

ABSTRACT
In this study, we sequenced the first full-length insect transcriptome using the Erthesina fullo Thunberg based on the PacBio platform. We constructed the first quantitative transcription map of animal mitochondrial genomes and built a straightforward and concise methodology to investigate mitochondrial gene transcription, RNA processing, mRNA maturation and several other related topics. Most of the results were consistent with the previous studies, while to the best of our knowledge some findings were reported for the first time in this study. The new findings included the high levels of mitochondrial gene expression, the 3′ polyadenylation and possible 5′ m7G caps of rRNAs, the isoform diversity of 12S rRNA, the polycistronic transcripts and natural antisense transcripts of mitochondrial genes et al. These findings could challenge and enrich fundamental concepts of mitochondrial gene transcription and RNA processing, particularly of the rRNA primary (sequence) structure. The methodology constructed in this study can also be used to study gene expression or RNA processing of nuclear genomes.
KEYWORD: Full-length transcriptome, gene transcription, mitochondrial genes, PacBio, RNA processing
Introduction
Mitochondria are pivotal for cellular adenosine triphosphate (ATP) production, numerous metabolic regulatory processes and the programming of cell death.1 Animal mitochondrial DNA is a small, circular and extrachromosomal genome, typically about 16 Kbp in size. Although the previous studies demonstrated that all animal mitochondrial genomes contain the same 37 genes: two for rRNAs, 13 for mRNAs and 22 for tRNAs,2, the exact mechanisms of the production and maturation of RNA species required for functioning of mitochondrial genomes are still not well understood.3 In the year of 1977, the first study into insect mitochondrial gene transcription identified 13 RNAs in Drosophila cells.4 It was not until after the complete Drosophila yakuba mitochondrial genome was published in 1985 5 that Berthier et al. identified the genetic identities of 11 mature mRNAs and two mature rRNAs in 1986.6 In 2009, Stewart et al. used the RACE and RT-PCR methods to characterize the 5′ and 3′ ends of the mature mRNA and rRNA transcripts and refined the mitochondrial transcription map constructed by Berthier et al in 1986.3 Although the previous studies partially revealed the mechanisms of mitochondrial gene transcription, RNA processing and mRNA maturation et al., these fundamental topics are still in the hypothesis stage.
The Next Generation Sequencing (NGS) technologies, particularly the Illumina platform now has produced high-throughput mitochondrial transcript sequences with quantification information, but the NGS short reads result in incompletely assembled transcripts which lack some important information (e.g. 5′ or 3′ end information). This limits better understanding of transcriptome data. Based on the single-molecule real-time (SMRT) sequencing technology, the PacBio platform can provide longer and even full-length transcripts that originate from observations of single molecules without assembly. The full-length transcripts can be used to improve the investigation of alternative splicing, alternative polyadenylation, fusion transcripts, non-coding RNAs, novel genes and etc. In 2015, the newest PacBio protocol IsoSeq™ was used to produce European cuttlefish, 7 tetraploid cotton 8 and fungi 9 full-length transcriptome, which provided insights in the complexity of gene transcription.
In this study, we sequenced the first insect (Erthesina fullo Thunberg) full-length transcriptome using the SMARTScribe reverse transcriptase with the newest PacBio reagents (P6/C4) on the PacBio RS II platform. By the observation of full-length transcripts, we were able to characterize animal mitochondrial transcripts with more comprehensive and accurate information. Our research objectives included: 1) to produce accurate full-length transcripts of the insect mitochondrial genome as a dataset for further studies; 2) to build a methodology for the investigation of mitochondrial gene transcription, RNA processing, mRNA maturation and several other related topics.
