of VP30 negatively regulates transcription activation and the strict VP30 dependency of viral transcription could be overcome by destabilizing a stem-loop structure at the transcription start site of the NP gene.8,9 Stem-loop structures were also predicted at the transcription initiation sites of the other EBOV genes, in line with the finding that VP30 is also involved in transcription reinitiation at the downstream genes.10 Previous studies have focused on the formation of such structures at the 5´-end of each mRNA,10,11 but they are also predicted by secondary structure calculation algorithms to form on the genomic RNA level (Fig. S1). It was previously proposed that the stability of these RNA structures on the mRNA level gradually increases from the first to the last EBOV gene,11 which may explain the declining gradient of successful transcriptional reinitiation along the viral genome.10
So far, evidence for RNA binding by VP30 was inferred from nitrocellulose filter binding and UV-crosslinking experiments.12 Yet, the authors analyzed only a few RNA substrates and neither addressed the dependency of binding on RNA length or base composition nor the position of VP30 binding within potential natural RNA substrates such as the 3′-leader. We also wanted to shed more light on the conclusion drawn in the previous study, namely that an RNA stem-loop supports binding to VP30 when placed in the context of single-stranded upstream and downstream sequences.12
Here, we investigated RNA binding by VP30 using an enlarged variety of RNA substrates and employing a newly developed electrophoretic mobility shift assay (EMSA) based on discontinuous native polyacrylamide gel electrophoresis (PAGE). Our results can be summarized as follows: (i) the optimal RNA substrate for VP30 is a single-stranded RNA of ∼ 40 nt and mixed base composition that is linked to a stem-loop structure, as found in the region of the replication promoter element 2 (PE2) of the Ebola genomic 3′-leader, (ii) RNA binding is impaired in the presence of a 5´-Cap(0) structure, (iii) DNA binds substantially weaker than RNA and (iv) heparin competes with RNA for binding to VP30, indicating a substantial electrostatic contribution to binding.
Results
As mentioned in the Introduction, we neither know much about RNA features recognized by VP30 in terms of RNA length, sequence/base composition and structure, nor is it clear whether VP30 interacts with genomic and/or antigenomic RNA. Potential binding partners of VP30 are the genomic 3′-leader or the mRNA transcript initiated 56 nucleotides upstream of the genome 3′-end. An alignment representing the major EBOV subtypes (Fig. 1A) indicates substantial sequence conservation in the ∼80 3′-terminal residues of the genomic leader, which then decreases in the second replication promoter element and even more further upstream (Fig. 1A). At the beginning of our study we considered the possibility that VP30 may bind to sequences in the promoter region and may have evolved an RNA binding mode adapted to the sequence and structural features in the genomic leader region. Thus, after an initial structure probing analysis, we first analyzed VP30 binding to RNA fragments derived from the genomic 3′-leader region of VP30.
Figure 1.

(A) Alignment of the 3′-leader region (∼154 nucleotides) of different Ebola species; the bipartite replication promoter is indicated by the green-shaded and the transcription start region (TSR) by the red-shaded bar regions above the sequences; 5 while the 3′-terminal ∼ 78 nt of the genomic (−) RNA are strongly conserved, the different EBOV sequences become more diverse further upstream (nt 79–154). The uracils of the 8 UN5 hexamers of the PE2 element of Zaire ebolavirus5 are marked in magenta. EBOV: Zaire ebolavirus; BDBV: Bundibugyo ebolavirus; SUDV: Sudan ebolavirus; RESTV: Reston ebolavirus; TAFV: Tai Forest ebolavirus. (B) Schematic comparison of Ebola VP30 and related M2–1 transcription factors of Paramyxoviridae. In VP30 proteins, the N-terminal domain, which is absent in M2–1 proteins, harbors the region of amino acids (aa) 26–46 involved in RNA binding and phosphorylation (white box; 12,30 the zinc finger domain (hatched with vertical lines, aa 72–90;31),the hexamerization motif (black box, aa 100–104; 32) and the region mediating VP30 dimerization and binding to NP (diagonally hatched box;17). For M2–1, the corresponding Zn finger domain and the core domain harboring the tetramerization helix and the phosphorylation targets S58 and S61 are depicted.23 For more details, see Fig. S7.
Secondary structure probing of the genomic 3′-leader and its antigenomic copy
As a first step, we analyzed the solution structure of the 3′-terminal 154 nt of the genomic (−) RNA leader using single strand-specific Pb2+-and RNase T1-induced hydrolysis to assess the potential structural complexity of this functionally important EBOV genomic region. Representative gel images are shown in Figs. 2A-D and the results are summarized in the proposed secondary structure in Fig. 2E. Protection against Pb2+-cleavage downstream of nt ∼ 17 and between nt 34 and 45 (Fig. 2A, lanes 7 and 8) indicated formation of a stem-loop structure at the 3′-end (nt 4–45; Fig. 2E). Pb2+-cleavage was also somewhat reduced at positions 23–27, pointing to some structure formation involving the apical loop nucleotides (Fig. 2A, lanes 7 and 8). The second structural element confirmed by Pb2+ probing and RNase T1 cleavage under native conditions is the hairpin between nt 56 and 78 (Fig. 2B and D, lanes 5–8). Downstream of position 78, we observed a general susceptibility to the single strand-specific probes, except for reduced accessibility around positions 130/131, 134/135 and 146/147 (Fig. 2B, lanes 5–8; Fig. 2C, lanes 7 and 8). The latter finding suggests some structure formation in the region of nt 130–150, although we could not assign this protection pattern to a specific secondary structure (see also RNAfold prediction in Fig. S1).
Figure 2.

Structure probing of the genomic 3′-leader (residues 154–1). 3′- or 5′-[32P]-endlabeled genomic RNA was probed at room temperature in 1 x TN buffer with Pb2+ ions (Pb2+) that induce hydrolysis in flexible single-stranded regions, and with RNase T1 at 37°C in 1 x TN buffer under non-denaturing conditions (T1 native), resulting in cleavage 3′ of single-stranded G residues. Control (con.): RNA incubated in 1 x TN buffer or ddH2O (no differences were observed between the 2 conditions) without Pb2+ ions or RNase T1 for 17 min at room temperature; OH−: alkaline ladder; T1 denat.: RNase T1 cleavage at 55°C in the presence of 4 M urea. Tandem lanes (e.g., lanes 3 and 4 in panel A, T1 native.) represent 2 incubation times, 3 and 5 min for T1 native and T1 denat., 15 and 17 min for Pb2+ hydrolysis lanes. (A) Probing of 3′-endlabeled RNA 154–1, analyzed by 15% denaturing PAGE. (B-D). Probing of 5′-endlabeled RNA 154–1, analyzed by 8% (panels B, D) or by 15% (panel C) denaturing PAGE. Nucleotide positions are marked at the left gel margins and colored vertical lines indicate flexible single-stranded regions. (E) Secondary structure inferred from the probing data using the numbering system and colored line code as in panels A-D. RNase T1 accessibilities are depicted by filled (denaturing conditions) and open (native conditions) triangles with different symbol sizes illustrating cleavage band intensity. Sites of Pb2+ hydrolysis are marked correspondingly by filled circles.
Probing of the antigenomic copy of the 3′-leader also indicated the formation of 2 stem-loop structures at the locations complementary to the 2 hairpin structures in the (−) RNA strand (Fig. S2E). Evidence for the 5′-proximal hairpin (nt 5–44) is based on protection from Pb2+ hydrolysis at positions 5′ of nt 16 and between nt 34–44 (Fig. S2A, lanes 7 and 8). Protection was also observed at positions 30–33 (Fig. S2A, B), either pointing to intraloop structures or interactions between loop residues and nucleotides elsewhere. The hairpin whose 5′-terminal nt (position 56) corresponds to the transcriptional start site is confirmed by strong protection of its stem region from Pb2+ hydrolysis (nt 56–78; Fig. S2B, lanes 7 and 8; Fig. S2C, lanes 5 and 6), consistent with previous chemical probing data.9 The remaining part of the sequence is largely accessible to single strand-specific probes, with some protection evident in the region of nt 131–148 in accordance with formation of the 3′-proximal hairpin (Fig. S2D, E) that is predicted by RNA secondary structure analysis tools such as RNAfold (Fig. S1).
