

Summary
Sepsis, also known as septicemia, is one of the 10 leading causes of death worldwide. The rising tide of sepsis due to bacterial, fungal and viral infections cannot be stemmed by current antimicrobial therapies and supportive measures. New paradigms for the mechanism and resolution of sepsis and consequences for sepsis survivors are emerging. Consistent with Benjamin Franklin's dictum ‘an ounce of prevention is worth a pound of cure’, sepsis can be prevented by vaccinations against pneumococci and meningococci. Recently, the NIH NHLBI Panel redefined sepsis as ‘severe endothelial dysfunction syndrome in response to intravascular and extravascular infections causing reversible or irreversible injury to the microcirculation responsible for multiple organ failure’. Microvascular endothelial injury underlies sepsis‐associated hypotension, edema, disseminated intravascular coagulation, acute respiratory distress syndrome and acute kidney injury. Microbial genome products trigger ‘genome wars’ in sepsis that reprogram the human genome and culminate in a ‘genomic storm’ in blood and vascular cells. Sepsis can be averted experimentally by endothelial cytoprotection through targeting nuclear signaling that mediates inflammation and deranged metabolism. Endothelial ‘rheostats’ (e.g. inhibitors of NF‐κB, A20 protein, CRADD/RAIDD protein and microRNAs) regulate endothelial signaling. Physiologic ‘extinguishers’ (e.g. suppressor of cytokine signaling 3) can be replenished through intracellular protein therapy. Lipid mediators (e.g. resolvin D1) hasten sepsis resolution. As sepsis cases rose from 387 330 in 1996 to 1.1 million in 2011, and are estimated to reach 2 million by 2020 in the US, mortality due to sepsis approaches that of heart attacks and exceeds deaths from stroke. More preventive vaccines and therapeutic measures are urgently needed.
Keywords: genome, infection, inflammation, microcirculation, septic shock
‘The main factor in all inflammatory states consists in a lesion of the vessels which are attacked by the irritating cause.’
(Elias Metchnikoff, 1845–1916) 1
Current and emerging definitions of sepsis
Consistent with Metchnikoff's prescience, the key event in inflammation as a mechanism of disease is damage to blood vessels attacked by an ‘irritating cause’. Runaway infections damage microcirculation, in which endothelium represents the main interface for blood‐tissue exchange. Microcirculation comprises the smallest blood vessels, the pre‐capillary arterioles, capillaries and post‐capillary venules, embedded in the body's organs, thereby regulating blood flow, tissue perfusion and oxygenation, blood pressure and tissue temperature. Microvascular endothelium constitutes the largest surface area of human circulation 2 (Fig. 1). Hence, infections that cause a lesion in microcirculation can potentially compromise the function of multiple organs, including the lungs, heart, liver, gut, kidneys and brain, leading to hypotension and myocardial dysfunction, microvascular leak, thrombocytopenia without or with disseminated intravascular coagulation (DIC), acute respiratory distress syndrome (ARDS), acute kidney injury (AKI) and acute brain injury. The common denominator for multi‐organ dysfunction in sepsis is endothelial microvascular injury 3, 4, 5, 6.
Figure 1.
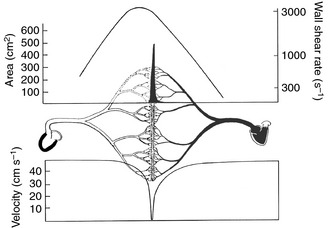
A global view of circulation. Arterial circulation is on the left and venous on the right. Microcirculation comprises the greatest endothelial surface area (left axis of the upper panel), as represented by the central solid peak. The upper panel right axis represents wall shear rate (s−1), with the curve reaching its highest values on the arterial side of microcirculation (pre‐capillary arterioles). The lower panel comprises the velocity of blood flow, and indicates that the slowest values correlate with post‐capillary venules, the terrain of white blood cell emigration (adapted from 2).
Therefore, the Working Group on Blood Systems Response to Sepsis convened by the NIH NHLBI Division of Blood Diseases and Resources in 2010 redefined sepsis as a severe endothelial dysfunction syndrome in response to intravascular and extravascular infections leading to reversible or irreversible injury to microcirculation responsible for multiple organ failure 7, 8. This definition emphasizes the central role of microvascular endothelial injury in systemic (intravascular) or localized (extravascular, e.g. urosepsis) infections by bacterial, fungal or viral agents that damage the integrity of microcirculation in multiple organs. It was developed amid growing dissatisfaction with the existing definition, proposed in 1992, which was ‘a systemic inflammatory response to infection’ (SIRS), popularly described as a ‘cytokine storm’ 9. The 1992 definition has been repeatedly criticized as non‐specific 10, 11, while a decade later an attempted change was not introduced 12. ‘Systemic inflammatory response’ is the mechanism of many acute and chronic diseases caused by non‐microbial ‘irritating causes’, such as autoimmune, metabolic or physical insults. Yet, these patients usually do not exhibit the hypotension characteristic of septic shock. Hence, SIRS does not adequately explain the fundamental nature of sepsis, which is caused by insufficiently controlled bacterial, fungal and viral infections manifested by impairment or collapse of microcirculation. The collapse, known as septic shock, underlies multiple organ failure, culminating in hypotension that is refractory to resuscitation measures recommended by the Surviving Sepsis Campaign 13.