Results
The quantitative transcription map of animal mitochondrial genomes
Total RNA was isolated from different tissues (antennae, heads, wings, legs and thoracic muscles) of one male and female adult E. fullo to construct one cDNA library. Then, the cDNA library was sequenced using the P6/C4 sequencing reagents on the PacBio RS II platform (Materials and Methods). Seven SMRT Cells were used to produce 381,394 raw reads with the average size of 16,262 bp. After removing low quality regions and SMRTbell adapters, raw reads were split into 4,900,485 high-quality (Accuracy >= 75%) subreads with the average size of 1,238 bp. All the subreads were processed into circular consensus sequencing (CCS) reads to further improve the data quality. Finally, CCS reads were used to produce 247,535 draft transcripts representing the cDNA library with the size of 1∼2 Kbp. Since mitochondrial genes do not contain introns, at least 37.65% (93,198/247,535) of draft transcripts could be contiguously aligned to the complete E. fullo mitochondrial genome (Supplementary file 1). This high mapping rate suggested a large part of insect gene expression could be attributed to the mitochondrial genome, although it contains only a few genes. This phenomenon was reported for the first time in this study, probably due to two reasons. The first reason is the RNA extraction and cDNA synthesis were conducted immediately after the insect's death to avoid RNA degradation. The second reason is the SMARTScribe reverse transcriptase has the ability to maintain the complexity of the original RNA (Materials and Methods). The alignment results showed the cDNA synthesis and amplification had successfully captured 11 mRNAs and 2 rRNAs (16S and 12S) by reverse-transcription primers containing 30 bp polyA sequences. The full-length transcripts of these mRNAs and rRNAs were identified and confirmed by at least 10 CCS reads for their fidelity (Supplementary file 2). Using the full-length transcriptome data, we built the first quantitative transcription map of animal mitochondrial genomes (Fig. 1). The transcripts sorted by expression levels from the highest to the lowest were 16S rRNA, COI, COII, ND5, COIII, Cytb, 12S rRNA, ND4/4L, ATP8/6, ND2, ND3, ND1 and ND6. This is similar to the order (16S rRNA = 12S rRNA, COIII, COI = COII, ATP8/6, Cytb, ND5 = ND4/4L = ND2 = ND3 = ND1, and ND6) estimated from densitometric data in the Drosophila study.6 In this study, the full-length transcripts of three scarce mRNAs (ND3, ND1 and ND6) were observed at different levels.
Figure 1.

The first quantitative transcription map of insect mitochondrial genomes. The Erthesina fullo genome was adjusted to the J(+) strand orientation. Alignments of transcripts on J(+) strand in red color were piled along the positive y-axis. Alignments of transcripts on N(−) strand in blue color were piled along the negative y-axis. tRNAs were represented using their amino acids in the green color.
Mature mRNAs and rRNAs
Basically, two mature transcripts (ATP8/6 and ND4/4L) were polycistronic mRNAs, while the other mature transcripts were monocistronic mRNAs or rRNAs (Table 1). Eight mRNA transcripts (ND2, COI, COII, ATP8/6, COIII, ND3, ND6 and Cytb) were produced to have the same nucleotide sequences (although thymine replaced by uracil) as the reference genome (major coding strand) has, while three mRNA transcripts (ND5, ND4/ND4L and ND1) and two rRNA transcripts were encoded by the minor coding strand (N strand) opposite to the major coding strand (J strand). Most of these results confirmed the results from the previous study,6 while we obtained full-length polyadenylated transcripts of two rRNAs (16S and 12S rRNA), which were originally identified as polyadenylated lrRNAs and nonpolyadenylated srRNAs in Drosophila.6 Later, short polyA tails were indirectly detected in srRNAs using the circularization and RT-PCR method, but the length of polyA tails could only be estimated to 3–4 or 6–7 bp.3 In this study, the full-length transcriptome data proved mature mRNA and rRNA transcripts have the same 3′ polyadenylation structure (Supplementary file 2). In addition, we observed some mature mRNA and rRNA transcripts with m7G caps (Fig. 2A).
Table 1.
Annotation of the E. fullo mitochondrial genome.
