

ABSTRACT
As well as encoding viral proteins, genomes of RNA viruses harbor secondary and tertiary RNA structures that have been associated with functions essential for successful replication and propagation. Here, we identified stem-loop structures that are extremely conserved among 1,884 M segment sequences of influenza A virus (IAV) strains from various subtypes and host species using computational and evolutionary methods. These structures were predicted within the 3′ and 5′ ends of the coding regions of M1 and M2, respectively, where packaging signals have been previously proposed to exist. These signals are thought to be required for the incorporation of a single copy of 8 different negative-strand RNA segments (vRNAs) into an IAV particle. To directly test the functionality of conserved stem-loop structures, we undertook reverse genetic experiments to introduce synonymous mutations designed to disrupt secondary structures predicted at 3 locations and found them to attenuate infectivity of recombinant virus. In one mutant, predicted to disrupt stem loop structure at nucleotide positions 219–240, attenuation was more evident at increased temperature and was accompanied by an increase in the production of defective virus particles. Our results suggest that the conserved secondary structures predicted in the M segment are involved in the production of infectious viral particles during IAV replication.
KEYWORDS: Influenza A virus, M segment, packaging signal, RNA secondary structure, stem loop
Introduction
Influenza A virus (IAV) is a member of the family Orthomyxoviridae and possesses a genome comprising 8 segmented, negative-strand RNA molecules (vRNAs), all of which are necessary for productive infection. Each vRNA sequence is composed of one or more open reading frames (ORFs) flanked by untranslated regions (UTRs) that are 19–58 nt long. The distal 12 and 13 nucleotides of the 3′ and 5′ UTRs (U12 and U13, respectively) are highly conserved among different IAV strains and different vRNA segments,1,2 and contribute to the formation of a panhandle structure via long-range interactions.3,4 The proximal parts of the UTRs contain segment-specific conserved nucleotide sequences that extend into the ORF termini.5 In complete virions, the vRNA exists in the form of viral ribonucleoprotein (vRNP), which comprises single molecules of the polymerase subunits PB2, PB1, and PA bound to the panhandle structure, while polymerized nucleoproteins (NPs) are attached to the phosphate-sugar backbone of vRNA, thereby leaving vRNA bases exposed.4,6–10
Fully infectious IAV virions are enveloped and approximately 100 nm in diameter and are thought to contain a single copy of each of the 8 different vRNAs (selective packaging).11-13 Reverse genetic studies of IAV that employ site-directed mutagenesis have shown that mutations in UTR sequences, as well as synonymous mutations in ‘segment-specific conserved sequences’, reduce the efficiency with which vRNAs are incorporated into progeny particles, suggesting that these sequences may contain the signals controlling the selective packaging (packaging signals).5,14–20 A recent study has further demonstrated that conserved sequences in the UTRs may be important for segment incorporation while those in ORF termini may regulate bundling of the 8 different vRNA segments.21 However, the mechanism by which these sequences contribute to the selective packaging of IAV segments is not fully understood.
Experimental studies based on RNA hybridization have demonstrated RNA/RNA interactions between different vRNA segments, although some of the interacting regions did not overlap with segment-specific conserved sequences described above.22-25 RNA-like string structures that interconnect 8 vRNPs were observed within the IAV particle by electron tomography.12,22 In addition, kissing loop structures have been predicted at the interacting regions between 2 vRNA segments, although these intersegment interactions may be strain specific.24 These observations suggest that the selective packaging of IAV may occur through RNA/RNA interactions between different vRNA segments mediated by vRNA secondary structures.
In this study we investigate vRNA secondary structures in the M segment of IAV. The M segment is the seventh RNA segment and encodes the M1 and M2 proteins, an mRNA3 and a variety of other proteins that are expressed only by certain IAV strains. M1 is translated from unspliced mRNA while M2 and mRNA3 RNAs are differentially spliced.26 Incorporation of the M segment vRNA into the IAV virion has been suggested to be critical to the packaging process18,27 and in silico analyses have predicted secondary RNA structures in the positive and negative sense RNA sequences of the M segment.28-31 Here, we use evolutionary and bioinformatic methods to identify stem loop structures in the M segment that are highly conserved among 1,884 IAV strains and that are located in regions previously proposed to contain packaging signals.18,20 These in silico findings were then followed up with reverse genetics experiments in order to investigate the functional significance of these regions. Specifically, we introduced synonymous mutations intended to disrupt the predicted stem loop structures. We observed that the infectivity of a mutant virus bearing mutations in one of the predicted RNA structures was significantly attenuated and this was accompanied by an increase in the production of defective virus particles. Our results suggest that secondary structures predicted within M segment vRNA may be involved in the production of infectious IAV particles.
Results
Identification and prediction of RNA secondary structures
The M segment nucleotide sequences of 1,884 IAV strains were analyzed to identify conserved secondary RNA structures (Supplementary Tables 1 and 2). These strains belonged to 88 subtypes and were isolated from human (n = 443), swine (n = 172), avian (n = 1,205), and other (n = 64) host species. We conducted a sliding window search of these sequences for regions that potentially form secondary structures in either positive (+RNA) or negative (-RNA) sense RNA sequences, using the method implemented in SSE with a window size of 150 nucleotides (see Materials & Methods for details).32 The windows with negative z-score values were predicted to form secondary structures. When we focused on windows with z-scores < −1, secondary structures were predicted to be formed in both strands at nucleotide positions 115–354 and 835–1014 (Fig. 1B and Table 1; position numbers are given for +RNA), which were located in the M1 and M2 coding regions, respectively. Further, positions 115–354 contained 2 subregions with z-scores < −1, at around positions 150 and 250 (Fig. 1B). Similar results were obtained when the sliding-window search was conducted with a window size of 300 nucleotides (Fig. S1). Notably, the regions described above were highly conserved among all 1,884 sequences (regions with >98% identity are shown in red in Fig. 1C). Synonymous substitutions also appeared to be more strongly suppressed in these regions (dS < 0.01) than in the surrounding regions (dS > 0.04; Fig. 1A), suggesting that functional constraints operate at the nucleotide sequence level. These observations are consistent with Moss et al.28
Figure 1.