How microbial agents damage microcirculation and cause sepsis – from Ebola to MRSA
Microbial injury to microcirculation can commence through direct invasion of endothelial cells by microbial agents or indirectly through attack by their products (‘virulence factors’). For example, the Ebola virus employs a virion glycoprotein that preferentially binds to endothelial cells, causing their death within 12–16 h 14. To add insult to injury, the virus deploys VP24 protein to disarm the host's innate immune response and sabotages antiviral antibody production by expressing a defensive decoy made of truncated virion glycoprotein 15, 16. Clearly, the Ebola virus, in this confrontation with the human host, outsmarts its genome. The Ebola virus 17, along with its ‘relative’, the Marburg virus, and rickettsiae, which cause epidemic typhus and Rocky Mountain spotted fever, demonstrates astonishingly effective virulence directed against microvascular endothelium to rapidly produce signs of septic shock.
Other microbes cause indirect damage to microvascular endothelium. In the absence of adequate antibiotics, bacteria rapidly proliferate to reach a quorum that bursts with virulence factors. These factors, such as anthrax toxins and cytolytic toxins from methicillin‐resistant Staphylococcus aureus (MRSA), penetrate the cell membrane of endothelial (and epithelial) cells and kill them. Other staphylococcal and streptococcal virulence factors (e.g. protein A, protein G, clumping factor and streptokinase) interact with plasma proteins (immunoglobulin G Fc fragment, fibrinogen, plasminogen and von Willebrand factor), disarming phagocytosis, counteracting antibody responses and causing tissue necrosis 18, 19, 20. Lipopolysaccharide (LPS, ‘endotoxin’) is a very potent proinflammatory virulence factor of Gram‐negative bacteria 21, the cause of sepsis in two‐thirds of patients either alone or in combination with other microbes 22. Fungi and viruses have also been found to play an increasingly important role in sepsis etiology 22. These microbial agents are sensed by toll‐like receptors (TLRs), which are the mainstay of innate immunity and inflammation 23 (see below).
The damage to microvascular endothelium is aggravated by host‐produced inflammatory mediators: complement, cytokines, chemokines, adhesion molecules, inducible cyclooxygenase 2 (COX2) and nitric oxide (NO) synthase metabolites. In addition, host endogenous products (HEPs) released from human blood and vascular cells (e.g. cell‐free hemoglobin 24, high mobility group box 1 protein 25, histones 26 and neutrophil extracellular traps 27) are detrimental. Cumulatively, they can inflict a deadly blow to microvascular endothelial cells through ‘anoikis’, apoptosis of endothelial cells detached from extracellular matrix. These ‘homeless’ cells can be detected and counted in the circulation 28, being a potential source of genomic information.
Persistent hypotension (vasoplegia) and microvascular leak in sepsis
A drop in blood pressure associated with incipient or ongoing infection, manifested by fever, chills and prostration, represents a hallmark of sepsis, prompting vigorous resuscitation measures and rapid administration of empiric antimicrobial therapy until the infectious agent is identified 29, 30. Hypotension in the setting of sepsis reflects endothelial instability manifested by hyporesponsiveness to catecholamines 31. Production of NO by endothelial NO synthase (eNOS), and especially inducible NO synthase (iNOS), and generation of prostacyclin by cyclooxygenase (COX) 2 are proposed as the main triggers of hypotension in sepsis. However, NOS inhibitors were not uniformly effective in improving sepsis‐induced hypotension 32.
The blood‒tissue barrier comprises microvascular endothelial cells and extracellular matrix. Tight junctions and adherence junctions keep endothelial cells together 33. Microvascular leak results from the direct action of microbial virulence factors (e.g. LPS 4, staphylococcal alpha toxin 34 or Ebola virus glycoprotein 14), which produce gaps in these junctions. Hence, blood plasma escapes and causes edema, one of the five cardinal signs of inflammation 33. In addition, overproduced pleiotropic cytokine interleukin (IL)‐6, chemokine monocyte chemotactic protein 1 (MCP‐1) 35 and vascular endothelial growth factor (VEGF), known as ‘vascular permeability factor’, open up endothelial adherence junctions by uncoupling the VE‐cadherin‐p120 catenin complex. The blood protein Slit, recognized by endothelial Robo receptor 4, stabilizes endothelial junctions 5. Recombinant Slit protein increased survival in polymicrobial sepsis whereas cytokines remained elevated. Similarly, selective targeting of transcription factor nuclear factor kappa B (NF‐κB) in endothelial cells prevented microvascular leak in polymicrobial sepsis and endotoxemia, thereby underscoring the central role of microvascular endothelial cytoprotection in experimental sepsis attenuation 4, 5.
The human genome's ‘weak spots’ for sepsis
The outcome of sepsis caused by the Sudan species of the Ebola virus has been linked to the human leukocyte antigen‐B locus 36. Alleles B*67 and B*15 were associated with fatal outcomes whereas B*07 and B*14 predicted survival, indicating that the polymorphic genome region, which encodes HLA proteins, may determine the outcome of sepsis. Mutations in human genes encoding IRAK‐4 (49 patients), MyD88 (22 patients), NF‐κB essential modulator (NEMO, 100 patients) or IkBα (five patients) are linked to microbial infections, some of them lethal 37. They encompassed a wide spectrum of bacteria, environmental mycobacteria, fungi (Candida), pneumocystis and viruses. The new paradigm of inborn errors of immunity formulated by J. L. Casanova and his collaborators was further corroborated by discovery of at least 34 new gene defects responsible for immunodeficiency 38. They teach us that not only defects in adaptive (‘specific’) immunity but also defects in innate (‘non‐specific’) immunity predispose to sepsis. Thus, genomic diagnosis, which is increasingly affordable, can be broadly applied to prevention and precise treatment of sepsis 39.