| Transcript | Strand | Start | End | Length |
|---|---|---|---|---|
| tRNA-Ile | J(+) | 1 | 66 | 66 |
| tRNA-Gln | N(−) | 64 | 132 | 69 |
| tRNA-Met | J(+) | 135 | 200 | 66 |
| ND2 | J(+) | 201 | 1,182 | 982 |
| tRNA-Trp | J(+) | 1,183 | 1,249 | 67 |
| tRNA-Cys | N(−) | 1,242 | 1,302 | 61 |
| tRNA-Tyr | N(−) | 1,303 | 1,366 | 64 |
| COI | J(+) | 1,373 | 2,907 | 1,535 |
| tRNA-Leu | J(+) | 2,910 | 2,974 | 65 |
| COII | J(+) | 2,975 | 3,651 | 677 |
| tRNA-Lys | J(+) | 3,654 | 3,724 | 71 |
| tRNA-Asp | J(+) | 3,725 | 3,789 | 65 |
| ATP8/6 | J(+) | 3,790 | 4,617 | 828 |
| COIII | J(+) | 4,619 | 5,409 | 791 |
| tRNA-Gly | J(+) | 5,411 | 5,475 | 65 |
| ND3 | J(+) | 5,476 | 5,829 | 354 |
| tRNA-Ala | J(+) | 5,828 | 5,896 | 69 |
| tRNA-Arg | J(+) | 5,900 | 5,962 | 63 |
| tRNA-Asn | J(+) | 5,965 | 6,030 | 66 |
| tRNA-Ser | J(+) | 6,030 | 6,101 | 72 |
| tRNA-Glu | J(+) | 6,101 | 6,166 | 66 |
| tRNA-Phe | N(−) | 6,165 | 6,231 | 67 |
| ND5 | N(−) | 6,230 | 7,949 | 1,720 |
| tRNA-His | N(−) | 7,951 | 8,018 | 68 |
| ND4/4L | N(−) | 8,018 | 9,632 | 1,615 |
| tRNA-Thr | J(+) | 9,635 | 9,698 | 64 |
| tRNA-Pro | N(−) | 9,699 | 9,760 | 62 |
| ND6 | J(+) | 9,771 | 10,236 | 466 |
| Ctyb | J(+) | 10,241 | 11,385 | 1,145 |
| tRNA-Ser | J(+) | 11,387 | 11,455 | 69 |
| ND1 | N(−) | 11,480 | 12,400 | 921 |
| tRNA-Leu | N(−) | 12,401 | 12,465 | 65 |
| 16S rRNA | N(−) | 12,464 | 13,742 | 1,279 |
| tRNA-Val | N(−) | 13,635 | 13,748 | 114 |
| 12S rRNA* | N(−) | 13,814 | 14,624 | 811 |
| N(−) | 13,865 | 14,624 | 760 | |
| N(−) | 14,051 | 14,624 | 574 |
J(+) and N(−) represents the major and minor coding strand of the mitochondrial genome, respectively.
Figure 2.
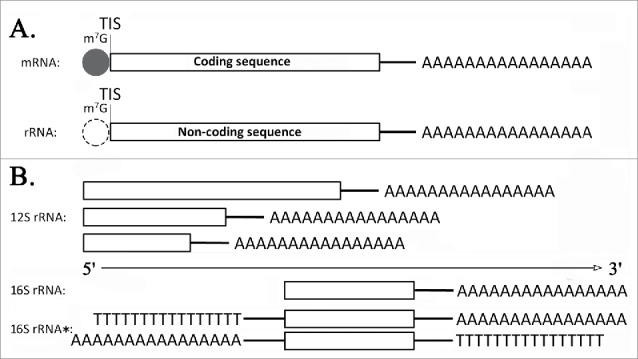
Transcript structures of mitochondrial mRNAs and rRNAs. (A) TIS represents the translation initiation sites (TISs). m7G represents 7-methylguanosine caps. (B) The orientation of 16S rRNA* transcripts was still not finally determined.