VP30:RNA binding analyzed by EMSA
For EMSA analysis, VP30 was expressed in Escherichia coli as a slightly shortened but fully functional (unpublished results) VP308–272 variant (full-length: 288 aa) N-terminally fused to the maltose binding protein (MBP). The MBP tag was cleaved off by TEV protease before RNA binding experiments to rule out steric interference of the MBP moiety with RNA binding (for purity of MBP-VP308–272 before and after TEV cleavage, see Fig. S3). This mixture was used for binding experiments to avoid any degradation of the released VP308–272 during further purification steps. MBP alone was used as a negative control in gel mobility assays. An EMSA example is shown in Fig. 3A. Two types of complexes were observed: the major fraction, defined here as C1 complexes, migrated into the 4% PAA layer of the gel and was assigned to VP30:RNA complexes whose formation depends on the ability of VP30 to form hexamers;33 and a minor fraction representing high molecular weight complexes, termed CHMW, (Fig. 3A). Apparent Kd determination was based on C1 complex formation (for further details, see Materials and Methods). We used 2.7μM VP30 as the highest concentration because the protein tended to aggregate at higher concentrations. We thus obtained values for the fraction of RNA as part of C1 complexes at 2.7 μM VP30 (termed correct to: F2.7μM). This F2.7μM value was in most cases lower than the calculated endpoint (saturation level) obtained by curve fitting. We will not only discuss appKd values in the following, but also the experimental F2.7μM values which are informative in terms of the fraction of RNA that binds to VP30 stably enough to resist irreversible dissociation under the conditions of gel electrophoresis.
Figure 3.
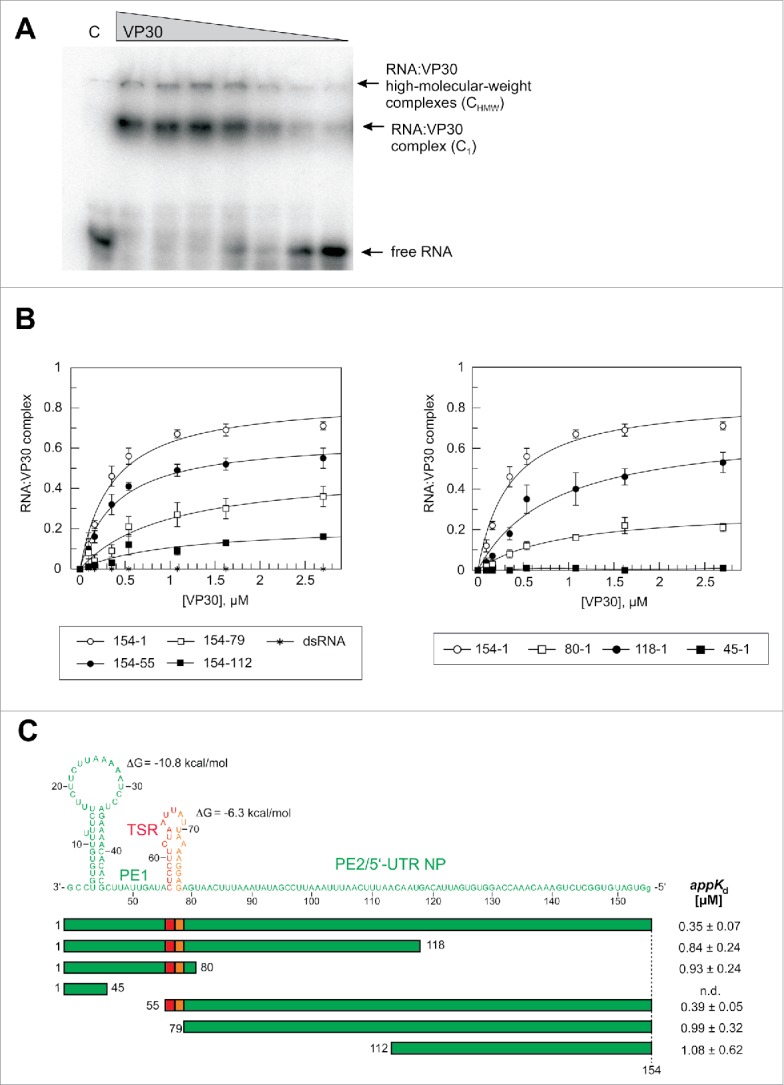
Effects of truncations of the genomic RNA 154–1 on VP30 binding. (A) Representative EMSA experiment for the genomic leader RNA 154–1. Two different RNA:VP30 complexes (C1 and CHMW; for details, see Materials and Methods) could be observed in addition to the free RNA. VP30 concentrations were from right to left: 0.1, 0.2, 0.4, 0.5, 1.1, 1.6 and 2.7 µM; C, 4 µM MBP only. (B) RNA binding curves of VP30 using the different derivatives of RNA 154–1 with deletions from the 3′- (left panel) or 5′-end (right panel). The left panel includes the dsRNA substrate (RNA 154–1 preannealed to its complementary strand 2–158). Each curve is based on at least 3 independent experiments (error bars are standard deviations of the mean); curves were fitted according to a one-ligand-binding-site model. (C) Correlation of RNA sequence and secondary structure (derived from the probing experiments in Fig. 2) with the RNA truncations; the ΔG values of the stem-loops are the minimum free energies of the centroid secondary structures predicted by RNAfold for the sequences of the hairpin structures plus 1 extra nt on each side using the default parameters (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). The sequence stretch responsible for transcription initiation by the viral polymerase (TSR) is colored in red, the spacer (5′-half of the hairpin) is marked in orange and the replication promoter elements 1 (PE1, nt 55–1) and 2 (PE2, nt 154–79) are shown in green (see also Fig. 1A); the 5′-terminal G residue, which is not encoded in the viral genome, was added for the purpose of efficient T7 transcription. appKd values (± SD) are based on the graphs shown in panel B (obtained by fitting to a one-ligand-binding-site model); n.d., appKd value not determinable owing to very low binding.
Deletion analysis of the genomic 3′-leader
In our EMSA setup, the 154-1 genomic RNA (Fig. 2E) bound to VP30 with an appKd of 0.35 μM (Fig. 3B, C; see also Table S1 for a summary of all appKd values determined in this study). Deletion of the 3′-terminal 54 nt (RNA 154–55) had only a minor effect on VP30 binding (appKd = 0.39 μM). Further deletion of the second hairpin in RNA 154–79, which included the transcription start site, caused an almost threefold drop (appKd = 0.99 μM) in affinity (Fig. 3B, C; a two-tailed P value of 0.004 relative to RNA 154–1 based on unpaired t test analysis, see Materials and Methods), indicating a role of the stem-loop in interaction with VP30. Further truncation from the 3′-end (variant 154–112) only resulted in a minor additional affinity reduction (appKd = 1.08 μM; P = 0.834; F2.7μM 0.17 vs. 0.36, P = 0.062; Table S1). Deletion of 36 nt from the 5′-end (RNA 118–1) increased appKd 2.4-fold (P = 0.004), while a further shortage by another 38 nt (RNA 80–1) had little additional effect on appKd, but caused a substantial drop in F2.7μM (from 0.53 to 0.21, P = 0.0029; Table S1). VP30 binding was abolished for the RNA fragment 45–1. These findings indicate that binding with relatively highest affinity can only be achieved if RNA substrates include an extended single-stranded region such as the genomic region of nt 154 to 79. This is in line with a double-stranded hybrid of the complementary RNAs 154–1 and 2–158 being completely unable to interact with VP30 (Fig. 3B, dsRNA). Generally, all truncated variants of RNA 154–1 resulted in lower F2.7μM values for C1 complex formation, although all binding curves appeared to approach the saturation range at this VP30 concentration (Fig. 3B). An explanation could be that shorter RNAs, which dissociate from VP30 at a higher rate than longer RNAs during electrophoresis (owing to a reduced interaction surface), also populate compact conformations that favor irreversible escape from gel cages to a larger extent than for the longer and bulkier RNAs.
Deletion analysis of antigenomic RNA complementary to the 3′-leader
For the antigenomic RNA 2–158, we observed a 1.4-fold higher appKd (0.49 μM; Fig. 4B, C) than for the complementary genomic 1–154 RNA. This difference (P = 0.034) may be attributed to reduced single-strandedness in the 3′-proximal region of RNA 2–158 (Fig. 4C). Deletion of the first 55 nt, resulting in RNA 56–158 whose 5′-terminus coincides with that of viral mRNA transcripts derived from the first NP gene but carrying a 5′-monophosphate end group,11 had no effect on VP30 affinity (appKd = 0.49 μM) but was associated with a trend toward a lower F2.7μM (from 0.7 to 0.45, P = 0.742; Table S1). Deletion of the hairpin (RNA 79–158) tended to impair binding relative to RNA 2–158 (appKd = 0.77 μM; P = 0.075) as observed for the genomic RNA. Variant 2–78 was essentially unable to bind to VP30 (Fig. 4B, C), which contrasts the considerable binding ability of the complementary genomic RNA 80–1 (appKd = 0.93 μM, F2.7μM of 0.21; Fig. 4C, Table S1). RNA 80–1 encompasses similar types of stem-loop structures as RNA 2–78, although with reversed polarity. One explanation could be that the residual RNA binding activity of RNA 80–1 is related to the lower stability (ΔG = −6.3 kcal/mol; Fig. 3C) of the genomic hairpin compared with the antigenomic one (ΔG = −9.1 kcal/mol; Fig. 4C). As a result, a substantial fraction of the genomic hairpin might be bound by VP30 in an unfolded state.
Figure 4.