Lifting the curse of sepsis: vaccinate!
It is clear that the best hope against the potentially deadly sepsis caused by the Ebola virus is vaccination. Such vaccines are imminent 40. Vaccines against other causes of sepsis are more difficult to come by. However, Streptococcus pneumoniae, which is responsible for pneumococcal sepsis, which complicates pneumonia, asplenia and sickle cell anemia, is being contained by a pneumococcal vaccine primarily administered to children 41. A striking reduction in hospitalization for pneumonia and its invasive complication, sepsis, was also observed among non‐vaccinated older adults, including the 65–74 and 75–84‐year‐old age groups, a compelling example of ‘herd immunity’! Similarly, patients with IRAK‐4 and MyD88 deficiencies should be immunized with S. pneumoniae conjugated and non‐conjugated vaccines, Haemophilus influenzae conjugated vaccine, and Neisseria meningitides conjugated and non‐conjugated vaccines 37. A vaccine against group B Neisseria meningitides reduced the incidence of meningococcal sepsis, manifested by Purpura fulminans, one of the most challenging forms of sepsis to treat. Despite antibiotic therapy, it often causes hearing loss, neurologic damage and loss of limbs due to amputation in young survivors 42.
An anti‐staphylococcal combination vaccine promises a new measure of protection against staphylococcal sepsis in immune‐compromised patients 34. Staphylococcal protein A subverts the ability of anti‐staphylococcal vaccines to mount an effective antibody response, again giving microbes the upper hand in this battle of the genome wars 18.
Genome wars in sepsis
Increasingly belligerent microbial agents unleash their genomic prowess to challenge potentially weak spots in human genome immune programming. The concept of genome wars in sepsis includes confrontation between the microbial genome that encodes virulence factors and the human genome that responds by immune reprogramming, and their stochastic interactions (Fig. 2) 8. These interactions encompass: (i) variable expression of host proteins and microbial virulence factors that subvert the host's innate immunity, and (ii) variable time elapsing from incipient infection to effective clearance of causative microbial agents. This clearance is executed by TLR‐mediated and antibody/complement‐mediated phagocytosis, supported by very early antimicrobial therapy 29, 30.
Figure 2.
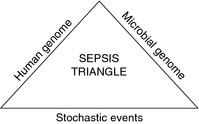
A conceptual depiction of the ‘sepsis triangle’. The outcome of the confrontation between the human genome and the microbial genome depends on stochastic events that represent ‘a black box’ in the sepsis conundrum. These events are influenced by variable expression of genes in both human and microbial genomes, as well as by variations in the time course of these genomic responses (adapted from 8).
The host genome's immune reprograming in response to infections is orchestrated by signaling to the nucleus. The first checkpoint in this signaling comprises TLRs responsible for activation of phagocytes, non‐phagocytic innate lymphoid cells and endothelial cells by LPS and other ligands (IL‐1β, IL‐18, bacteria, fungi and viral nucleic acids) (Fig. 3). The ‘family of five’ adaptors led by the myeloid differentiation primary response 88 (MyD88) ‘staffs’ this checkpoint 23, 43. Signals generated by different TLRs are integrated into the second checkpoint, downstream of the MyD88 family. It is a nuclear transport ‘relay station’ comprised of nuclear transport adaptors, termed importins/karyopherins alpha and beta (importins α and β). They shuttle NF‐κB, activator protein‐1 (AP‐1) and other stress‐responsive transcription factors (SRTFs) to the nucleus. Once inside the nucleus, SRTFs are freed to bind their cognate promoters and initiate gene transcription, leading to genome reprogramming from a resting to an activated state. SRTFs, either alone or in various combinations, regulate the genomic response to microbial agents, as well as to signaling pathways emanating from cytokine/chemokine receptors 44. In addition, the nuclear transport checkpoint integrates metabolic signals conveyed by sterol regulatory element‐binding proteins (SREBPs) shuttled solely by importin β1 45.
Figure 3.
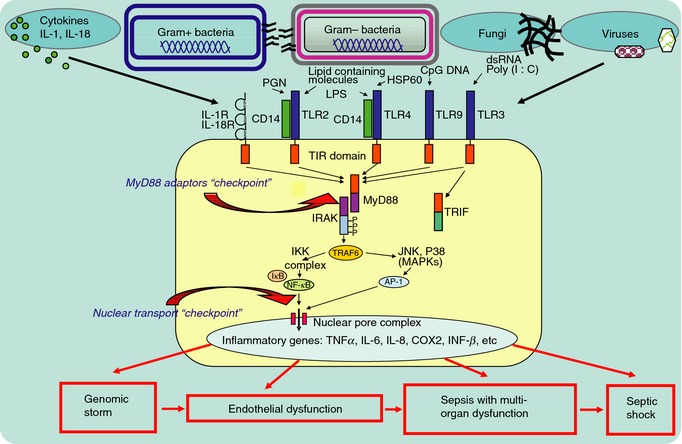
Sepsis‐causing microbes are sensed by toll‐like receptors (TLRs), the mainstays of innate immunity. These receptors are expressed on immune and non‐immune cells (endothelial and epithelial cells). Signals generated upon encounter with invading microbes are conveyed by innate immunity adaptors that belong to the MyD88 family of five (the first signaling checkpoint). Subsequent activation of proinflammatory stress responsive transcription factors (SRTFs), including nuclear factor kappa B (NF‐κB), activator protein‐1 (AP‐1, comprised of cFOS and cJUN) and others, allows them to be ferried to the nucleus by nuclear transport shuttles (a second signaling checkpoint). These specialized proteins, termed importins/karyopherins alpha and beta, loaded with their SRTF cargos, are poised to cross nuclear pores. In the nucleus, SRTFs find their cognate sites in the promoter regions of multiple genes that encode mediators of inflammation and apoptosis. Massive activation of these genes, termed a ‘genomic storm’, follows. The wave of mediators of endothelial damage hits the microcirculation, causing endothelial dysfunction, injury and apoptosis. Sepsis with multi‐organ dysfunction ensues, culminating, in some cases, in septic shock.