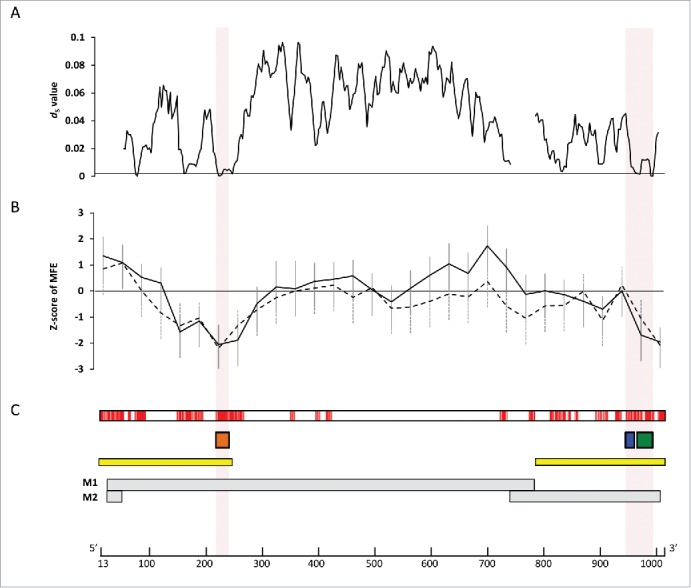
Conserved secondary structures among 1,884 M segment vRNA sequences. (A) Average dS values in the non-overlapping coding regions of M1 and M2, calculated using ADAPTSITE.50 (B) Average z-scores of the minimum free energy for the secondary structures potentially formed by the nucleotide sequences in the windows, calculated using SSE.32 Solid and dashed lines represent the z-scores and the standard deviations for –RNA and +RNA, respectively. Windows with negative z-scores contain sequences that potentially form stable RNA secondary structures. (C) Nucleotide sites with high sequence identity. Nucleotide sites are colored red where >6 contiguous sites with >98% identity are present. Orange, blue, and green boxes show the locations of the SL3–10(+)/(−), SL5-2(+), and SL5-3B(+)/(−) structures, respectively. Yellow boxes represent regions of the M segment where packaging signals have previously been inferred to be located.18,20 Gray boxes represent coding regions for M1, and M2. A scale bar for nucleotide positions in M vRNA is shown below. Nucleotide positions are given relative to the +RNA sequence of the M segment A/hvPR8/34(H1N1) (accession number EF190977).
Table 1.
M segment positions predicted to form secondary structures.
| z-score |
||||
|---|---|---|---|---|
| coding region | nt positionsa | strand | average | SE |
| M1 | 115–264 | – | −1.5666 | 0.6246 |
| + | −1.3270 | 0.8544 | ||
| 145–294 | – | −1.1529 | 0.5390 | |
| + | −1.0313 | 0.6243 | ||
| 175–324 | – | −2.0586 | 0.7688 | |
| + | −2.2007 | 0.7180 | ||
| 205–354 | – | −1.8819 | 0.7828 | |
| + | −1.3244 | 0.5862 | ||
| M2 | 835–984 | – | −1.6945 | 0.7812 |
| + | −1.0636 | 0.7549 | ||
| 865–1014 | – | −1.9571 | 0.5580 | |
| + | −2.0936 | 0.5514 | ||
Nucleotide positions of the windows with z-score of -1.
Consensus secondary structures that may be formed in these regions were predicted using RNAalifold.33 Analyses were undertaken 4 times separately for human, swine, and avian IAVs by randomly choosing 10 strains from each host species (Table S3). Within positions 115–354, a stem-loop structure with multi-branch loops was predicted to be formed at positions 130–217 of +RNA in human, swine, and avian IAV strains (Fig. S2). This structure has been reported to be involved in the splicing of M segment mRNAs.28,29,31 In addition, we newly identified stem-loop structures (named SL3–10(+) in +RNA and SL3-10(−) in –RNA) at positions 219–240 in M segment sequences from all 3 host species (Figs. 1, 2A and 3A; Figs. S2 and 3). When secondary structures at positions 219–240 were predicted for each of the 1,884 M sequences using RNAfold,34 we found an exceptional level of structural sequence conservation; every set of complementary base-pairs that comprise the stems of SL3-10(+) and SL3-10(−) was observed in >99.79% of sequences with minimum free energies ranging from −9.2 to −10.6 kcal/mol and from −12.6 to −15 kcal/mol, respectively (Figs. 2B and 3B). Further, the frequency at which the entire structures of SL3-10(+) and SL3-10(−) were identified in human, swine, and avian IAV strains was 96.7 – 99.6% (Table 2).
Figure 2.

Stem loop structure of SL3–10(+) predicted at nucleotide positions 219–240 of M segment +RNA. (A) Secondary structures were predicted using the M nucleotide sequence of England/195/2009(H1N1) (accession number GQ166660) at temperature setting of 37°C. The background red color of each base represents nucleotide identities of > 98% among 1,884 sequences. Third codon positions are circled in blue. The red arrows and nucleotide sequences indicate the position and nature of synonymous mutations that were introduced to produce SL3–10 mutants. (B) The number (percentage) of sequences that were predicted to form base-paring (BP) at the stem of SL3–10(+) among 1,884 sequences. (C) Secondary structure predicted at positions 219–240 of SL3–10 M sequence. Bases with synonymous mutations are circled in red.