Effects of truncations of the antigenomic RNA 2–158 on VP30 binding. (A) Top: Schematic comparison of the end regions in the (−) RNA genome and (+) RNA antigenome. Bottom: sequence similarity between replication promoter sequences in the genomic leader and the antigenomic trailer (ctrailer) regions. Sequence similarity is most pronounced within the first 50 nucleotides. (B) RNA binding curves of VP30 using derivatives of RNA 2–158 with deletions from the 5′- or 3′-end. We also included a ctrailer RNA (178 nt in length) substrate (see panel A) for comparison and further tested RNAs that mimic the first viral mRNA transcript (RNA 56–158) with different 5′-end groups (5′-monophosphate, 5′-triphosphate or a 5′-cap(0) moiety). Each curve is based on at least 3 independent experiments (error bars are standard deviations of the mean); curves were fitted according to a one-ligand-binding-site model. (C) Correlation of RNA sequence and secondary structure (derived from the probing experiments in Fig. S2) with the RNA truncations. The color code corresponds to that in Fig. 3. The first nucleotide complementary to the 3′-terminal residue of the genome was omitted for reasons of efficient T7 transcription. The ΔG values of the stem-loops are the minimum free energies of the centroid secondary structures predicted by RNAfold for the sequences of the hairpin structures plus 1 extra nt on each side using the default parameters (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). AppKd values (± SD) are based on the graphs shown in panel B (obtained by fitting to a one-ligand-binding-site model); n.d., appKd values not determinable owing to very low binding or abnormal curve shapes.
Antigenomic replicates of the EBOV genome carry the replication promoter in their 3′-region complementary to the 5′-trailer of the genome (termed “ctrailer” for “complementary trailer”). As replication of (+) RNA antigenomes occurs in the absence of VP30,7 we used the 178-nt ctrailer RNA as a control RNA that has not been evolved for VP30 binding. The ctrailer RNA bound VP30 with similar but slightly higher affinity (appKd = 0.23 μM; Fig. 4C) relative to RNAs 154–1 (P = 0.043) and 2–158 (P = 0.006), which may be attributed to its somewhat increased length. Good binding of the ctrailer RNA, which is not expected to be a natural substrate of VP30, is in line with a rather broad RNA binding specificity of VP30.
5′-terminal end groups on the RNA substrate mimicking NP mRNA transcripts
Ebola mRNA 5′-ends are equipped with a Cap 0 (m7GpppN) or Cap 1 (m7GpppN2'-O-methyl) structure by the viral polymerase.11,13 As VP30 might interact with nascent mRNA transcripts, we further compared VP30 binding to the mRNA-mimicking substrate RNA 56–158 carrying either a 5′-monophosphate, a 5′-triphosphate or a 7-methylguanylate cap (Cap 0). Whereas the 5′-mono- and triphosphorylated RNAs showed similar binding behavior, the 5′-capped variant was bound with markedly reduced efficiency (Fig. 4B, C). This finding indicates that VP30 contacts the 5′-end of RNA ligands and is sensitive to the presence of a 5′-cap structure, while the presence of a 5′-tri- or monophosphate makes little difference.
Hairpin swap between the 3′-leader and its antigenomic copy
Our deletion studies revealed that the internal hairpins of the genomic 154–1 and antigenomic 2–158 RNAs contribute to VP30 binding. Moreover, the antigenomic RNA 2–78 RNA failed to yield significant amounts of gel-resolvable complexes with VP30, while the genomic counterpart RNA 80–1 resulted in substantial complex formation, although both RNAs were of very similar size but with inverse terminal positioning of the hairpin structure (Fig. 3C and 4C). As a first step, we tested if the genomic hairpin with 15 single-stranded nucleotides at the 3′-end (RNA 80–46, carrying 5 additional single-stranded nucleotides at the 3′-end, see Supplementary material) may be able to confer binding affinity. However, only very weak residual binding was observed (F2.7μM = 0.08; Fig. 5B). We then mutually swapped the genomic and antigenomic RNA hairpins in RNAs 154–55 (appKd = 0.39 μM) and P_56–158 (appKd = 0.49 μM) (Fig. 5A). When we replaced the genomic hairpin with the antigenomic one, yielding RNA 154–55(+hp), the appKd remained unchanged (0.39 μM) and the difference in F2.7μM was insignificant (0.55 ± 0.09 versus 0.62 ± 0.07, P = 0.348; Table S1). Conversely, replacing the antigenomic with the genomic RNA hairpin, resulting in RNA (−hp)56–158, left appKd unaffected as well (0.48 μM; Fig. 5B), but F2.7μM increased by 26% (Fig. 5B, Table S1; 0.71 ± 0.06 vs. 0.45 ± 0.1, P = 0.018). We conclude that addition of a 5′- or 3′-terminal stem structure positively affects the interaction with VP30. The stability of the hairpin structure was neutral at the 3′-end of the genomic RNA substrate, while a less stable hairpin (or some other feature of the hairpin) appeared to be beneficial at the 5′-end of the RNA mimicking the mRNA transcript.
Figure 5.

Swapping of the genomic and antigenomic hairpin structures at the transcriptional start site. (A) Scheme of hairpin swapping (nucleotides 56–78; see also Fig. 3 and 4) between the genomic and antigenomic substrates. The parental substrates were (−) RNA 154–55 and (+) RNA P_56–158 and the hybrid substrates were termed RNA 154–55(+hp) and RNA (-hp)56–158. Note that in the chimeric RNAs the (−) RNA hairpin was transplanted from the 3′- to the 5′-end and the (+) RNA hairpin vice versa. (B) EMSA-based VP30 binding curves and derived appKd values (± SD) for the RNA substrates illustrated in panel A as well as additional control RNAs for comparison. Each curve is based on at least 3 independent experiments (error bars are standard deviations of the mean); curves were fitted according to a one-ligand-binding-site model. Note that RNA 80–46 had 5 additional single-stranded nucleotides at the 3′-end owing to linearization of the T7 transcription template with EcoR1 (see Supplementary material).
Hybridization scanning of RNA 154–1
To better assess the relative contribution of sequence elements in RNA 154–1 to VP30 binding, we designed 4 DNA oligonucleotide probes (17–21 nt, mediating comparable RNA/DNA duplex stability) to different parts of RNA 154–1 (Fig. 6A) and analyzed VP30 binding after their annealing. The extent of probe annealing to the RNA under EMSA conditions was analyzed by native PAGE. Probes 2–4 quantitatively increased gel mobility (Fig. 6B, lanes 2, 4 and 5) which we attribute to some compaction of RNA 154–1 caused by DNA:RNA duplex formation. Probe 1 annealed to roughly 80% under the test conditions and gel mobility was retarded (Fig. 6B, lane 6). We attribute this to a loss of compactness owing to disruption of the 3′-terminal hairpin which is apparently not compensated by the compaction originating from probe 1:RNA duplex formation. Annealing of probe 1 did not interfere with VP30 binding (Fig. 6C), confirming the finding that deletion of the 3′-terminal 54 nt had little effect on VP30 binding (Fig. 3). Probes 2 and 3 impaired VP30 binding affinity by 2.6-fold and 3.3-fold (P = 0.004 and 0.005), respectively. In addition, these 2 probes reduced F2.7μM. This finding supports the notion that the extended single-stranded stretch in the center of RNA 154–1 makes a major contribution to VP30 binding. Probe 4 had no substantial effect on binding (appKd = 0.44 μM relative to 0.35 μM for the free RNA 154–1, P = 0.162; Table S1) suggesting little contribution of the 5′-terminal 25 nt to VP30 binding.
Figure 6.

Hybridization of DNA probes to the genomic RNA 154–1. (A) Secondary structure (as experimentally defined, see Fig. 2) of the genomic RNA and position of DNA probes that were annealed to the RNA before VP30:RNA gel shift assays. (B) Native PAA gel to verify stable annealing of the different probes to the RNA substrate. The RNA (154–1) without any DNA oligonucleotide bound (RNA alone) showed the same mobility as the RNA preincubated with a better: non-complementary DNA oligonucleotide (control). The DNA probes complementary to single-stranded RNA region resulted in accelerated mobility of the RNA (probes 2, 3 and 4), whereas probe 1 disrupting a double-stranded RNA regions led to retarded RNA mobility, also suggesting that hybridization was not complete. (C) VP30:RNA binding curves and derived appKd values (± SD) of the different preannealed RNA:DNA probe complexes. Each curve is based on at least 3 independent experiments (error bars are standard deviations of the mean); curves were fitted according to a one-ligand-binding-site model.
Binding of non-viral RNAs to VP30
Based on the requirement of elongated single-stranded RNA (ssRNA) stretches for VP30 binding (Fig. 3–6), we extended our analysis to 2 natural non-coding RNAs (ncRNAs) with mixed structures, which were considered as being potentially informative. The first is a (precursor-)tRNAGly (ptRNAGly, Fig. S4A) with 3 stem-loops and ssRNA stretches at the 5′- and/or 3′-end of the terminal stem. Surprisingly, the ptRNA with stretches of 14 nt at the 5′- and 7 nt at the 3′-end bound with moderate affinity to VP30 (appKd = 2.7 μM; Fig. S4B). Deleting the 14-nt precursor segment (tRNA) largely reduced binding, manifesting as a reduction of F2.7μM to 0.05 (relative to 0.49 for the ptRNA, Table S1, P = 0.009) (Fig. S4B). Deletion of the 7 3′-terminal nt (variant ΔCCA-ptRNA) impaired VP30 binding to an almost similar extent (F2.7μM of 0.09; Fig. S4B). These findings suggest that terminal ssRNA stretches, either at the 5′- or 3′-end and even better at both ends, are required for a highly structured tRNA molecule to stably interact with VP30.