The ‘genomic storm’ and its consequences
In response to virulence factors encoded by microbial genomes, the human genome expresses or represses a plethora of genes. Up‐regulated genes encode inflammatory cytokines and chemokines, signal transducers (COX2 and NOS, both mediators of vascular hyporeactivity) and cell adhesion molecules. The concept of a ‘genomic storm’, originally based on the response of white blood cells to trauma and burns in critically injured patients was extended to encompass human volunteers responding to the extremely low doses of endotoxin (2–4 ng kg−1). Therefore, it represents a fundamental human response to severe inflammatory stress 46.
A tidal wave of up‐regulated gene products raises their levels in blood and vascular cells. They include cytokines and chemokines and other mediators of microvascular injury. Subsequently, IL‐6, IL‐1β and TNFα induce an ‘acute phase’ protein response characterized by a burst of protein synthesis in the liver involving C‐reactive protein (CRP), serum amyloid proteins, coagulation proteins, fibrinogen and factor VIII, and complement proteins. CRP is a widely used biomarker of systemic and localized inflammation.
Chemokines induce increased trafficking of neutrophils, monocytes and macrophages to infected tissue. Activated neutrophils and monocytes penetrate the blood‒tissue barrier and produce reactive oxygen intermediates (‘free radicals’) and isoprostanes 47. They potentiate damage to endothelium, which then loses its barrier function and anticoagulant mechanism. The latter is mediated by the thrombomodulin–protein C axis 48. Thus, a ‘genomic storm’ disturbs the microvascular homeostasis maintained in endothelial cells by physiologic suppressors of inflammation and coagulation. Multiple organ dysfunction ensues, culminating in persistent hypotension despite adequate fluid resuscitation along with vasopressors (septic shock). Perfusion abnormalities include lactic acidosis, oliguria and an acute alteration in mental status 49. Some of these changes are linked to mitochondrial dysfunction 50.
Intracellular signaling pathways in endothelial cells, as well as immune cells involved in the sepsis mechanism, are also controlled by short, non‐coding RNAs, known as microRNAs (miRNAs). They regulate gene expression by repressing translation or degrading mRNA. The extracellular miR‐223 transported by high‐density lipoproteins is delivered to endothelial cells to reduce expression of intercellular adhesion molecule 1 and granulocyte‐macrophage colony stimulating factor 2 51. Other miRNAs, such as miR‐181b, have been linked to regulation of expression of nuclear transport shuttles (e.g. importin α3) in sepsis 52.
Calming the ‘genomic storm’
Rapid initiation of antimicrobial therapy in patients with hypotension, fever and other signs of infection is crucial in limiting its runaway course 29, 30. This is accompanied by fluid replacement and vasopressors 13. Novel methods of microbial detection based on fluorescent in situ hybridization, matrix‐assisted laser desorption/ionization‐time‐of‐flight mass spectrometry and real‐time polymerase chain reaction allowed a turnaround time of 30–150 min for staphylococci and their antibacterial resistance elements 53. Thus, antimicrobial treatment in the first ‘golden hour’, including pre‐hospital settings, is the goal to avert widespread microvascular changes in patients suspected of harboring sepsis‐causing infections. Insidiously, virulence factors (e.g. LPS, staphylococcal and streptococcal toxins, anthrax toxins) can remain in microcirculation following initiation of antimicrobial therapy and continue to target blood cells and microvascular endothelial cells 54, 55. Disarming the microbial genome with small molecule compounds to suppress expression of its virulence factors, such as inhibiting streptokinase expression in group A streptococci, is a new paradigm in antimicrobial genomic therapy 56.