Figure 3.

Stem loop structure of SL3–10(−) predicted at nucleotide positions 219–240 of M segment -RNA. (A) Secondary structures were predicted using the M nucleotide sequence of England/195/2009(H1N1) (accession number GQ166660) at temperature setting of 37°C. The background red color of each base represents nucleotide identities of > 98% among 1,884 sequences. Third codon positions are circled in blue. The red arrows and nucleotide sequences indicate the position and synonymous mutations, which were introduced to produce SL3–10 mutant. (B) The number (percentage) of sequences that were predicted to form base-paring (BP) at the stem positions of SL3–10(−) among 1,884 sequences. (C) Secondary structure predicted at positions 219–240 of SL3–10 M sequence. Bases with synonymous mutations are circled in red.
Table 2.
Frequencies of stem-loop structures predicted in M segment RNA.
| frequency(%)b |
|||||
|---|---|---|---|---|---|
| stem loop | nt positionsa | strand | human | swine | avian |
| SL3–10 | 219–240 | – | 99.6 | 98.9 | 99.5 |
| + | 99.4 | 98.3 | 96.7 | ||
| SL5–3B | 967–994 | – | 97.1 | 94.9 | 98.7 |
| + | 97.4 | 99 | 98.7 | ||
| SL5–2 | 950–964 | + | 99.6 | 92 | 96.3 |
Nucleotide positions refer to those of M segment +RNA in A/hvPR8/34(H1N1) (accession number: EF190977).
Frequency of entire stem-loop structures predicted in M segment RNAs of human (n = 443), swine (n = 172), and avian (n = 1,250) IAV strains.
The above procedure to identify consensus secondary structures was repeated for positions 835–1014. In this region we newly identified a conserved stem-loop structure, named SL5–3B(+), at positions 967–994 in the +RNA of human, swine, and avian IAV strains (Figs 1B and 4A, Fig. S4). A consensus structure in −RNA in the same sequence location as SL5–3B(+) was observed only in human and swine IAV strains, named SL5–3B(−) (Fig. 5A and Fig. S5). However, when secondary structures were predicted at positions 967–994 (the location at which SL5–3B(−) was predicted in human and swine strains) using 1,884 M sequences, SL5–3B(−) was also predicted to be present in avian strains, suggesting that surrounding sequences could affect the formation of SL5–3B(−). Each complementary set of base-pairs that comprise the stems of SL5–3B(+) and SL5–3B(−) was observed in >99.6% of 1,884 sequences (Figs. 4B and 5B). In the prediction of SL5-3B(+)/(−) using M sequences at nucleotide positions 967–994, the frequency at which the entire structures of SL5–3B(+) and SL5–3B(−) were observed in human, swine, and avian IAV strains was 94.9 – 99% (Table 2). In addition, a small stem loop structure named SL5–2(+) was newly identified at positions 950–964 in the +RNA of IAVs from all 3 host species (Fig. 6A and Fig. S4). Each pair of bases constituting the stem of SL5–2(+) was observed in >95.54% of 1,884 sequences (Fig. 6B), and the frequency at which the entire SL5–2(+) structure was seen in human, swine, and avian strains was 92–99.6% (Table 2).
Figure 4.

Stem loop structure of SL5–3B(+) predicted at nucleotide positions 967–994 of M segment +RNA. (A) Secondary structures were predicted using the M nucleotide sequence of England/195/2009(H1N1) (accession number GQ166660) at temperature setting of 37°C. The background red, pink and white colors of each base represents nucleotide identities of > 98%, 90–98%, and< 90% among 1,884 sequences, respectively. Third codon positions are circled in blue. The red arrows and nucleotide sequences indicate the position and nature of synonymous mutations that were introduced to produce SL5–3B mutants. (B) The number (percentage) of sequences that were predicted to form base-paring (BP) at the stem positions of SL5–3B(+) among 1,884 sequences. (C) Secondary structure at positions 967–994 of SL5–3B M sequence. Bases with synonymous mutations are circled in red.
Figure 5.

Stem loop structure of SL5–3B(−) predicted at nucleotide positions 967–994 of M segment -RNA. (A) Secondary structures were predicted using the M nucleotide sequence of England/195/2009(H1N1) (accession number GQ166660) at temperature setting of 37°C. The background red, pink and white colors of each base represents nucleotide identities of > 98%, 90–98%, and< 90% among 1,884 sequences, respectively. Third codon positions are circled in blue. The red arrows and nucleotide sequences indicate the position and nature of synonymous mutations that were introduced to produce SL5–3B mutants. Green-filled circles flanked by nucleotide bases denote the synonymous sites at which mutations affect M segment packaging in A/PR/8/34.18 (B) The number (percentage) of sequences that were predicted to form base-paring (BP) at the stem positions of SL5–3B(−) among 1,884 sequences. (C) Secondary structure at positions 967–994 of SL5–3B M sequence. Bases with synonymous mutations are circled in red.
Figure 6.