Polycistronic transcripts, RNA precursors and RNA processing
The polycistronic transcripts existed widespread among mitochondrial transcripts, although they were much less than the amount of monocistronic transcripts in total. In classical concepts, polycistrons are a common feature of prokaryotes, but are relatively rare in eukaryotes. Using the PacBio full-length fungi transcriptome, Gordon et al. first reported the polycistronic transcripts in eukaryotic fungi.9 In this study, we found two types of polycistronic transcripts (Supplementary file 2) in the E. fullo mitochondrial genome. The first type of polycistronic transcripts could be mapped to more than two mRNAs with tRNAs in between, while the second type could be mapped to not more than one mRNA with neighboring tRNAs preceding it. One typical example in the first type of polycistronic transcripts was ND2/COI containing ND2, tRNA-Trp, tRNA-Cys, tRNA-Tyr and COI. One example in the second type of polycistronic transcripts was Tyr/COI containing tRNA-Tyr and COI. These sequences containing incomplete cleaved tRNAs were RNA precursors. In mitochondrial RNA processing, primary transcripts are processed into RNA precursors. Then, tRNAs encoded between the mRNAs or rRNAs are recognized and removed from their RNA precursors following a “tRNA punctuation” model, and the remaining mRNA and rRNA subunits are processed to their functional forms.3 The mechanism of “tRNA punctuation” was interpreted as the removal of tRNAs by 3′ to 5′ cleavage on the RNA precursors in the previous study.3 Here, we named it “reverse cleavage” model, which was supported by observed polycistronic transcripts of ND3 (Fig. 3A) and COII (Fig. 3B) ended with tRNAs. In this study, we propose a “forward cleavage” model, based on observed polycistronic transcripts ended with mRNAs or rRNAs. In this model, tRNAs preceding mRNAs or rRNAs in the polycistronic transcripts are removed by 5′ to 3′ cleavage (Fig. 3C). These findings suggested the 3′ polyadenylation of mRNAs or rRNAs could be tightly coupled with the tRNA cleavage. This explained why all the polycistronic transcripts obtained in this study ended with mRNAs or rRNAs except Ctyb/Ser, but no other polycistronic transcripts ended with tRNAs since they lacked or had very short 3′ polyA tails. In addition, we obtained a large number of polycistronic transcripts of Lys/Asp/ATP8/6, which were against the putative cleavage model in Fig. 3B.
Figure 3.

Models of “tRNA punctuation.” (A) A model of processing ND3/Glu transcripts assumed in the previous study. (B) A model of processing COII/ATP8/6 transcripts assumed in the previous study. (C) The “forward cleavage” model of processing ND2/COI and COI/COII transcripts proposed in this study. * are putative transcripts, which were not observed.
The polycistronic transcripts were also used to identify transcription start sites (TSSs) and transcription termination sites (TTSs). The results supported the five primary transcripts assumed in the Drosophila mitochondrial study,6 but did not support the two primary transcripts assumed in the human mitochondrial study.10 In this study, five primary transcripts were mapped to unit 1 (ND2, COI, COII, ATP8/6, COIII and ND3), unit 2 (ND5 and ND4/4L), unit 3 (ND6 and Cytb), unit 4 (ND1, 16S rRNA and 12S rRNA) and unit 5. In addition, we proved unit 5 is the antisense transcript of ND2 (discussed below). Therefore, the insect mitochondrial genome could have four primary transcripts with several antisense transcripts.
Antisense transcripts and isoforms of 12S rRNA
In this study, we found a few natural antisense transcripts (NATs), which were anti-ND2 (ChrM: 135–1241), anti-ND5/ND4/4L (ChrM: 7354–9249), anti-ND5 (ChrM: 7117–7844) and anti-ND6/Cty6 (ChrM: 9766–10800) and etc. NATs are reverse-complementary to the known mitochondrial mRNA or rRNA transcripts and have been identified in eukaryotes including humans, mice, yeast and Arabidopsis thaliana,11 their existence in prokaryotes has only been predicted.12 Among these NATs, anti-ND2 was confirmed by at least 10 CCS reads for their fidelity (Supplementary file 2). ND2 and anti-ND2 with the same transcript structure composed a pair which has the full overlap orientation. Another potential NAT was 16S rRNA* which could be reverse-complementary to 16S rRNA (Fig. 2B). But the sequence analysis could not ruled out the possibility that 16S rRNA* were chimeric sequences, since 16S rRNA* had polyT and ployA sequences at both ends (Supplementary file 2).
In this study, we found the 12S rRNA gene was transcribed in three isoforms. The isoform1 (ChrM: 13814–14624), isoform2 (ChrM: 13865–14624) and isoform3 (ChrM: 14051–14624) were mapped to the reference genome with the same 5′ ends but different 3′ ends. Three isoforms were confirmed by at least 10 CCS reads for their fidelity (Supplementary file 2). They were verified in full-length by observation of the complete 5′ PCR primers, 3′ PCR primers with polyA sequences.