A parallel concept is to calm the host's ‘genomic storm’ by targeting the nuclear transport checkpoint (Fig. 3), 21, 45. This approach would counteract uncontrolled production of potentially harmful inflammatory mediators (TNFα, interferon gamma, IL‐6, IL‐8 and MCP‐1) that appear in plasma within 90–120 min after LPS administration in human volunteers 57. As shown in Fig. 3, signaling pathways emanating from LPS‐stimulated TLR4, other TLRs, and from subsequently stimulated cognate receptors for cytokines and chemokines converge at the nuclear transport checkpoint 44. Importins α and β shuttle SRTFs and SREBPs through nuclear pores. In the nucleus, SRTFs bind and activate the regulatory elements in at least 46 human genes encoding mediators of inflammation 21. SREBPs, master regulators of lipid homeostasis, activate >30 genes encoding proteins responsible for synthesis and uptake of cholesterol, fatty acids and triglycerides 45. The concept of targeting this key checkpoint for SRTFs and SREBPs has been tested by using nuclear transport modifiers (NTMs) such as cSN50.1, a highly soluble 28 amino acid cell‐penetrating peptide 45 with dual specificity toward importin α5 and importin β1 58. NTMs sufficiently reduce nuclear translocation of transcription factors, thereby attenuating the hyperinflammatory and hypermetabolic responses underlying microvascular injury 21, 45, 59, 60. NTM is able to extinguish production of multiple inflammatory mediators at once. In contrast, monoclonal antibodies can neutralize only a single target (e.g. TNFα), which is not enough to attenuate human sepsis though effective in some animal models 32. Significantly, NTM attenuated plasma levels of 23 out of 26 LPS‐induced proinflammatory cytokines, chemokines and growth factors, and dramatically increased survival in a murine model of LPS‐induced systemic microbial inflammation (lethal endotoxic shock) (Fig. 4). Some of these suppressed mediators (e.g. GM‐CSF) are produced by innate response activator B cells that either protect from or, paradoxically, contribute via IL‐3 to experimental and clinical sepsis 61, 62. Moreover, NTM reduced neutrophil trafficking to lungs and suppressed production of chemokines, cytokines and VEGF in LPS‐challenged lungs 21. In other studies, NTM (cSN50) reduced intravascular platelet thrombi, improved thrombocytopenia and normalized fibrin‐related markers and plasminogen activator inhibitor‐1 in LPS‐induced hemorrhagic necrosis and apoptosis of the liver 60, and reduced microvascular leak caused by staphylococcal superantigen SEB 59. The concept of targeting nuclear transport of SRTFs was extended to the control of experimental sepsis associated with airway infection by anthrax spores 54.
Figure 4.
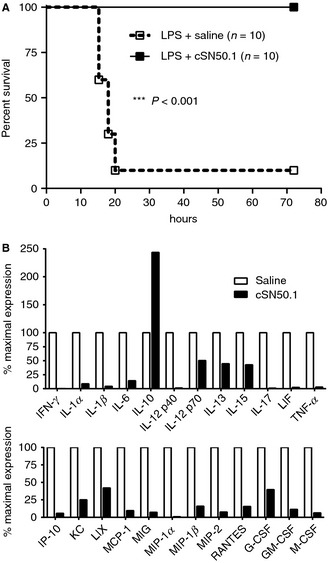
Calming the ‘genomic storm’ induced by lipopolysaccharide (LPS). Nuclear transport modifier (NTM) was administered intraperitoneally to mice before and after a high dose of LPS. (A) Modulation of nuclear transport of SRTFs by NTM enhances survival from lethal endotoxic shock. (B) Twenty‐three cytokines, chemokines and hemopoietic growth factors in plasma are reduced by NTM. Notably, expression of the anti‐inflammatory cytokine IL‐10 was enhanced by NTM (adapted from 21).
The metabolic profile of sepsis non‐survivors is marked by metabolites linked to fatty acid transport and β‐oxidation, and gluconeogenesis 63. These pathways depend on transcriptional regulation, not only by peroxisome proliferator‐activated receptors α, β and γ, but also by SREBPs, which are reduced in the nucleus by NTM 45. Thus, attenuating genomic derangements of inflammatory, coagulant and metabolic pathways with NTMs offers a new strategy for combating sepsis (Fig. 5).
Figure 5.
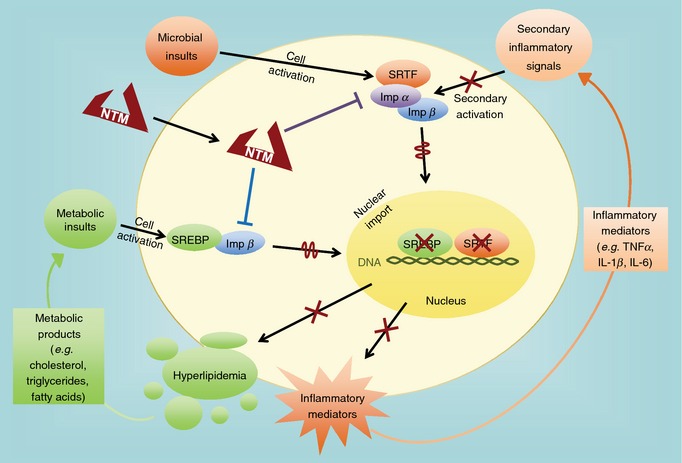
Nuclear transport of stress‐responsive transcription factors (SRTFs) and sterol regulatory element‐binding proteins (SREBPs) is modulated by a dual specificity NTM, cSN50.1 peptide, which binds nuclear import proteins (importin α5 and importin β1, respectively). Attenuation of nuclear transport of SRTFs (as shown by wiggly line) reduces production of inflammatory mediators (cytokines, chemokines as shown by X). Expression of their target genes, induced by a feed‐forward activation loop, is likewise suppressed (as shown by X). Attenuation of nuclear transport of SREBPs (as shown by wiggly line) reduces expression of their target genes (as shown by X) that encode proteins involved in synthesis of cholesterol, triglycerides and fatty acids. Both groups of transcription factors, SRTFs and SREBPs, contribute to the ‘genomic storm’ induced by endotoxin and other microbial virulence factors involved in sepsis (for details see 21 and 45).