Stem loop structure of SL5–2(+) predicted at nucleotide positions 950–964 of M segment +RNA. (A) Secondary structures were predicted using the M nucleotide sequence of England/195/2009(H1N1) (accession number GQ166660) at temperature setting of 37°C. The background red, pink and white colors of each base represents nucleotide identities of > 98%, 90–98%, and < 90% among 1,884 sequences, respectively. Third codon positions are circled in blue. The red arrows and nucleotide sequences indicate the position and nature of synonymous mutations that were introduced to produce SL5-2 mutant. (B) The number (percentage) of sequences that were predicted to form base-paring (BP) at the stem positions of SL5–2(+) among 1,884 sequences. (C) Secondary structure at positions 950–964 of SL5-2 M sequence. Bases with synonymous mutations are circled in red.
Effect on virus growth of predicted stem loop disruption
SL3-10(−) and SL5-3B(−) were identified as highly-conserved secondary RNA structures in the –RNA sequence of the M segment of IAV. Interestingly, SL5-3B(−) and SL3-10(−) were identified within sequence regions where packaging signals have been inferred by reverse genetic studies to be located (yellow boxes in Fig. 1C).18,20 In order to examine whether these secondary structures were indeed involved in replication of IAV, 3 mutant viruses, SL3-10, SL5-3B and SL5-2 were constructed from A/England/195/2009(H1N1) (hereafter termed 195E) by introducing synonymous mutations in the M1 or M2 coding region that disrupt the structures of SL3-10(+)/(−), SL5-3B(+)/(−) and SL5-2(+), respectively (Figs. 2C-6C). It should be noted that SL3-10(+)/(−), SL5-3B(+), and SL5-2(+) were observed in 195E even when nearly full length of M nucleotide sequences (positions 13-1015) were used for the secondary structure prediction (Figs. S6 and 7), while SL5-3B(−) was observed with the prediction using local nucleotide sequences at positions 967-994 (Fig. 5A).
MDCK cells were infected at low multiplicity (MOI = 0.001) with SL3-10, SL5-3B, SL5-2, or 195E. The multicycle growth curves of these viruses were compared by titrating the infectious virus in infected cell supernatants collected at 24 h, 48 h, and 74 h post infection (pi). We found that titers for SL3-10 and SL5-3B viruses were approximately 5-fold lower than those for 195E after 24 h pi when incubated at 37°C (Fig. 7A), while the titer of SL5-2 was comparable to 195E, indicating that the secondary structures SL3-10 and SL5-3 disrupted in the mutant viruses were important for productive infection of IAV. The differences in titers between SL3-10 and 195E were even greater and statistically significant when the incubation temperature was raised from 37°C to 39°C (Fig. 7B; 2-way ANOVA; p < 0.05). Similarly, we observed significant attenuation of SL3-10 in a single step growth curve obtained at 39°C (Figs. 7C and 7D). Since the most significant phenotypic effect was observed in SL3-10, all following analyses were performed using SL3-10 only.
Figure 7.

Growth rates of SL3-10, SL5-3B, SL5-2 and 195E in MDCK cells at different temperatures. PFU counts in MDCK infections (MOI = 0.001) with SL3-10, SL5-3B, SL5-2 and 195E at (A) 37°C and (B) 39°C. PFU counts in single-step MDCK infections (MOI = 1) with SL3-10 and 195E at (C) 37°C and (D) 39°C.
Increase in defective particle production of SL3-10 infection
Bearing in mind the location of the SL3-10 structure, we reasoned that attenuation of the SL3-10 mutant may be due to incomplete packaging of vRNA segments, which in turn would lead to production of semi – infectious (SI) particles, which are non-infectious particles with fewer vRNA segments.35-37 When the total number of virus particles released from MDCK cells infected with SL3-10 or 195E at 39°C was quantified using a hemagglutination assay (HA), both viruses produced similar numbers of particles (Fig. 8A), but 195E particles exhibited almost 10 times greater infectivity than SL3-10 (Figs. 7B and 7D). For the same PFU count, the HA titer of SL3-10 was Log24, while 195E did not contain sufficient virus particles to register in the assay (Fig. 8B), indicating that SL3-10 produced defective particles more abundantly than 195E.
Figure 8.

Quantification of virus particles produced by infection with isolates SL3-10 and 195E. Viral particle counts assessed by hemagglutination of chicken red blood cells in supernatant harvested 9 hours post infection following MOI=1 infection of MDCK cells incubated at 39°C. (A) HA titer of unadjusted supernatants. HA titration performed in triplicate showed no variation between triplicates, data representative of 2 independent experiments. (B) HA titer normalized to the PFU of the harvested samples of SL3-10 and 195E. HA titration performed in triplicate showed no variation between triplicates, data representative of 2 independent experiments. (C) Fetuin ELISA to detect virus particles using normalized PFU samples of SL3-10 and 195E. y-axis indicates the OD values following capture of virus particles onto the ELISA plate coated with fetuin, and detection using HA antibody, and thus indicates the quantity of virus particles in each viral sample. x-axis indicates the dilution of SL3-10 and 195E viruses with DMEM.
When the virus particles with the same PFU count were quantified using a fetuin-based capture ELISA using HA antibodies, the OD values in SL3-10 were 3 times higher than those in 195E (Fig. 8C), and a >50 fold dilution of SL3-10 was required to give an absorbance reading equivalent to that of 195E. Taken together, these data suggest that SL3-10 virus preparations produced in MDCK cells at 39°C contained a high number of noninfectious particles, indicating that the conserved secondary structure SL3-10 contributes to the production of infectious IAV virions.