Discussion
In this study, we sequenced the first full-length insect transcriptome and constructed the first quantitative transcription map of animal mitochondrial genomes. The methodology was proved to be effective to investigate mitochondrial gene transcription, RNA processing, mRNA maturation and several other related topics. The new findings included the high levels of mitochondrial gene expression, the 3′ polyadenylation and possible 5′ m7G caps of rRNAs, the isoform diversity of 12S rRNA, the polycistronic transcripts and natural antisense transcripts of mitochondrial genes et al. These findings raised three tops for future studies.
The first topic is how to explain the high expression levels of a few mitochondrial genes, which were not noticed in the previous studies. One possible reason is mitochondrial transcripts are shot-lived and degrade more quickly than nuclear genes do, due to they have very short 3′ polyA tails and lack 5′ untranslated regions (UTRs) and 3′ UTRs. The second topic is we hypothesize both mitochondrial mRNA and rRNA transcripts have the same primary (sequence) structure (Fig. 2A). This raises another hypothesis that mitochondrial mRNAs and rRNAs could arise from the same origin. It is an interesting question to know if these rRNAs still have potential abilities to encode proteins or how they lost the function to encode proteins. The third topic is about the antisense transcripts. Are they coding or non-coding sequences? What is their function and do they regulate their corresponding sense transcripts? We hypothesize four significantly low-expressed transcripts (ND2, ND3, ND1 and ND6) could be down-regulated by their antisense transcripts directly or indirectly (Fig. 1), although we only found two of them (anti-ND2 and anti-ND6/Cty6) in our data.
Materials and methods
Full-length transcriptome sequencing using PacBio
The nymph insects (Erthesina fullo Thunberg) were purchased from Shen Nong Biological Technology Co. Ltd (Jiangxi, China) in May 2015 and fed 6 months on a 20% sucrose solution in a growth chamber at the temperature of 27 ± 1°C, with 75% relative humidity and a photoperiod of 13.5 hours/day and 10.5 hours/night. One male and female adult E. fullo were selected to remove abdomen and the remaining tissues were immediately used to extract total RNA with the UNIQ-10 Total RNA Extraction Kit (Biotech, China). The agarose gel electrophoretic analysis showed the RNA had not been degraded and the RNA concentration was read as 1.72 μg/μL using Qubit 2.0 (Life Tech, USA). Finally, 34.64 μg of total RNA was washed with 75% ethanol and then resuspended into nuclease-free water.
The E. fullo cDNA was synthesized using the SMARTScribe reverse transcriptase with 3′ SMART CDS Primer II A (5′-AAGCAGTGGTATCAACGCAGAGTAC-T(30)-3′) and SMARTer II A Oligonucleotide (5′-AAGCAGTGGTATCAACGCAGAGTACATGGG-3′). Then, partial cDNA (30% or less) was used to optimize PCR parameters and the agarose gel electrophoretic analysis showed 12 PCR cycles had reached the best results. Finally, the remaining cDNA (70% or more) were amplified for library construction by 12 PCR cycles. The size distribution (1∼2 Kbp) of amplified cDNA product was close to that of the E. fullo mRNA measured by Agilent 2100 Bioanalyzer (Agilent, USA).
The cDNA products of one male and one female insect were used to construct one SMRTbell library following the manual of the DNA Template Prep Kit 3.0 (Pacific Biosciences, USA): The fragmented cDNA was concentrated by AMPure® PB beads and the ends were repaired. Then, blunt hairpin adapters were ligated to the cDNA and exonucleases were added to remove failed ligation products. SMRTbell templates containing cDNA inserts were purified by AMPure® PB beads. The sequencing primers and the polymerase were then sequentially annealed to the SMRTbell templates using the DNA/Polymerase Binding Kit P6 v2 (Pacific Biosciences, USA). The MagBead loading Kit (Pacific Biosciences, USA) was used to load the annealed templates onto a Pacific Biosciences RS II sequencer. The sequencing was performed using 7 SMRT Cells with the DNA Sequencing Reagent Kit 4.0 v2 (Pacific Biosciences, USA).