Thrombocytopenia in sepsis
A drop in blood platelets to less than 150 000 uL−1 has long been recognized as a hallmark of Gram‐negative bacteremia and ensuing sepsis 55. Thrombocytopenia in sepsis may be centered in bone marrow suppression of platelet‐producing megakaryocytes with a decreased immature platelet fraction, or peripherally be due to trapping of circulating platelets in zones of endothelial injury 64. Platelets are also susceptible to cytolytic microbial toxins or the membrane attack complex of complement activated by microbial agents 65. Heparin may induce antibodies against platelet factor 4, a hallmark of heparin‐induced thrombocytopenia, and antimicrobial therapy may lead to drug‐induced thrombocytopenia 66. In a retrospective study of 304 patients (mean age 68.8 ± 15.8 years) with severe sepsis or septic shock, 47.6% developed thrombocytopenia, which was drug induced in 17.9% of patients 66. Significantly, thrombocytopenic patients suffered more episodes of bleeding and were more prone to AKI and ARDS. Moreover, they had elevated serum lactic acid and prolonged requirement for vasopressors, suggesting more severe microvascular endothelial dysfunction.
DIC in sepsis
Rapid and widespread microbial injury to microvascular endothelium in sepsis sets the stage for DIC, reported in 37% of 145 patients with thrombocytopenia as a complication of severe sepsis and septic shock, diagnosed in 304 patients 66. In another study using the International Society on Thrombosis and Haemostasis DIC score to evaluate 40 patients with severe sepsis or septic shock, 95% had fibrin‐related markers (fibrin monomer and D‐dimer) 67. Therefore, the importance of prolonged prothrombin time and thrombocytopenia in predicting disease severity and survival was highlighted.
While the mechanism of DIC remains enigmatic, recent advances indicate that injury to endothelium directly activates the coagulation cascade through assembly of a prothrombinase complex linked to phosphatidyl serine exposed on injured endothelial cell membrane 68. This mechanism of thrombin formation in microcirculation seems to obviate the need for tissue factor pathway that was unsuccessfully targeted in clinical sepsis trials 32. LPS‐induced thrombocytopenia and DIC with microvascular fibrin deposition were independent of thrombin‐induced platelet stimulation and the associated procoagulant activity, as was observed in mice deficient in protease activated receptor (PAR)‐1, ‐2 and ‐4 and PAR‐2/PAR‐4 and PAR‐1/PAR‐2 69. Thrombin generated on the surface of injured endothelium activates an physiologic anticoagulant mechanism that is vested in the thrombomodulin‐protein C axis 48. Surprisingly, in humans, a supra‐normal dose of protein C zymogen concentrate had no significant effect on LPS‐induced biomarkers of coagulation, fibrinolysis and inflammation, whereas significant protein C activation and production of tumor necrosis factor alpha (TNFα) were detected 70. Likewise, heparin administration remains controversial, awaiting a well‐designed prospective study. A retrospective propensity‐matched cohort study indicated that early intravenous administration of unfractionated heparin was associated with a 12% relative risk reduction in mortality 71. As only a fraction of patients with severe sepsis and septic shock displays signs of DIC, this level of effectiveness is not surprising. Similarly, clinical trials of antithrombin and thrombomodulin were ineffective 32.
Thrombin activates microvascular endothelial cells through PAR1, leading to a loss of barrier function. This signaling pathway culminates in nuclear transport of NF‐κB and AP‐1 72. They mediate production of barrier‐disrupting cytokine IL‐6 and MCP‐1 21, 35. PAR1‐derived cell‐penetrating, membrane‐tethered peptide (‘pepducin’) reduced lung vascular leakage and increased survival in a murine polymicrobial sepsis model 73. However, mice deficient in PAR‐1, PAR‐2, PAR‐4, PAR‐2/PAR‐4 and PAR‐1/PAR‐2 succumbed to LPS‐induced lethal shock 69. Imatinib, an inhibitor of the tyrosine kinase Abl‐related gene, attenuated thrombin‐induced endothelial barrier dysfunction by stabilizing cell‒matrix interactions and reduced organ edema in a murine polymicrobial sepsis model 74, while exacerbating ventilator‐induced lung injury in a mouse model 75.
Why physiologic anti‐inflammatory regulators fail in sepsis
Proinflammatory signaling in response to microbial virulence factors activates feedback systems designed to ‘put on the brakes’, encompassing extracellular and intracellular regulators. Extracellular anti‐inflammatory cytokines IL‐4, IL‐10, IL‐11, IL‐13 and IL‐1 receptor antagonist counteract the deleterious action of proinflammatory cytokines and chemokines. Their production represents a compensatory anti‐inflammatory response syndrome (CARS), which is linked to the ‘immunoparalysis’ seen in the later stage of sepsis 76. Transition to an immunosuppressive phenotype is mediated in part by transcription factor hypoxia‐inducible factor 1α in human monocytes derived from patients with sepsis 77.
Intracellular inhibitory proteins, such as IL‐1 receptor‐associated kinase (IRAK)‐M, inhibitors of NF‐κB (IκB), suppressors of cytokine signaling (SOCS) and A20 protein, which regulates the NF‐κB pathway, have evolved to limit the duration and strength of proinflammatory signaling pathways emanating from TLRs and cytokine receptors in immune and non‐immune cells (e.g. endothelial cells). Their role in the host response to endogenous or exogenous microbial insults in experimental models is emerging. Selective expression of degradation‐resistant transgenic IκBα in endothelial cells was remarkably protective in a polymicrobial sepsis model 4.