Discussion
Many RNA viruses with segmented genomes are believed to package a single copy of each genomic segment into the virus particle and the contribution of vRNA secondary structures to the selective packaging of vRNA segments has been suggested for several segmented RNA viruses.38-42 In IAVs, many studies have shown that selective packaging may occur through direct interactions between different vRNAs.22-25 The proposal that vRNAs form secondary structures in vRNPs was previously controversial because NP can melt secondary structures in vRNAs.8 However, several studies have indicated the presence of RNA secondary structures in vRNPs7,10,25 and the observation of intermolecular connections among the 8 vRNPs within IAV virions suggests that vRNA secondary structures may be involved in RNA/RNA interactions during the selective packaging of IAV segments.22
In our analysis of 1,884 M nucleotide sequences belonging to 88 IAV subtypes, we predicted the existence of highly conserved stem-loop structures (SL3-10(+)/(−) and SL5-3B(+)/(−)), which were important for productive infection of IAV. SL3-10(+)/(−) and SL5-3B(+)/(−) were present in the 5′ and 3′ ends of the coding regions for M1 and M2, respectively, where packaging signals have been inferred to be located.18,20 Since the body temperatures at which IAVs replicate vary between ∼33°C and ∼40°C among host species,43 the stability of a particular vRNA structure may vary in different hosts.30,45,46 However, stem base-pairings in SL3-10(+)/(−) and SL5-3(+)/(−) are retained in >99% of 1,884 sequences, and in silico analysis predicts these stem-loop structures are stable under different temperature settings (37°C and 40°C) (data not shown), suggesting that they are common and conserved structures within the M segment among IAVs, and may play a role in productive IAV infection even in avian hosts whose body temperature is higher than in mammals. Indeed, our experiments demonstrate that synonymous mutations that disrupt SL3-10(+)/(−) impair the replication of IAV isolate 195E. The attenuation of SL3-10 was accompanied by an increase in the production of defective virus particles, probably SI particles. Similar phenomena were observed when synonymous mutations were introduced into other nucleotide sites in the packaging signals, including those of M segment.18,21,25
In addition, some nucleotide positions in SL5-3B(−) have been shown to be important for the packaging and accumulation of vRNA segments in A/PR/8/34.18 Specifically, synonymous mutations in conserved codons encoding H90-V92 in M2 appear to reduce virus replication and vRNA packaging, and these are located within the stem and loop of SL5-3B(−) (bases with green-filled circles in Fig. 5A). We introduced into 195E synonymous mutations at different nucleotide positions to those reported previously and which disrupt SL5-3B(+)/(−). This resulted in a reduction of infectivity in the mutant virus. Our results thus support the idea that formation of secondary structures in vRNA may be involved in the genome packaging of IAV.
Defects in the M segment might impair the incorporation of M or other specific vRNA segments into virus particles.18,27 Some previous studies report that the M segment interacts directly with NA and HA segments, although the interacting regions between segments may vary depending on IAV strain,22,24,46 and neither SL3-10(−) nor SL5-3B(−) overlap with previously described interacting regions. Further, it is possible that SL3-10(+) affects the expression level of M1 and M2 proteins, which may have an effect on assembly and budding of virus particles.47 It would be interesting to analyze the relative copy numbers of all 8 vRNA segments within virus particles to further elucidate the functions of the highly-conserved SL5-3B(−) and SL3-10(−) structures.
Materials and methods
Influenza virus sequences
A total of 12,984 complete nucleotide sequences for M segment RNA (∼1002 nt long) were retrieved from the Influenza Virus Resource on 1st March 2012.48 After eliminating multiple sequences isolated from the same host individual and sequences containing ambiguous bases and minor gaps, 1,884 sequences were retained for analysis (Supplementary Tables 1 and 2). Most sequences were isolated from human (n = 443), avian (n = 1,205) or swine (n = 172) hosts and the data set included a total of 88 different IAV subtypes (Supplementary Table 1). Multiple alignment of the data set was undertaken using MAFFT.49 Nucleotide positions corresponding to U12 and U13 were removed because they are known to form a panhandle structure.
Searching for nucleotide sequence conservation
Sequence identity at each nucleotide position was calculated from the base frequencies (A, T, C, or G) calculated from the abovementioned multiple alignment. For the non-overlapping coding regions of M1 and M2, synonymous site diversity (dS) within a sliding window was calculated using the program ADAPTSITE50; the window was 5 codons wide and was moved in steps of 1 codon. The dS value for each window was calculated by dividing the total number of synonymous differences by the number of synonymous sites between each pair of sequences, and the average of these pairwise values was shown in Fig. 1A.
Identification and prediction of RNA secondary structures
Nucleotide positions in the M segment RNAs that possibly contain secondary structural elements were predicted by calculating folding energies along the sequence. For both the negative and positive sense sequences of all M segments, we calculated the minimum free energy (MFE) within a sliding window using the UNAFOLD program implemented in the SSE package.32,51 The window sizes for this analysis were 150 or 300 nucleotides, and the step size was 30 nucleotides. To obtain z-scores of MFE for each window, we randomized the nucleotide sequence within each window. Randomization was replicated 100 times using the dinucleotide model.
In order to identify putative secondary structures that were conserved among different IAV strains, we randomly chose 10 sequences each from human, swine, and avian hosts. This subset of sequences was then used to predict consensus secondary structures with RNAalifold.33 Further, the secondary structures of each of the 1,884 M segments were predicted using RNAfold.34
Tissue culture
Madin Darby canine kidney (MDCK) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal calf serum (FCS), 1% penicillin-streptomycin (P/S) mix, and 1% of nonessential amino acids (NEAA) (Gibco-Life technologies) and incubated at either 37°C or 39°C in a 5% CO2 atmosphere.