Bacterial 6S RNA, the second tested ncRNA, contains 2 extended stem regions flanking an enlarged internal loop (Fig. S4A;14,15).This RNA was completely unable to form gel-resolvable complexes with VP30 (Fig. S4B). Combined with the binding results for the tRNA variants we conclude that VP30 requires single-stranded RNA overhangs to thread the RNA into its RNA binding interface.
VP30 binding to single-stranded nucleic acids: influence of length, base composition and RNA vs. DNA
The data outlined in the following are summarized in Fig. 7, S5 and S6. As the ptRNA with a 14-nt ssRNA 5′-flank was able to form gel-resolvable complexes with VP30, we tested if the 5′-flank itself may be able to confer binding. However, this RNA (ssRNA 14 nt CU-rich) failed to stably bind to VP30. We then tested a 28-nt ssRNA, whose sequence almost corresponded to a tandem of the 14-nt 5′-flank (ssRNA 28 nt CU-rich). This variant showed trends (Table S1) toward reduced VP30 affinity (appKd = 5.26 μM) and decreased F2.7μM values relative to the ptRNA which only carried a total of 21 single-stranded nucleotides on its termini. Thus, some features of the tRNA stem-loop elements apparently contributed to binding, similar to what we observed with the genomic and antigenomic EBOV leader truncations (Fig. 3 and 4). Extending the ssRNA from 28 to 33 nt improved binding, and addition of another 4 nt (ssRNA 37 nt CU-rich) further increased affinity almost threefold (appKd = 0.76 vs. 2.12 μM; P = 0.016). A length extension to 42 nt failed to raise affinity (ssRNA 42 nt CU-rich, appKd = 1.20 μM), suggesting that a ssRNA length of approx. 37 nt confers maximum affinity to VP30 in the absence of any structured RNA elements.
Figure 7.

Binding of VP30 to single-stranded RNA and DNA substrates. Lengths and sequences of ssRNA oligonucleotides analyzed for VP30 binding are shown on the left. Table on the right: appKd and F2.7μM (fraction of C1 complexes at 2.7μM VP30) values, derived from experiments shown in Figs. S5 and S6, also including the values for ptRNA (Fig. S4) for comparison. AppKd (± SD) values are based on at least 3 independent experiments; curves were fitted according to a one-ligand-binding-site model. Standard deviations are also given for F2.7μM values; n.d., not determinable, either due to no or very low complex formation, or because no reasonable fit to the “one-ligand-binding-site” model (see Materials and Methods) could be obtained.
We next tested if the prevalence of pyrimidines in ssRNA 37 nt CU-rich might have a substantial impact on binding to VP30. This was addressed by the variant ssRNA 37 nt GA-rich, which bound with very similar affinity to VP30 as its CU-rich length counterpart (appKd = 0.61 μM vs. 0.76 μM, P = 0.216). Another variant, ssRNA 37 nt GA-repeat, which lacked any pyrimidines and had an increased proportion of G residues, bound less efficiently (appKd = 1.47 μM, F2.7μM of 0.4) than the GA-rich 37-mer (P = 0.012 based on appKd values), suggesting that an increased proportion of guanosines or the entire absence of pyrimidines is less favorable for binding. At present we do not know if there are position-specific effects where certain base identities are preferred.
37-meric homopolymers, poly(A), (U) or (C) resulted in binding curves that were less hyperbolic than the curves obtained for the mixed oligomer sequences (Fig. S5). For this reason, we only obtained a reasonable fit to a “one-ligand-binding-site model” with the ssRNA 37 nt poly(A) (appKd of 3.49 μM) but not with the poly(U) and poly(C) variants, although the binding curves were very similar for all 3 homooligomers (Fig. S5, lower right). All three as well as the ssRNA 37 nt GA-repeat had F2.7μM values between 0.34 and 0.46, thus substantially lower than for the CU-rich and GA-rich 37-mers (P = 0.0238 for F2.7μM of the poly(C) versus GA-rich 37-mer). These observations indicate that VP30 prefers mixed sequences, tolerating a large but not absolute disproportion of pyrimidines and purines.
DNA counterparts of 3 selected ssRNAs, ssDNA 42 nt GU-rich, ssDNA 37 nt GA-repeat and ssDNA 37 nt poly(A) showed impaired binding to VP30 relative to their RNA versions (Fig. 7 and S6A). For example, ssRNA 37 nt GA-repeat had a F2.7μM of 0.4 relative to 0.06 for its DNA counterpart (P = 0.0067). We conclude that ribose 2'-hydroxyls contribute to VP30 binding.
Electrostatic component of VP30:RNA binding
In view of the relaxed RNA binding specificity of VP30, we performed EMSA experiments in the presence of the polysaccharide heparin. With its negative charge density (∼1.5 negative charges per monosaccharide unit above pH 7;16) being similar to single-stranded nucleic acid backbones (1 negative charge per nucleotide), heparin is commonly used in RNA binding assays to inhibit unspecific RNA:protein interactions that are caused by electrostatic interactions of positive protein surface charges and nucleic acid phosphates. Here we analyzed the effect of heparin when present in equimolar amounts to the genomic leader RNA 154–1 (each 12 nM). Heparin substantially weakened RNA binding to VP30, as inferred from the less hyperbolic binding curve and reduced complex formation (Fig. S6B), suggesting a considerable electrostatic component involved in VP30:RNA interactions.
Discussion
Several features of nucleic acid binding by VP30 can be extracted from our analysis. The basic RNA substrate is a single-stranded RNA of optimally ∼ 40 nt with a mixed base composition (Fig. 8A). There seems to be a further gain in affinity if the ssRNA stretch is linked to a stem(-loop) structure (Fig. 8B; see discussion below). Homopolymeric poly(U), poly(C) and poly(A) RNAs show weaker binding than mixed sequences of identical length and RNA binding is impaired by introduction of a 5´-Cap(0) structure. DNA binds considerably weaker than RNA and heparin competes with RNA for binding to VP30, indicating a substantial contribution of ribose 2′-hydroxyls and polar contacts to binding. Furthermore, analysis of non-viral model substrates (Fig. S4) provided evidence that 5′- and 3′-terminal single-stranded extensions strengthen VP30 binding, whereas substrates with only internal loops, despite extended single-strandedness (6S RNA, Fig. S4), are unable to bind to VP30 at all. Based on oligonucleotide interference with VP30 binding (Fig. 6), the internal expanded single-stranded region (∼125–80) was deduced to be a major binding element of the genomic 154–1 substrate. This region corresponds to the replication promoter element 2 (PE2; Fig. 1A).
Figure 8.
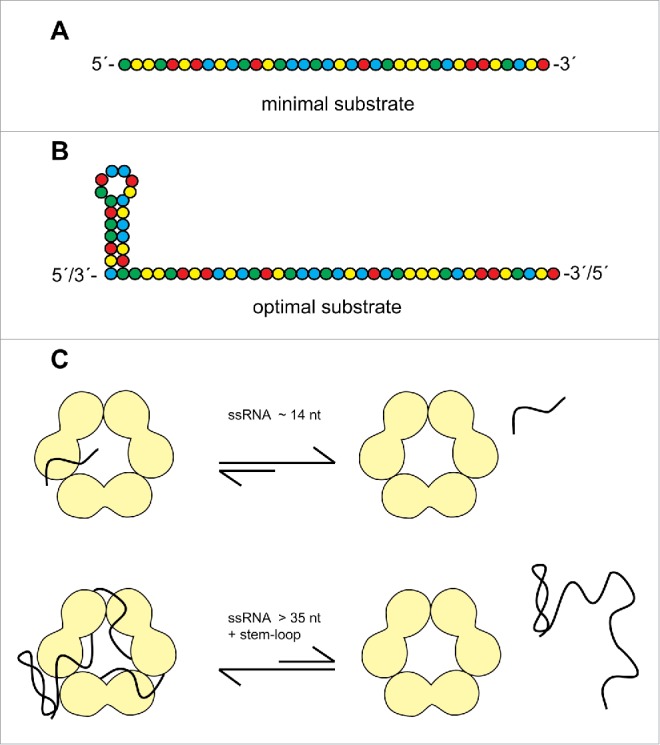
Features of (A) minimal and (B) optimal RNA substrates for Ebola VP30. (C) Sketch of the proposed RNA binding interface. VP30 is shown as a trimer of dimers, taking into account that the protein's capability to form hexamers is essential for gel-resolvable RNA binding.33 As a consequence, the RNA binding path is tentatively indicated at the interface between dimers. While short ssRNAs (∼14 nt) rapidly dissociate from VP30, as inferred from the absence of gel-resolvable complexes in the case of a ssRNA 14-mer (Fig. 7), longer RNAs bind more stably, further supported by a stem-loop structure particularly at the 3′-end.