DNA sequencing of E. fullo mitochondrial genome
One side of legs weighing 5 mg were excised from one female adult insect. The material was homogenized 30 times using manual glass tissue grinders within 10 mL SE buffer (30 mM Tris-HCl, 10 mM Na2-EDTA, 2.5 mM CaCl2, pH 7.6). The homogenized sample was centrifuged at 1,000 × g for 5 min at 4°C to remove nuclei, cell walls, intact cells and other debris. The supernatant was transferred into a new EP tube and centrifuged at 12,000 × g for 30 min at 4°C to obtain crude mitochondrial fraction. The mitochondria pellet was resuspended in 100 μL TE, and then the DNA was extracted following classical phenol-chloroform procedure.
After whole genome amplification with the REPLI-g Mitochondrial DNA Kit (QIAGEN, Germany), the enriched mitochondrial DNA was sequenced with Ion 318 Chip Kit v2 on the Ion Torrent PGM platform to produce 130,543 reads (Max = 615 bp, Min = 8 bp, N50 = 284 bp, Total = 28,575,952 bp). Using the software MIRA v4.0, 5% (6,527/130,543) of total reads were randomly selected to assemble a draft genome. Then, all of reads were aligned to the draft genome to produce a consensus sequence. To remove the individual differences, we aligned the full-length transcriptome sequences to the consensus sequence and produced the final consensus sequence as the complete E. fullo mitochondrial genome v1.0 (Supplementary file 1). The annotation of E. fullo mitochondrial mRNAs and rRNAs was conducted using the full-length transcripts and the annotation of tRNAs was conducted using MITOS server (http://mitos.bioinf.uni-leipzig.de).
Data analysis
The IsoSeq™ protocol (Pacific Biosciences, USA) was used to process the sequenced reads to circular consensus sequencing (CCS) reads with parameters (Minimum Full Passes = 1, Minimum Predicted Accuracy = 75), then to produce draft transcripts with parameters (Minimum Sequence Length = 300) by removing the 5′ end cDNA primers, 3′ end cDNA primers and 3′ ployA sequences, which had been identified by the pipeline Fastq_clean.13 Fastq_clean is a Perl based pipeline to clean DNA-seq,14 RNA-seq 15 and sRNA-seq data 16 with quality control and had included some tools to process Pacbio data in the version 2.0. The software BWA v0.7.12 was used to align draft transcripts to the complete E. fullo mitochondrial genome. Alignment quality control and filtering were performed using in-house Perl programs to remove errors in draft transcripts from the IsoSeq™ protocol. To filter out alignments with poor quality, the filtering program allowed aligned transcripts with query coverage more than 90% and identities more than 90%. Statistics and plotting were conducted using the software R v2.15.3 with the package ggplot2.17 The 5′ and 3′ ends of mature transcripts, polycistronic transcripts, antisense transcripts and the positions of 5′ m7G caps were observed and curated using the software Tablet v1.15.09.01.18
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We appreciate the help equally from the people listed below. They are Professor Guoqing Liu and graduate student Penglei Guo from College of Life Sciences, Nankai University. The data analysis in this study was supported by National Scientific Data Sharing Platform for Population and Health Translational Cancer Medicine Specials.
Funding
This work was supported by grants from by the 2015 Graduate Research Innovation Fund of Nankai University, Natural Science Foundation of China (31371974 and 31201738) and Fundamental Research Funds for the Central Universities.