Construction of reverse genetics viruses
The reverse genetics viruses were generated using a 12 plasmid reverse genetics system synthesized (GeneArt) directly from the A/England/195/2009 whole-genome sequence as previously described.52 The mutants were generated by site-directed mutagenesis (Stratagene Lightening mutagenesis kit) of A/England/195/2009 segment 7 plasmid sequence. The plasmids were sequenced to confirm the presence of the desired mutations. The 12 plasmids were transfected into 293T cells which were subsequently co-cultured with MDCK cells. Viruses generated were passaged one time in MDCK cells at 37°C in DMEM in the presence of TPCK treated trypsin (Worthinton), 1% NEAA and 1% P/S mix and titrated in triplicate by plaque assay on MDCK cells.
Cell infection
Confluent MDCK cells were washed with PBS, then inoculated with 100 µl of virus at the appropriate MOI (0.001 or 1) and incubated at 37°C for 1 h. After incubation the inoculum was removed and cells were washed with PBS (for single-step growth curves an additional acid wash was included with PBS at pH = 4.8). The cells were incubated with DMEM supplemented with 1% NEAA and 1% P/S at appropriate temperature. 300 µl of cell supernatant was taken from each well at required time point and replaced by 300 µl of DMEM. The viral yield was assessed by plaque assay on MDCK cells.
Plaque assay
Confluent MDCK cells were washed with PBS then inoculated with 100 µl of tenfold serial dilutions in DMEM of the supernatant from infected cells and incubated at 37°C for 1 h. Subsequently, the inoculum was removed and agar overlay was applied to each well as previously described.53 The plaques formed after 3 d were then counted.
Hemagglutination assay
Virus stocks were serially 2-fold diluted in 50 µl of PBS in 96 well v bottomed plate starting at 1:2 (21). Chicken red blood cells (Harlan laboratories UK Ltd) were diluted to 0.5% in PBS and 100 µl were applied to each well. Plates were incubated for 1 h before pelleted and suspension cells were counted.
Fetuin ELISA
This method was modified from Gambaryan and Matrosovich.54 Twenty-five ng/ml fetuin solution (Sigma-Aldrich) was prepared in 0.1 M carbonate/bicarbonate buffer (Sigma-Aldrich) and 96-well Greinier medium binding plates were coated with the solution. 195E and SL3-10 viruses normalized to be of equal PFU were diluted 2-, 25-, 50-, and 100-fold with DMEM along the plate and incubated for 1 h at 4°C. Wells were washed 3 times with PBS/Tween (0.01%) and incubated with mouse anti 195E HA monoclonal antibody (a gift from PHE, Colindale, UK) (1:250) at 4°C for 1 h. Washing and incubation steps were repeated with secondary anti mouse-goat horseradish peroxidase antibody (AbD Serotec™) (1:1000). 50 µl of TMB liquid ELISA substrate (Thermo Fisher Scientific) were added to each well, incubated for 30 min, and plates were read at 405 nm wave length on a FluoStar Omega (BMG Labtech).
Supplementary Material
Disclosure of potential confllicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a Japan Society for the Promotion of Science grant to YK (24-448). OGP was supported by the European Research Council under the European Commission Seventh Framework Program (FP7/2007-2013)/European Research Council grant agreement 614725- PATHPHYLODYN.
References
- 1.Robertson JS. 5′ and 3′ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res 1979; 6:3745-57; PMID:493121; http://dx.doi.org/ 10.1093/nar/6.12.3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desselberger U, Racaniello VR, Zazra JJ, Palese P. The 3′ and 5′-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 1980; 8:315-28; PMID:7358274; http://dx.doi.org/ 10.1016/0378-1119(80)90007-4 [DOI] [PubMed] [Google Scholar]
- 3.Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc Natl Acad Sci U S A 1987; 84:8140-44; PMID:2446318; http://dx.doi.org/ 10.1073/pnas.84.22.8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arranz R, Coloma R, Chichón FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortín J, Martín-Benito J. The structure of native influenza virion ribonucleoproteins. Science 2012; 338:1634-37; PMID:23180776; http://dx.doi.org/ 10.1126/science.1228172 [DOI] [PubMed] [Google Scholar]
- 5.Gog JR, Afonso Edos S, Dalton RM, Leclercq I, Tiley L, Elton D, von Kirchbach JC, Naffakh N, Escriou N, Digard P. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res 2007; 35:1897-907; PMID:17332012; http://dx.doi.org/ 10.1093/nar/gkm087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murti KG, Webster RG, Jones IM. Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling. Virology 1988; 164:562-6; PMID:3369093; http://dx.doi.org/ 10.1016/0042-6822(88)90574-0 [DOI] [PubMed] [Google Scholar]
- 7.Yamanaka K, Ishihama A, Nagata K. Reconstitution of influenza virus RNA-nucleoprotein complexes structurally resembling native viral ribonucleoprotein cores. J Biol Chem 1990; 265:11151-5; PMID:2358455. [PubMed] [Google Scholar]
- 8.Klumpp K, Ruigrok RW, Baudin F. Roles of the influenza virus polymerase and nucleoprotein in forming a functional RNP structure. EMBO J 1997; 16:1248-57; PMID:9135141; http://dx.doi.org/ 10.1093/emboj/16.6.1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elton D, Medcalf L, Bishop K, Harrison D, Digard P. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J Virol 1999; 73:7357-67; PMID:10438825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng W, Olson J, Vakharia V, Tao YJ. The crystal structure and RNA-binding of an orthomyxovirus nucleoprotein. PLoS Pathog 2013; 9:e1003624; PMID:24068932; http://dx.doi.org/ 10.1371/journal.ppat.1003624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006; 439:490-2; PMID:16437116; http://dx.doi.org/ 10.1038/nature04378 [DOI] [PubMed] [Google Scholar]