Interestingly, VP30:RNA binding was positively influenced by a hairpin structure (capped by a short loop) flanking an extended ssRNA region. This finding was based on the observation that deletion of the 3′-terminal 54 nt of RNA 154–1, yielding RNA 154–55, impaired VP30 binding to a lesser extent than deletion of the internal hairpin from RNA 154–55, yielding RNA 154–79 (Fig. 3). Deletion of the complementary more stable hairpin on the (+) RNA level (RNA 79–158 vs. P_56–158, see Fig. 4 and Table S1) affected VP30 binding only to a minor extent. This raises the possibility that the positioning of the stem structure plays a role, suggesting that it is more beneficial at the 3′-end of the substrate. Also, the stability of the hairpin (nt 56–78) on the (−) versus (+) RNA level might influence VP30:RNA binding. The hairpin swap experiments (Fig. 5) suggest that either hairpin has a comparably beneficial effect on VP30 binding as part of the genomic substrate, while the (−) RNA hairpin results in a higher F2.7μM than the (+) RNA hairpin in the context of the antigenomic substrate. Thus, when positioned on the 5′-end, a less stable secondary structure is beneficial, which might suggest that VP30 binds to the hairpin in its unfolded state. However, at least for the genomic substrate (hairpin at the 3′-end) it is unlikely that VP30 captures the hairpin upon transient unfolding in view of equal binding affinity of substrates 154–55 and 154–55(+hp) (Fig. 5). As the (−) hairpin in RNA 154–55 is less stable than the (+) hairpin in RNA 154–55(+hp), it should have been more efficiently captured by VP30 in its transiently unfolded state and this should have resulted in more efficient binding, which we did not observe. We also like to note that both types of hairpin yielded strong protection against Pb2+−induced hydrolysis (Fig. 2 and S2) and dsRNA is not a substrate for VP30 (Fig. 3B). Thus, we do not favor the idea that the nt 56–78 (+) and (−) hairpins are bound by VP30 in an unfolded state and rather conclude that the beneficial effect of a hairpin structure, particularly at the 3′-end, primarily reflects another function than simply increasing the number of ssRNA contacts to VP30.
A second line of evidence for the beneficial effect of a stem(−loop) structure comes from comparison of VP30 binding to the tRNA derivatives and ssRNAs. VP30 binding of a tRNA with 14 5′-terminal and 7 3′-terminal single-stranded nucleotides was more efficient than binding of a single-stranded 28-mer almost identical to a tandem of the 14 nt long 5′-flank of the tRNA (Fig. 7 and S4). One possibility is that a stem(-loop) structure, either positioned internally or terminally, might direct binding of RNAs in a defined register, metaphorically in the sense of a traffic bollard, as suggested in Fig. 8C. The working model in Fig. 8C is based on the finding that VP30 forms hexamers 17 and that mutations in VP30 that prevent hexamerization of the protein, abolish RNA binding in the mobility shift assay.33 Thus, we assume that VP30 hexamerization generates a composite RNA binding interface and is thus a prerequisite for stable (gel-resolvable) RNA binding.
The ssRNA oligonucleotides tended to gain affinity when extended from 28 to ∼ 40 nt, and the CU-rich 37- and 42-mers gave the highest F2.7μM values among all tested substrates (Fig. 7; Table S1). The appKd values of these ssRNA oligonucleotides were only twofold though significantly higher than that for RNA 154–1 (P = 0.001 in comparison with the CU-rich 37-mer), clearly indicating that single-strandedness is a major feature of RNA binding by VP30. The base composition was found to be permissive, as the CU-rich 37-mer with only 7 purines and the GA-rich 37-mer with only 7 pyrimidines allowed efficient binding (Fig. 7). However, purine- or pyrimidine-only substrates including the GA repeat 37-mer showed trends toward impaired binding (Fig. 7). A possible interpretation is that the VP30 binding interface includes sites that are sterically inaccessible to purines and others where a pyrimidine is too small for stable nucleoside docking. This may be combined with some contact sites specific to the functional groups of C, U, G or A bases (see discussion below on the related M2-1 protein). The preference for mixed sequences and the tolerance toward underepresentation of purines or pyrimidines suggests VP30 binding to many viral RNA regions as long as single-strandedness is preserved (illustrated in Fig. 8A). Assuming that the PE2 region (∼ nt 120–80), identified here as a potential VP30 binding domain (Fig. 3 and 6), may also be transiently accessible to VP30 binding during viral infection cycles, then the promiscuous binding mode of VP30 might reflect an evolutionarily reasonable adaptation, considering that the PE2 region has a mixed sequence and shows little conservation among the different Ebola isolates (Fig. 1A). The major constraint for PE2 is to be unstructured and to maintain a register of consecutive UN5 hexamers (8 in EBOV; Fig. 1A;5). Thus, PE2 is prone to mutational change (which creates mixed sequences), but random mutations can be ruled out to lead to the formation of stable secondary structures in a region that is entirely devoid of structure. As a result, quasi species of the PE2 region will always be single-stranded mixed sequences that fully match the RNA recognition profile of VP30. In our study presented here, we started from rather large naked RNA molecules (∼150 nt) as substrates for VP30 and identified ssRNAs of about ≥ 40 nt to bind to VP30 with higher affinity than shorter ssRNAs. However, these findings obtained in vitro raise questions as to how this relates to RNA binding during the viral infection cycle. According to current models of replication/transcription of negative strand non-segmented RNA viruses, the template RNA is covered by N protein (NP) and only 2–3 NP molecules are assumed to be displaced simultaneously at the site of transcription, giving access to not more than 30 nucleotides.18,19 Yet, at present it cannot be excluded that larger regions of RNA become transiently accessible during EBOV transcriptional initiation (e.g. by VP30 displacing NP), also taking into account that the leader promoter is bipartite in EBOV, and thus more extended, than in vesicular stomatitis virus (VSV) or respiratory syncytial virus (RSV); furthermore, Ebola VP30 is an exceptionally large and unique viral transcription factor within the Mononegavirales, which leaves the possibility open that temporary RNA accessibilities are different in the EBOV system. Finally, Reguera et al.20 recently hypothesized based on studies of Dengue virus polymerase 21 that the combination of sequence- and secondary structure-specific binding of vRNA by the polymerase is likely a general feature for template recognition among RNA virus polymerases. However, these are still open questions and future studies have to reveal how our findings on VP30 binding to naked RNA can be reconciled with RNA binding in the context of nucleocapsids during infection cycles.
We further observed a 5´-Cap(0) structure to be unfavorable for VP30 binding relative to a 5′-mono- or 5′-triphosphate in the context of an mRNA-mimic substrate (Fig. 4B). First of all, this provides evidence that VP30 contacts the 5′-end of RNA substrates and is sensitive to this functionality. Furthermore, the negative effect of the 5´-Cap(0) structure implies that VP30 binding to nascent mRNA transcripts is disfavored upon cap addition by the viral polymerase. This could be a mechanistic component that contributes to preventing VP30 from binding to viral mRNA transcripts, or may contribute to the release of viral mRNA transcripts from transcription complexes after their synthesis in infected cells. As another possibility, preferential binding of VP30 to 5′-triphosphates relative to 5′-cap structures may point to an independent function in dampening cellular antiviral responses by masking 5′-triphosphates at viral RNA ends. Here, it is interesting to note that VP35 with intact dsRNA binding activity can protect 5′-triphosphates of dsRNAs from interacting with RIG-I, thereby inhibiting innate immune responses.22 Thus, it is a possibility that both VP30 and VP35 share the task of counteracting RIG-I activation, VP30 by masking 5′-triphosphates on ssRNAs and VP35 those on dsRNAs.
A previous study analyzed VP30 binding by nitrocellulose filter binding and UV crosslinking,12 using a few Ebola-derived RNA substrates (45 nt and shorter) and an unrelated control ssRNA (31 nt). The authors came to the conclusion that decent VP30 binding affinity is conferred by a stem-loop when placed in the context of (single-stranded) upstream and downstream sequences, which is in line with our findings. Low binding affinity was inferred for an Ebola-unrelated 31 nt long ssRNA substrate from binding competition experiments, leading the authors to conclude that VP30 RNA binding is more specific to EBOV RNAs. This conclusion is not supported by our data showing that ssRNAs of mixed base composition are affine substrates, irrespective of their origin. Based on our data (Fig. 7), the used ssRNA 31-mer may have been a few nucleotides too short to confer good VP30 binding.