References
- 1.Shiota T, Imai K, Qiu J, Hewitt VL, Tan K, Shen H-H, Sakiyama N, Fukasawa Y, Hayat S, Kamiya M, et al.. Molecular architecture of the active mitochondrial protein gate. Science 2015; 349:1544-8; PMID:26404837; http://dx.doi.org/ 10.1126/science.aac6428 [DOI] [PubMed] [Google Scholar]
- 2.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res 1999; 27:1767-80; PMID:10101183; http://dx.doi.org/ 10.1093/nar/27.8.1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart JB, Beckenbach AT. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 2009; 445:49-57; PMID:19540318; http://dx.doi.org/ 10.1016/j.gene.2009.06.006 [DOI] [PubMed] [Google Scholar]
- 4.Spradling A, Pardue ML, Penman S. Messenger RNA in heat-shocked Drosophila cells. J Mol Biol 1977; 109:559-87; PMID:403287; http://dx.doi.org/ 10.1016/S0022-2836(77)80091-0 [DOI] [PubMed] [Google Scholar]
- 5.Clary DO, Wolstenholme DR. The mitochondrial DNA molecule ofDrosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol 1985; 22:252-71; PMID:3001325; http://dx.doi.org/ 10.1007/BF02099755 [DOI] [PubMed] [Google Scholar]
- 6.Berthier F, Renaud M, Alziari S, Durand R. RNA mapping on Drosophila mitochondrial DNA: precursors and template strands. Nucleic acids Res 1986; 14:4519-33; PMID:3086843; http://dx.doi.org/ 10.1093/nar/14.11.4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worley KC. European Cuttlefish Whole Transcriptome Sequencing: A Single-Molecule Full Length Transcript Survey with Iso-Seq Method. Plant and Animal Genome XXIII Conference: Plant and Animal Genome, 2015. [Google Scholar]
- 8.van Eijk M. Genome assembly and Iso-Seq transcriptome sequencing of tetraploid cotton. Plant and Animal Genome XXIII Conference: Plant and Animal Genome, 2015. [Google Scholar]
- 9.Gordon S, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev IV, Figueroa M, et al.. Widespread polycistronic transcripts in mushroom-forming fungi revealed by single-molecule long-read mRNA sequencing. PLoS One 2015; 10:e0132628; PMID:26177194; http://dx.doi.org/ 10.1371/journal.pone.0132628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez Sanchez MI, Mercer TR, Davies SM, Shearwood A-MJ, Nygård KK, Richman TR, Mattick JS, Rackham O, Filipovska A. RNA processing in human mitochondria. Cell Cycle 2011; 10:2904-16; PMID:21857155; http://dx.doi.org/ 10.4161/cc.10.17.17060 [DOI] [PubMed] [Google Scholar]
- 11.Vanhée-Brossollet C, Vaquero C. Do natural antisense transcripts make sense in eukaryotes? Gene 1998; 211:1-9; PMID:9573333; http://dx.doi.org/ 10.1016/S0378-1119(98)00093-6 [DOI] [PubMed] [Google Scholar]
- 12.Osato N, Suzuki Y, Ikeo K, Gojobori T. Transcriptional interferences in cis natural antisense transcripts of humans and mice. Genetics 2007; 176:1299-306; PMID:17409075; http://dx.doi.org/ 10.1534/genetics.106.069484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Sun H, Fei Z, Zhan F, Gong X, Gao S. Fastq_clean: An optimized pipeline to clean the Illumina sequencing data with quality control. Bioinformatics and Biomedicine (BIBM), 2014 IEEE International Conference on: IEEE, 2014:44-8; Conference 2014; 2014:44-8; http://dx.doi.org/ 10.1109/BIBM.2014.6999309 [DOI] [Google Scholar]
- 14.Wang Y, Wang Z, Chen X, Zhang H, Guo F, Zhang K, Feng H, Gu W, Wu C, Ma L, et al.. The complete genome of brucella suis 019 provides insights on cross-species infection. Genes 2016; 7:7; PMID:26821047; http://dx.doi.org/ 10.3390/genes7020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao Q, Li A, Chen J, Sun Y, Tang J, Zhang A, et al.. Transcriptome sequencing of the sweet potato progenitor (Ipomoea Trifida (HBK) G. Don.) and discovery of drought tolerance genes. Trop Plant Biol 2016; 9:63-72; http://dx.doi.org/ 10.1007/s12042-016-9162-7 [DOI] [Google Scholar]
- 16.Wang F, Sun Y, Ruan J, Chen R, Chen X, Chen C, Kreuze JF, Fei Z, Zhu X, Gao S. Using small RNA deep sequencing to detect human viruses. BioMed Res Int 2016; 2016:2596782; PMID:27066498; http://dx.doi.org/ 10.1155/2016/2596782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wickham H. ggplot2: elegant graphics for data analysis. Springer Science & Business Media, 2009. [Google Scholar]
- 18.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, et al.. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 2012. Apress Media LLC, 233 Spring St, New York, NY 10013, USA; 14:193-202; PMID:22445902; http://dx.doi.org/ 10.1093/bib/bbs012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.