- 12.Noda T, Sugita Y, Aoyama K, Hirase A, Kawakami E, Miyazawa A, Sagara H, Kawaoka Y. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat Commun 2012; 3:639; PMID:22273677; http://dx.doi.org/ 10.1038/ncomms1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou YY, Vafabakhsh R, Doğanay S, Gao Q, Ha T, Palese P. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc Natl Acad Sci U S A 2012; 109:9101-6; PMID:22547828; http://dx.doi.org/ 10.1073/pnas.1206069109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci U S A 2003; 100:2002-7; PMID:12574509; http://dx.doi.org/ 10.1073/pnas.0437772100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujii K, Fujii Y, Noda T, Muramoto Y, Watanabe T, Takada A, Goto H, Horimoto T, Kawaoka Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J Virol 2005; 79:3766-74; PMID:15731270; http://dx.doi.org/ 10.1128/JVI.79.6.3766-3774.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang Y, Hong Y, Parslow TG. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J Virol 2005; 79:10348-55; PMID:16051827; http://dx.doi.org/ 10.1128/JVI.79.16.10348-10355.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Y, Huang T, Ly H, Parslow TG, Liang Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J Virol 2008; 82:229-36; PMID:17959657; http://dx.doi.org/ 10.1128/JVI.01541-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J Virol 2008; 82:11869-79; PMID:18815307; http://dx.doi.org/ 10.1128/JVI.01634-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsh GA, Rabadán R, Levine AJ, Palese P. Highly conserved regions of influenza a virus polymerase gene segments are critical for efficient viral RNA packaging. J Virol 2008; 82:2295-304; PMID:18094182; http://dx.doi.org/ 10.1128/JVI.02267-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozawa M, Maeda J, Iwatsuki-Horimoto K, Watanabe S, Goto H, Horimoto T, Kawaoka Y. Nucleotide sequence requirements at the 5′ end of the influenza A virus M RNA segment for efficient virus replication. J Virol 2009; 83:3384-8; PMID:19158245; http://dx.doi.org/ 10.1128/JVI.02513-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goto H, Muramoto Y, Noda T, Kawaoka Y. The genome-packaging signal of the influenza A virus genome comprises a genome incorporation signal and a genome-bundling signal. J Virol 2013; 87:11316-22; PMID:23926345; http://dx.doi.org/ 10.1128/JVI.01301-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fournier E, Moules V, Essere B, Paillart JC, Sirbat JD, Isel C, Cavalier A, Rolland JP, Thomas D, Lina B, et al.. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res 2012; 40:2197-209; PMID:22075989; http://dx.doi.org/ 10.1093/nar/gkr985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fournier E, Moules V, Essere B, Paillart JC, Sirbat JD, Cavalier A, Rolland JP, Thomas D, Lina B, Isel C, et al.. Interaction network linking the human H3N2 influenza A virus genomic RNA segments. Vaccine 2012; 30:7359-67; PMID:23063835; http://dx.doi.org/ 10.1016/j.vaccine.2012.09.079 [DOI] [PubMed] [Google Scholar]
- 24.Gavazzi C, Yver M, Isel C, Smyth RP, Rosa-Calatrava M, Lina B, Moulès V, Marquet R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc Natl Acad Sci U S A 2013; 110:16604-9; PMID:24067651; http://dx.doi.org/ 10.1073/pnas.1314419110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavazzi C, Isel C, Fournier E, Moules V, Cavalier A, Thomas D, Lina B, Marquet R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: comparison with a human H3N2 virus. Nucleic Acids Res 2013; 41:1241-54; PMID:23221636; http://dx.doi.org/ 10.1093/nar/gks1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson D, Lamb RA. The influenza A virus spliced messenger RNA M mRNA3 is not required for viral replication in tissue culture. J Gen Virol 2008; 89:3097-101; PMID:19008398; http://dx.doi.org/ 10.1099/vir.0.2008/004739-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Q, Chou YY, Doğanay S, Vafabakhsh R, Ha T, Palese P. The influenza A virus PB2, PA, NP, and M segments play a pivotal role during genome packaging. J Virol 2012; 86:7043-51; PMID:22532680; http://dx.doi.org/ 10.1128/JVI.00662-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moss WN, Priore SF, Turner DH. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 2011; 17:991-1011; PMID:21536710; http://dx.doi.org/ 10.1261/rna.2619511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moss WN, Dela-Moss LI, Kierzek E, Kierzek R, Priore SF, Turner DH. The 3′ splice site of influenza A segment 7 mRNA can exist in two conformations: a pseudoknot and a hairpin. PLoS One 2012; 7:e38323; PMID:22685560; http://dx.doi.org/ 10.1371/journal.pone.0038323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Priore SF, Moss WN, Turner DH. Influenza A virus coding regions exhibit host-specific global ordered RNA structure. PLoS One 2012; 7:e35989; PMID:22558296; http://dx.doi.org/ 10.1371/journal.pone.0035989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang T, Kennedy SD, Moss WN, Kierzek E, Turner DH. Secondary structure of a conserved domain in an intron of influenza A M1 mRNA. Biochemistry 2014; 53:5236-48; PMID:25026548; http://dx.doi.org/ 10.1021/bi500611j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmonds P. SSE: a nucleotide and amino acid sequence analysis platform. BMC Res Notes 2012; 5:50; PMID:22264264; http://dx.doi.org/ 10.1186/1756-0500-5-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008; 9:474; PMID:19014431; http://dx.doi.org/ 10.1186/1471-2105-9-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuker M. Mfold web sever for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406-3415; PMID:12824337; http://dx.doi.org/ 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooke CB, Ince WL, Wrammert J, Ahmed R, Wilson PC, Bennink JR, Yewdell JW. Most influenza a virions fail to express at least one essential viral protein. J Virol. 