RNA binding by VP30 and M2-1
It is instructive to compare VP30 with the structurally and functionally related transcription factor M2-1 of the Paramyxoviridae family. M2-1differs from VP30 in that it lacks the N-terminal RNA binding and phosphorylation domain (Fig. 1B and S7A), and it forms stable tetramers instead of hexamers.17,23 In a study reporting the crystal structure of M2-1 from human respiratory syncytial virus (hRSV;23), RNA binding by M2-1 was analyzed using a series of 13-meric RNAs. This revealed a preference for oligo(A) (Kd ∼20 nM), whereas Kd values for oligo(C), (U) or (G) 13-mers were estimated to be > 100 μM. Thirteen-mers of mixed base composition were bound with Kd's in the range of 0.05 to 2 μM, with a trend toward better affinity with increasing A content. The authors proposed that M2-1 preferentially binds to internal and 3′-terminal A-rich stretches in nascent mRNAs during viral transcription.23 NMR-based analysis of RNA binding of the RSV M2-158-177 core confirmed the preference for oligo(A) sequences, but also showed binding of a 12-bp RNA duplex with even higher affinity than the single strands of the same duplex,24 a finding that remains enigmatic at present. Leyrat et al. 25 proposed a model explaining the preference of M2-1 for A-rich RNA ∼13-mers, according to which the interaction of A5 with the Zn finger domain is the main specificity determinant (further stabilized by an adenosine at position 6), and A resides at positions 9–13 are favorable for interaction with the core domain.
Our results indicate that RNA specificity of VP30 is clearly different. VP30 requires longer sequences for efficient RNA binding, and there is no obvious preference for A-rich sequences, although this does not exclude a role of single A residues in binding. Yet, it is a possibility that VP30 binds to the conserved gene end motifs 3′-UAAUUC(U)5/6 or their reverse complement in the mRNA, which would conform to the specificity pattern inferred from the data in Fig. 7, i.e. sequences mainly consisting of pyrimidines or purines, but combined with a few bases of the other nucleobase type, are good substrates. One possibility is that the RNA binding interface of VP30 is more extended than that of M2-1 and intimate base-specific contacts are likely rare, in line with the best appKd values for VP30:RNA interactions being in the range of 0.2 to 0.5 µM, although such appKd values are not directly comparable with the at least tenfold lower Kd of ∼20 nM observed for equilibrium binding of the oligo(A) 13-mer to hRSV M2-1.23 An expanded RNA binding interface of VP30 may be structurally related to the fact that VP30 carries an additional domain upstream of the Zn finger, which is thought to contribute to RNA binding by VP30 (aa 26–46 in VP30;12), and also to its different oligomerization state (hexamer instead of tetramer). It appears that the RNA binding interface of VP30, relative to that of M2-1, has evolved toward a more expanded one with decreased RNA specificity and affinity. This difference in RNA binding between M2-1 and VP30 points to large mechanistic differences between the 2 proteins, which are in line with recent findings. A structural study of M2-1 of human metapneumovirus (hMPV) revealed formation of an asymmetric tetramer in which 3 subunits are in a closed conformation and the fourth protomer is in an open conformation (defined by the relative orientation of the Zn finger and core domains;25). The authors provided evidence that simultaneous RNA binding to residues in the Zn finger and core domain stabilizes the closed conformation and favors formation of tetramer aggregates. Each protomer of the tetramer can bind RNA or the polymerase cofactor P (the homolog of Ebola VP35;26). Based on their finding of an asymmetric M2-1 tetramer,25 the authors suggested that in hMPV transcription complexes only one protomer of the M2-1 tetramer binds RNA while the other 3 protomers interact with the P protein. This model reconciles the finding that binding of RNA and P protein to M2-1 is mutually exclusive, but the P protein is required to recruit M2-1 to the viral polymerase. Such a competition between RNA and VP35 binding has not been observed for VP30, in line with the different RNA binding properties of VP30 and M2-1 proteins. Whereas M2-1 was proposed to bind to internal and 3′-terminal A-rich stretches in nascent viral mRNAs to prevent premature transcription termination,23 we favor a working model according to which VP30 interacts primarily with the (−) RNA template (such as the PE2 region in the 3′-leader) to stabilize binding of the VP35/L polymerase on the template RNA as a prerequisite for productive transcription initiation in the presence of termination-active RNA secondary structures. However, as discussed before, the accessibility of vRNA to protein interaction within viral nucleocapsids is an issue that has to be illuminated in future studies.
Material and methods
Plasmids
For producing the leader RNA of the Zaire EBOV wild type (wt) (−) genome and (+) antigenome as well as the antigenomic trailer regions, T7 transcription cassettes (T7 class III promoter; 27) of the just mentioned EBOV-derived coding sequences were constructed by PCR and cloned via BamHI and EcoRI restriction sites into pUC19 (3′-leader genome: pUC19_154-1: 5´-gGU GAU GUG GCU CUG AAA CAA ACC AGG UGU GAU UAC AGU AAC AAU UUC AAU UUA AAU UCC GAU AUA AAU UUC AAU GAG AGG AAA AUU AUU AAU CUU CCU CAU AGU UAU UCG CAC ACA AAA GAU CCU AAA AAU UCU UCU UUC UUU UUG UGU GUC CG-3′; leader antigenome: pUC19_2-158: 5′- GGA CAC ACA AAA AGA AAG AAG AAU UUU UAG GAU CUU UUG UGU GCG AAU AAC UAU GAG GAA GAU UAA UAA UUU UCC UCU CAU UGA AAU UUA UAU CGG AAU UUA AAU UGA AAU UGU UAC UGU AAU CAC ACC UGG UUU GUU UCA GAG CCA CAU CAC AAA G-3′; trailer antigenome: pUC19_ctrailer: 5′gGA AAU ACC UUC UUU ACA AUA UAG CAG ACU AGA UAA UAA UCU UCG UGU UAA UGA UAA UUA AGA CAU UGA CCA CGC UCA UCA GAA GGC UCG CCA GAA UAA ACG UUG CAA AAA GGA UUC CUG GAA AAA UGG UCG CAC ACA AAA AUU UAA AAA UAA AUC UAU UUC UUC UUU UUU GUG UGU CCA-3′; additional G residues added to the 5′-ends of T7 transcripts for reasons of transcription efficiency are indicated in lower case and italics; most T7 transcripts additionally carried either a 3′-terminal 5’-GAAUU extension owing to linearization of transcription templates with EcoRI, or a 5’-GUC extension due to transcribing the RNA as a fusion transcript with a 3′-terminal self-cleaving hammerhead ribozyme; 28 all RNA sequences are listed at the end of the Supplementary Material). For the production of truncated variants of the 3′-leader genomic and the complementary antigenomic RNA (see above), the entire pUC derivative plasmid (pUC19_154-1 or pUC19_2-158) purified from Dam+ E. coli DH5α bacteria was amplified (by Pfu or Phusion DNA polymerase, Thermo Fisher Scientific) with 5′-phosphorylated primers (Tm values between 54 and 59°C (calculated with the OligoAnalyzer tool, Integrated DNA Technologies; length 17–61 nt) which flanked the region to be deleted, followed by DpnI digestion of parental DNA strands. 29 This mixture was loaded onto a preparative 1% agarose gel, and the double-stranded linear PCR product was excised and eluted from the gel, followed by circularization using T4 DNA ligase and transformation of E. coli DH5α cells.
Chemically synthesized oligonucleotides
RNA oligonucleotides were purchased from Integrated DNA Technologies (IDT) and DNA oligonucleotides from Metabion.
ssRNA 14 nt CU-rich: 5′-GGAUUUUCCCUUUC-3′
ssRNA 28 nt CU-rich: 5′-GGAUUUUCCCUUUCCCAUUUUCCCUUUC-3′
ssRNA 33 nt CU-rich: 5′-GGAUUUUCCCUUUCCCAUUUUCCCUUUUCGUCC-3′
ssRNA 37 nt CU-rich: 5′-GGAUUUUCCCUUUCCCAUUUUCCCUUUUCGUCCGGUC-3′
ssRNA 42 nt CU-rich: 5′-GGAUUUUCCCUUUCCCAUUUUCCCUUUCCCAUUUUCCCUUUC-3′
ssRNA 37 nt GA-rich: 5′-CCUAAAAGGGAAAGGGUAAAAGGGAAAAGCAGGCCAG-3′
ssRNA 37 nt GA repeat: 5′-GGAGAGAGAGAGAGAGAGGAGAGAGAGAGAGAGAGAG-3′
ssRNA 37 nt poly(A): 5′-AAAAAAAAAAAAAAAAAAGAAAAAAAAAAAAAAAAAA-3′
ssRNA 37 nt poly(U): 5′-UUUUUUUUUUUUUUUUUUCUUUUUUUUUUUUUUUUUU-3′
ssRNA 37 nt poly(C): 5′-CCCCCCCCCCCCCCCCCCUCCCCCCCCCCCCCCCCCC-3′
Expression and purification of VP30
VP30 was overexpressed as an N-terminal fusion with the maltose binding protein (MBP). The VP30 moiety, named VP308-272, lacked the N-terminal 7 and C-terminal 16 aa of wild type VP30 (VP30_wt), as these minor terminal truncations enhance stability and solubility of the recombinant protein.17 VP308-272 supports viral transcription with equal efficiency as VP30_wt, and also the entire MBP-VP308-272 fusion protein activates viral transcription (twofold less efficiently than VP30_wt;33). MBP-VP308-272 was overexpressed in Escherichia coli BL21 (DE3) cells and the VP30 moiety physically separated from the MBP-tag by TEV protease cleavage to rule out any steric interference of the MBP moiety with RNA binding (for more details, see the Supplementary material). Subsequent RNA binding experiments were conducted in the presence of both proteolytic products, MBP and VP30.