A20 protein protects endothelial and epithelial cells from microbial inflammation in multiple organs. A20 protein deficiency causes dissemination of commensal intestinal microbes (gut microbiome) through the intestinal barrier due to uncontrolled proinflammatory signaling from TLRs and the MyD88 adaptor axis 78. This signaling culminates in nuclear transport of NF‐κB and other SRTFs that are responsible for a ‘genomic storm’ 21.
Another newer endothelial ‘rheostat’, caspase and receptor interacting protein adaptor with death domain (CRADD/RAIDD), controls signaling from TLRs and G protein‐coupled receptors activated by LPS and thrombin, respectively. This signaling is dependent on cytoplasmic B‐cell lymphoma/leukemia 10 (BCL10) in immune and non‐immune cells 35.
SOCS 1 and SOCS 3 target cytoplasmic segments of cytokine receptors and/or Janus kinases for ubiquitin‐mediated proteosomal degradation and are consumed in the process by the same mechanism 79. Intracellular protein therapy with bioengineered cell‐penetrating (CP)‐SOCS3 was developed to replenish intracellular stores of endogenous SOCS3. Microvascular injury (apoptosis and hemorrhagic necrosis) caused by LPS or superantigenic staphylococcal enterotoxin B in murine liver was suppressed, proving the concept of relative depletion of physiologic anti‐inflammatory regulators 80. CP‐SOCS3, with an extended half‐life (up to 29 h) 81, further expands the potential of intracellular protein therapy for sepsis.
The brain in sepsis and long‐term consequences for survivors
Scenes from the sepsis battlefield in intensive care units (ICUs) are especially harrowing when patients display delirium and fall into coma. These signs of acute brain dysfunction are linked to microvascular injury that may contribute to alterations in cerebral blood flow and impairment of the blood‒brain barrier. In brain endothelial cells, transforming growth factor β‐activated kinase 1 controls fever and lethargy mediated by IL‐1β 82. The measure of impaired endothelial function, known as the reactive hyperemia index, correlates with the number of days with delirium or coma 83. Likewise, disruption of the integrity of brain white matter analyzed by magnetic resonance imaging correlated with duration of delirium in the ICU and persisted for at least 3 months, leading to lower cognitive scores at 3 and 12 months 84. A groundbreaking study of 1194 sepsis survivors revealed moderate to severe cognitive impairment as compared with non‐sepsis patients 85. This outcome has profound consequences for patients, their families and societal resources for long‐term care, adding immeasurably to the high cost of sepsis.
Late‐life sepsis
Increased susceptibility to sepsis is observed in physiologic and accelerated aging, as indicated by the highest incidence and mortality above 65 years of age 49. In a study monitoring accelerated aging in neonatal progeroid syndrome, four of five patients followed for 1–7 years died due to sepsis or aspiration pneumonia 86. Accelerated aging is caused by mutations in nuclear lamins. Therefore, laminopathies, as well as defective lamin processing associated with vascular aging in the normal population, are correlated with transcriptional dysregulation, oxidative stress, inflammatory signaling, vascular smooth muscle apoptosis and accelerated atherosclerosis 87. Innate immunity adaptor sterile alpha‐ and armadillo‐ motif‐containing protein protects lamins from inflammation‐induced apoptotic degradation 88.
Sepsis resolution
The emerging concept of sepsis resolution by lipid mediators, such as resolvin D1, is linked to regulation of inflammation, reduction of apoptosis and restoration of homeostasis 89. Notably, resolvin D1 inhibits endoplasmic reticulum stress‐induced apoptosis of liver cells by reducing SREBP‐1 expression and caspase 3 activity 90. Expression of SREBPs, the transcription factors responsible for lipid homeostasis in a model of hyperlidemia, was reduced by NTM 45. Thus, NTM‐regulated expression and action of SREBPs 45, as well as reduction of endotoxin‐induced caspase 3/7 activity 60, offer potential mechanisms for cytoprotective effects of NTM in late‐stage sepsis, characterized by immunosuppression 76. Further investigation may provide a missing link between sepsis resolution and transcriptional control.
Animal models of sepsis: ‘The best laid schemes of mice and men, go oft astray’
Robert Burns in his ode ‘To a Mouse’ reflected on the relationship between himself and the rodents living in his field. Mice are widely popular for sepsis studies due to the many genetically modified strains available and their ease of handling. The surgical model of sepsis known as cecal ligation and puncture (CLP) is frequently employed 4, 5. A non‐surgical murine model of sepsis evolving from peritonitis avoids surgical wounding and the uncontrolled spillage of cecal contents into the peritoneal cavity that is inherent to CLP 91. These models have generated significant new findings, such as attenuation of multiple organ injury and improved survival by genetic ablation of NF‐κB signaling in endothelial cells 4.
In 2013, the field of experimental sepsis was stunned by a comparison of human and murine transcriptomes obtained from peripheral blood leukocytes after severe injury due to trauma and burns. The conclusion of this study, highlighted in the title, was that ‘Genomic responses in mouse models poorly mimic human inflammatory diseases’ 92. The mainstream media rapidly heralded a mass exodus of mice from biomedical research institutions, evoking deep concern among basic and translational sepsis investigators 93. A reexamination by scientists from Japan of the same datasets came to a diametrically opposed conclusion: that ‘Genomic responses in mouse models greatly mimic human inflammatory diseases’ 94, quelling the kerfuffle. However, a lack of convincing explanation from the authors of the first report for this scientific dichotomy confirms the wisdom of Robert Burns that ‘the best laid schemes of mice and men, go oft astray’.