2013; 87:3155-62; PMID:23283949; http://dx.doi.org/ 10.1128/JVI.02284-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooke CB. Biological activities of 'noninfectious' influenza A virus particles. Future Virol 2014; 9:41-51; PMID:25067941; http://dx.doi.org/ 10.2217/fvl.13.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooke CB, Ince WL, Wei J, Bennink JR, Yewdell JW. Influenza A virus nucleoprotein selectively decreases neuraminidase gene-segment packaging while enhancing viral fitness and transmissibility. Proc Natl Acad Sci U S A 2014; 111:16854-49; PMID:25385602; http://dx.doi.org/ 10.1073/pnas.1415396111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol 2010; 91:313-28; PMID:19955561; http://dx.doi.org/ 10.1099/vir.0.017608-0 [DOI] [PubMed] [Google Scholar]
- 39.Jouvenet N, Lainé S, Pessel-Vivares L, Mougel M. Cell biology of retroviral RNA packaging. RNA Biol 2011; 8:572-80; PMID:21691151; http://dx.doi.org/ 10.4161/rna.8.4.16030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burkhardt C, Sung PY, Celma CC, Roy P. Structural constraints in the packaging of bluetongue virus genomic segments. J Gen Virol 2014; 95:2240-50; PMID:24980574; http://dx.doi.org/ 10.1099/vir.0.066647-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fajardo T Jr, Sung PY, Roy P. Disruption of Specific RNA-RNA Interactions in a Double-Stranded RNA Virus Inhibits Genome Packaging and Virus Infectivity. PLoS Pathog 2015; 11:e1005321; PMID:26646790; http://dx.doi.org/ 10.1371/journal.ppat.1005321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyce M, McCrae MA, Boyce P, Kim JT. Inter-segment complementarity in orbiviruses: A driver for co-ordinated genome packaging in the Reoviridae? J Gen Virol 2016; 97:1145-57; PMID:26763979; http://dx.doi.org/ 10.1099/jgv.0.000400 [DOI] [PubMed] [Google Scholar]
- 43.Massin P, Kuntz-Simon G, Barbezange C, Deblanc C, Oger A, Marquet-Blouin E, Bougeard S, van der Werf S, Jestin V. Temperature sensitivity on growth and/or replication of H1N1, H1N2 and H3N2 influenza A viruses isolated from pigs and birds in mammalian cells. Vet Microbiol 2010; 142:232-41; PMID:19926410; http://dx.doi.org/ 10.1016/j.vetmic.2009.10.012 [DOI] [PubMed] [Google Scholar]
- 44.Brower-Sinning R, Carter DM, Crevar CJ, Ghedin E, Ross TM, Benos PV. The role of RNA folding free energy in the evolution of the polymerase genes of the influenza A virus. Genome Biol 2009; 10:R18; PMID:19216739; http://dx.doi.org/ 10.1186/gb-2009-10-2-r18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chursov A, Kopetzky SJ, Leshchiner I, Kondofersky I, Theis FJ, Frishman D, Shneider A. Specific temperature-induced perturbations of secondary mRNA structures are associated with the cold-adapted temperature-sensitive phenotype of influenza A virus. RNA Biol 2012; 9:1266-74; PMID:22995831; http://dx.doi.org/ 10.4161/rna.22081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Essere B, Yver M, Gavazzi C, Terrier O, Isel C, Fournier E, Giroux F, Textoris J, Julien T, Socratous C, et al.. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc Natl Acad Sci U S A 2013; 110:E3840-8; PMID:24043788; http://dx.doi.org/ 10.1073/pnas.1308649110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen BJ, Leser GP, Jackson D, Lamb RA. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 2008; 82:10059-70; PMID:18701586; http://dx.doi.org/ 10.1128/JVI.01184-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the National Center for Biotechnology Information. J Virol 2008; 82:596-601; PMID:17942553; http://dx.doi.org/ 10.1128/JVI.02005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002; 30:3059-66; PMID:12136088; http://dx.doi.org/ 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki Y, Gojobori T, Nei M. ADAPTSITE: detecting natural selection at single amino acid sites. Bioinformatics 2001; 17:660-1; PMID:11448887; http://dx.doi.org/ 10.1093/bioinformatics/17.7.660 [DOI] [PubMed] [Google Scholar]
- 51.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 2008; 453:3-31; PMID:18712296; http://dx.doi.org/ 10.1007/978-1-60327-429-6_1 [DOI] [PubMed] [Google Scholar]
- 52.Shelton H, Smith M, Hartgroves L, Stilwell P, Roberts K, Johnson B, Barclay W. An influenza reassortant with polymerase of pH1N1 and NS gene of H3N2 influenza A virus is attenuated in vivo. J Gen Virol 2012; 93:998-1006; PMID:22323532; http://dx.doi.org/ 10.1099/vir.0.039701-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elleman CJ, Barclay WS. The M1 matrix protein controls the filamentous phenotype of influenza A virus. Virology 2004; 321:144-53; PMID:15033573; http://dx.doi.org/ 10.1016/j.virol.2003.12.009 [DOI] [PubMed] [Google Scholar]
- 54.Gambaryan AS, Matrosovich MN. A solid-phase enzyme-linked assay for influenza virus receptor-binding activity. J Virol Methods 1992; 39:111-23; PMID:1430058; http://dx.doi.org/ 10.1016/0166-0934(92)90130-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.