In vitro transcription and capping reaction
Standard T7 in vitro transcription was performed as described.27 Since EBOV mRNAs are 5′-capped in infected cells, we analyzed how VP30 binding is affected by different 5′-end groups on the antigenomic RNA 56–158, mimicking the first part of the NP mRNA 5′-UTR. While a 5′-monophosphate was introduced by including excess 9 mM GMP (relative to 4 mM GTP) as initiator nucleotide in transcription reactions,27 a 5′-triphosphate was generated by omitting GMP (only 4 mM each NTP present). For introducing a 7-methylguanylate cap (Cap 0) at the 5′-end, the RNA carrying a 5´-triphosphate was used as the substrate for the Vaccinia Capping System (NEB) which was applied according to the manufacturer's instructions, with minor changes. Briefly, 30 pmol RNA were heated to 95°C for 3 min and kept on ice for 5 min. The RNA solution was then adjusted to 1 x Capping buffer, 0.5 mM GTP, 0.1 mM S-adenosylmethionine (SAM; freshly diluted from a 32 mM stock solution) and 10 U Vaccinia Capping Enzyme. For radioactive labeling of the capped RNA, reaction mixtures additionally contained 3.3 pmol [α-32P]-GTP. Capping reactions were incubated at 37°C for 1 h. The success of the capping reaction was analyzed by denaturing 20% PAGE of unlabeled capped next to uncapped RNA, where gel mobility of the capped RNA was reduced relative to the 5′-triphosphate counterpart. Capped RNA was purified by denaturing 20% PAGE.
Endlabeling of RNAs
5´-[32P]-endlabeling was performed using T4 polynucleotide kinase (T4 PNK; Thermo Scientific). 30 pmol of RNA were incubated with [γ-32P]-ATP (Hartmann Analytic) and 10 U T4 PNK for 1 h at 37 °C. For EMSA experiments, the labeled products were purified using Illustra™ Microspin G-25 columns (GE healthcare), for secondary structure analysis the labeled RNAs were purified using denaturing PAGE, eluted in 1 M NaOAc (pH 4.9) and precipitated with isopropanol (1 vol. for RNAs > 100 nt; 2 vol. for RNAs < 100 nt) at room temperature (30 min) and centrifugation at 4° C for 30 min. For 3′-endlabeling of RNAs used in probing experiments, 2',3′-cyclic phosphate ends derived from 3′-hammerhead self-cleavage to generate homogeneous 3′-ends had to be removed before labeling. This was performed in 1 x buffer P (50 mM MES pH 6.5, 100 mM MgCl2) in the presence of 10 U T4 PNK (Thermo Scientific) for 4 h at 37 °C. 3′-endlabeling was carried out in the presence of 30 pmol RNA, 0.15 mM ATP, 1 x T4 RNA ligase buffer (50 mM Tris/HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP), [5′-32P]pCp (Hartmann Analytic) and 20 U T4 RNA ligase (Thermo Scientific). The mixture was incubated at 8°C overnight. The purification was performed either using Illustra™ Microspin G-25 columns (GE healthcare) or by denaturing PAGE.
Electrophoretic mobility shift assay
5′- or 3′-32P-endlabeled RNAs were diluted to 24 nM RNA in 50 mM Tris/HCl pH 8.0, 200 mM NaCl, followed by unfolding at 95°C for 1 min and cooling on ice for 2 min. RNAs were then incubated at room temperature for another 20 min, followed by mixing 5 μL with an equal volume of VP30 (final concentrations: 0.1 to 2.7 µM VP30, 12 nM RNA) in buffer A (50 mM Tris/HCl pH 8.0, 200 mM NaCl, 25% glycerol) and further incubation at room temperature for 10 min. For competition assays with heparin, see Fig. S6. After the final incubation step, reaction mixtures were immediately loaded (without addition of extra sample buffer) on a native PAA gel consisting of 2 gel layers of different PAA percentages (the upper one-fourth to one-third was 4% PAA, the lower two-thirds to three-fourths were 10% PAA, and the casting buffer composition was 50 mM Tris/HCl, 200 mM NaOAc, pH 8.0). As electrophoresis buffer, 25 mM Tris/HCl, 100 mM NaOAc (pH 8.0) was used. After electrophoresis, mobility shift gels were scanned with a Bio-Imaging Analyzer FLA3000–2R (Fujifilm), and 32P-endlabeled bands were marked by boxes and quantified using the analysis software PCBAS/AIDA (Raytest, Straubenhardt, Germany). RNAs were shifted into 2 types of complexes, termed C1 for complexes migrating into the 4% PAA zone and, to a low extent, CHMW for high molecular weight complexes that remained in the gel pockets. For quantification, the intensity of the radioactive signal for C1 or CHMW was divided by the total intensity (free RNA + C1 + CHMW) after substraction of the corresponding background in reference lanes using 4 µM MBP protein alone (instead of the equimolar MBP and VP30 mixture after TEV cleavage of the fusion protein). For calculating apparent Kd values (appKd), mean values and standard errors of the mean (SEM) for C1 complex formation at each VP30 concentration, derived from at least 3 independent experiments, were fitted using the software Grafit (Version 5.0.139) and applying a one ligand binding site model. All appKd values and experimental endpoints (capacity) are given with standard deviations (± SD) in Table S1. Values for the fraction of RNA as part of C1 complexes at the highest VP30 concentration used here (2.7 μM; termed F2.7μM) are also provided in Table S1 (± SD).
Statistics
Two-tailed P values were calculated from comparison of 2 appKd or F2.7μM values using the unpaired t test (http://www.graphpad.com/quickcalcs/ttest1/?Format=SD); the mean appKd or F2.7μM value and the corresponding standard deviation as well as the number of independent experiments (N) were used as input (summarized in Table S1).
DNA oligonucleotide interference with VP30:RNA binding
We hybridized different DNA probes (probe 1: 5′-GGATCTTTTGTGTGCGAA-3′; probe 2: 5′-ATTGAAATTTATATCGG-3′; probe 3: 5′-AATTTAAATTGAAATTGTTAC-3′; probe 4: 5′-CCTGGTTTGTTTCAGAG-3´; Metabion) to the genomic substrate (154–1). For this purpose, 0.06 pmol of 5´-32P-endlabeled RNA were mixed with 0.5 pmol of the respective DNA probe, followed by probe annealing in a thermocycler (95°C 5 min, 90°C 5 min, 80°C 5 min, 70°C 5 min, 60°C 5 min, 50°C 5 min, 40°C 5 min, 30°C 5 min, 20°C 5 min, 10°C 5 min). The efficiency of annealing was confirmed by oligonucleotide-induced RNA mobility changes in a native 10% PAA/1 x TBE gel system. After DNA oligonucleotide annealing to the RNA, the VP30 EMSA was performed as described above.
Secondary structure analysis
The secondary structures of the genomic (154-1) and antigenomic (2–158) RNA were probed by Pb2+−induced and RNase T1 hydrolysis using 5′- or 3′-32P-endlabeled RNA as described.15 Briefly, 1 µg of RNA containing trace amounts of the same 32P-labeled RNA (∼30,000 Cherenkov cpm) in 1 x refolding buffer (10 mM Tris, 1 mM EDTA, 20 mM ammonium acetate, pH 6.5; 8 µL final volume) was denatured at 95°C for 3 min, followed by cooling on ice for 5 min and incubation at room temperature for about 15 min. 1 µL of such an RNA mixture was used for a single probing reaction (final volume: 5 µL). For generating an OH− ladder, the RNA solution was adjusted to 100 mM Na2CO3/100 mM NaHCO3 (pH 9.0) and incubated at 95°C for 3 min. The native RNase T1 ladder was generated in 1 x TN buffer (20 mM Tris-acetate, 100 mM NaCl, pH 7.5) using 1 µL RNase T1 (0.02 U, Thermo Scientific; freshly diluted 1: 50,000 in ddH2O from a stock solution of 1000 U/µL) at 37°C for 3–5 min. A denaturing RNase T1 ladder was generated under the same conditions, except that the mixture was adjusted to 4 M urea and the incubation temperature was raised to 55°C. Pb2+-induced hydrolysis was carried out at room temperature by incubating the RNA in 1 x TN buffer in the presence 2 mM lead(II) acetate (freshly diluted from a 50 mM lead(II) acetate stock) for 15 or 17 min. All reactions were stopped with 2 x PPF sample buffer (0.02% (w/v) bromophenol blue, 0.02% (w/v) xylene cyanol blue, 2.6 M urea, 66% (v/v) formamide). Hydrolysis patterns were analyzed by denaturing PAGE using (8 or 15%).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Marcus Lechner for the structural comparison (Fig. S7) of VP30 and M2-1 proteins and Sara Doll for technical assistance in the probing experiments.
Funding
This work was supported by the German Research Foundation (DFG), grant CRC 1021.
References
- 1.Kiley MP, Bowen ET, Eddy GA, Isaacson M, Johnson KM, McCormick JB, Murphy FA, Pattyn SR, Peters D, Prozesky OW, et al.. Filoviridae: a taxonomic home for Marburg and Ebola viruses? Intervirol 1982; 18:24-32; PMID:7118520; http://dx.doi.org/24906522 10.1159/000149300 [DOI] [PubMed] [Google Scholar]
- 2.Adams MJ, Lefkowitz EJ, King AM, Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol 2014; 159:2831-41; PMID:24906522; http://dx.doi.org/ 10.1007/s00705-014-2114-3 [DOI] [PubMed] [Google Scholar]
- 3.Mahanty S, Bray M. Pathogenesis of filoviral haemorraghic fevers. Lancet Infect Dis 2004; 4:487-98; PMID:15288821; http://dx.doi.org/ 10.1016/S1473-3099(04)01103-X [DOI] [PubMed] [Google Scholar]
- 4.Sanchez A, Kiley MP, Holloway BP, Auperin DD. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res 1993; 29:215-40; PMID:8237108; http://dx.doi.org/ 10.1016/0168-1702(93)90063-S [DOI] [PubMed] [Google Scholar]
- 5.Weik M, Enterlein S, Schlenz K, Mühlberger E. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J Virol 2005; 79:10660-71; PMID:16051858; http://dx.doi.org/ 10.1128/JVI.79.16.10660-10671.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volchkov VE, Volchkova VA, Chepurnov AA, Blinov VM, Dolnik O, Netesov SV, Feldmann H. Characterization of the L gene and 5´ trailer region of Ebola virus. J Gen Virol 1999; 80:355-62; PMID:10073695; http://dx.doi.org/ 10.1099/0022-1317-80-2-355 [DOI] [PubMed] [Google Scholar]
- 7.Mühlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 1999; 73:2333-42; PMID:9971816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modrof J, Mühlberger E, Klenk HD, Becker S. Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem 2002; 277:33099-104; PMID:12052831; http://dx.doi.org/12163572 10.1074/jbc.M203775200 [DOI] [PubMed] [Google Scholar]
- 9.Weik M, Modrof J, Klenk HD, Becker S, Mühlberger E. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J Virol 2002; 76:8532-9; PMID:12163572; http://dx.doi.org/ 10.1128/JVI.76.17.8532-8539.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martínez MJ, Biedenkopf N, Volchkova V, Hartlieb B, Alazard-Dany N, Reynard O, Becker S, Volchkov V. Role of Ebola virus VP30 in transcription reinitiation. J Virol 2008; 82:12569-73; PMID:18829754; http://dx.doi.org/8806574 10.1128/JVI.01395-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mühlberger E, Trommer S, Funke C, Volchkov V, Klenk HD, Becker S. Termini of all mRNA species of Marburg virus: sequence and secondary structure. Virology 1996; 223:376-80; PMID:8806574; http://dx.doi.org/ 10.1006/viro.1996.0490 [DOI] [PubMed] [Google Scholar]
- 12.John SP, Wang T, Steffen S, Longhi S, Schmaljohn CS, Jonsson CB. Ebola virus VP30 is an RNA binding protein. J Virol 2007; 81:8967-76; PMID:17567691; http://dx.doi.org/ 10.1128/JVI.02523-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogino T, Banerjee AK. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol Cell 2007; 25:85-97; PMID:17218273; http://dx.doi.org/ 10.1016/j.molcel.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 14.Steuten B, Hoch PG, Damm K, Schneider S, Köhler K, Wagner R, Hartmann RK. Regulation of transcription by 6S RNAs: Insights from the Escherichia coli and Bacillus subtilis model systems. RNA Biol 2014; 11:508-21; PMID:24786589; http://dx.doi.org/25771336 10.4161/rna.28827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köhler K, Durchardt-Ferner E, Lechner M, Damm K, Hoch PG, Salas M, Hartmann RK. Structural and mechanistic characterization of 6S RNA from the hyperthermophilic bacterium Aquifex aeolicus. Biochimie 2015; 117:72-86; PMID:25771336; http://dx.doi.org/ 10.1016/j.biochi.2015.03.004 [DOI] [PubMed] [Google Scholar]
- 16.Heuck CC, Schiele U, Horn D, Fronda D, Ritz E. The role of surface charge on the accelerating action of heparin on the antithrombin III-inhibited activity of α-thrombin. J Biol Chem 1985; 260:4598-603; PMID:3988727 [PubMed] [Google Scholar]
- 17.Hartlieb B, Muziol T, Weissenhorn W, Becker S. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc Natl Acad Sci USA 2007; 104:624-9; PMID:17202263; http://dx.doi.org/ 10.1073/pnas.0606730104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan SP. Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 2015; 162:314-27; PMID:26144317; http://dx.doi.org/ 10.1016/j.cell.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox R, Plemper RK. The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front Microbiol. 2015; 6:459; PMID:26029193; http://dx.doi.org/ 10.3389/fmicb.2015.00459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reguera J, Gerlach P, Cusack S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr Opin Struct Biol. 2016; 36:75-84; PMID:26826467; http://dx.doi.org/ 10.1016/j.sbi.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 21.Filomatori CV, Iglesias NG, Villordo SM, Alvarez DE, Gamarnik AV. RNA sequences and structures required for the recruitment and activity of the dengue virus polymerase. J Biol Chem 2011; 286:6929-39; PMID:21183683; http://dx.doi.org/25142601 10.1074/jbc.M110.162289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen B, Mulder LCF, Martinez O, Basler CF. Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J Virol 2014; 88:12500-10; PMID:25142601; http://dx.doi.org/ 10.1128/JVI.02163-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanner SJ, Ariza A, Richard CA, Kyle HF, Dods RL, Blondot ML, Wu W, Trincao J, Trinh CH, Hiscox JA, et al.. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci USA 2014; 111:1580-5; PMID:24434552; http://dx.doi.org/ 10.1073/pnas.1317262111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blondot ML, Dubosclard V, Fix J, Lassoued S, Aumont-Nicaise M, Bontems F, Eléouet JF, Sizun C. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. Plos Pathog 2012; 8(5):e1002734; PMID:22675274; http://dx.doi.org/ 10.1371/journal.ppat.1002734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. Elife 2014; 3:e02674; PMID:24842877; http://dx.doi.org/ 10.7554/eLife.02674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esperante SA, Noval MG, Altieri TA, de Oliveira GA, Silva JL, de Prat-Gay G. Fine modulation of the respiratory syncytial virus M2-1 protein quaternary structure by reversible zinc removal from its Cys(3)-His(1) motif. Biochem 2013; 52:6779-89; PMID:23984912; http://dx.doi.org/19036794 10.1021/bi401029q [DOI] [PubMed] [Google Scholar]
- 27.Gößringer M, Helmecke D, Köhler K, Schön A, Kirsebom LA, Bindereif A, Hartmann RK. Enzymatic RNA Synthesis using Bacteriophage T7 RNA Polymerase In: Handbook of RNA Biochemistry, second edition (eds. Hartmann RK, Bindereif A, Schön A, Westhof E), WILEY-VCH, Weinheim, Germany, 2014:pp. 3-27 [Google Scholar]
- 28.Mörl M, Hartmann RK. Production of RNAs with Homogeneous 5′- and 3′-Ends In: Handbook of RNA Biochemistry, second edition (eds. Hartmann RK, Bindereif A, Schön A, Westhof E), WILEY-VCH, Weinheim, Germany, 2014:pp. 29-44 [Google Scholar]
- 29.Li D, Willkomm DK, Hartmann RK. Minor changes largely restore catalytic activity of archaeal RNase P RNA from Methanothermobacter thermoautotrophicus. Nucleic Acids Res 2009. January; 37(1):231-42; PMID:19036794; http://dx.doi.org/ 10.1093/nar/gkn915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 2013; 288:11165-74; PMID:23493393; http://dx.doi.org/ 10.1074/jbc.M113.461285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Modrof J, Becker S, Mühlberger E. Ebola virus transcription activator VP30 is a zinc-binding protein. J Virol 2003; 77:3334-8; PMID:12584359; http://dx.doi.org/ 10.1128/JVI.77.5.3334-3338.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartlieb B, Modrof J, Mühlberger E, Klenk HD, Becker S. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem 2003; 278:41830-6; PMID:12912982; http://dx.doi.org/ 10.1074/jbc.M307036200 [DOI] [PubMed] [Google Scholar]
- 33.Biedenkopf N, Schlereth J, Grünweller A, Becker S, Hartmann RK RNA-binding of Ebola virus VP30 is essential for activating viral transcription. J Virol. 2016. Jun 8. pii: JVI.00271-16; PMID:27279615 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.