Non‐human primates (baboons and vervets) display exceptionally high intrinsic resistance to endotoxin (>5 mg per kg body weight), whereas human volunteers respond to 2–4 ng per kg body weight, a dose that is one million times lower 57! This enormous difference in sensitivity to endotoxin, the common virulence factor of Gram‐negative bacteria that cause two‐thirds of human sepsis cases 22, may explain the therapeutic incongruence between baboons and humans in some recently tested sepsis treatments 32. In contrast, chimpanzees, like humans, are extremely susceptible to endotoxin, offering a relevant preclinical model for sepsis studies 95.
The Shock Society and the International Sepsis Forum have addressed the challenges of translational research in shock and sepsis 96 and strongly recommended greater standardization of preclinical models. This includes gender and age matching, akin to the measures employed for clinical studies of sepsis, and reflecting its most adverse impact on extreme age groups (neonatal and late‐life sepsis). Ultimately, to quote Ian Anderson, ‘but a mouse is a mouse for all that’; hence, the verification of preclinical sepsis studies must come from investigations in humans.
The rising tide and cost of sepsis
Sepsis is a major public health problem in the United States (US) and worldwide, affecting up to 19 million patients per year 49. Approximately 21 000 patients were diagnosed with sepsis in US hospitals each week in 2011 97, and approximately 20–25% of those with severe sepsis were facing death within 28 days 49. Thus, the number of sepsis victims approaches that of acute myocardial infarction and exceeds deaths from stroke. An analysis of 192 980 patients with severe sepsis in the US found that sepsis arose from a medical condition in 71% and a surgical condition in 29% 98. Cumulatively, severe sepsis undermines advances in the medical and surgical management of diseases and counteracts the trend for increased life expectancy in the US and worldwide.
Sepsis is responsible for the most expensive hospital stays in the US, often costing more than $500 000 per hospitalization. In 2011, the estimated annual cost of sepsis in the US exceeded $20 billion 97. This mounting cost, corresponding to two‐thirds of the annual US National Institutes of Health budget, did not include expenses for long‐term care of sepsis survivors who suffer life‐long developmental abnormalities (neonates) and incapacitating cognitive decline (older patients) 85, 99. Both extreme age‐groups have an unacceptably high mortality (up to 45%), and the incidence of severe sepsis in newborns doubled between 1995 and 2005. Thus, sepsis is one of the most challenging problems to prevent and treat in modern hospitals.
Conclusion
Over the centuries, the specter of plagues manifested by sepsis has been an inseparable part of human existence. Human immune defenses are continually challenged by microbiomes from within and without human body. Recent impressive advances in understanding the relationship between human and microbial genomes bring us closer to the development of new preventive vaccines and therapeutic countermeasures to be deployed for containment of sepsis. They are needed in the face of (i) widespread emergence of microbes that escape antimicrobial therapy (e.g. MRSA), (ii) drift from immunization‐acquired immunity (e.g. influenza viruses), and (iii) unsuccessful outcomes of over 100 trials in sepsis 32. Table 1 summarizes the proven and experimental antisepsis regimens discussed above. The prospect of new pan‐specific vaccines that surpass antigenic drift and vigilant precautions to reduce both hospital‐ and community‐acquired infections, combined with new cytoprotective measures for microvascular endothelium, suggest that the estimated rise of sepsis cases to 2 million in the US by 2020 100 can be reversed. Hopefully, the same will apply to stemming the rise of sepsis worldwide.
Table 1.
Compilation of antisepsis regimens used currently as preventive (A) and proven therapeutic measures (B). In addition, some experimental therapeutic measures (C) and proof‐of‐concept measures awaiting preclinical testing (D) are listed (for details see text and references cited)
| Cause of sepsis | Regimen | Outcome | Ref. |
|---|---|---|---|
| (A) Preventive measures | |||
| Streptococcus pneumoniae | Vaccination | Reduced incidence | 41 |
| Neisseria meningitides | Vaccination | Reduced incidence | 42 |
| Haemophilus influenzae | Vaccination | Reduced incidence | 37 |
| (B) Proven therapeutic measures | |||
|
Undetermined (blood culture negative) |
Empiric antimicrobial therapy + fluid resuscitation/vasopressors + respiratory therapy | Increased survival | 13 |
| Bacterial | Pathogen‐directed antimicrobial therapy + fluid resuscitation/vasopressors + respiratory therapy | Increased survival | 13 |
| Fungal | Pathogen‐directed antimicrobial therapy + fluid resuscitation/vasopressors + respiratory therapy | Increased survival | 13 |
| Viral | Pathogen‐directed antimicrobial therapy + fluid resuscitation/vasopressors + respiratory therapy | Increased survival | 13 |
| (C) Experimental therapeutic measures | |||
|
Viral (Ebola virus) |
Survivors’ blood plasma/monoclonal Ab ZMapp + fluid resuscitation/vasopressors + respiratory therapy | Increased survival | 16 |
| Polymicrobial peritonitis (mice) | Recombinant SLIT protein | Increased survival | 5 |
| Polymicrobial peritonitis (mice) | Pepducin (protease‐activated receptor 1 peptide agonist) | Increased survival | 73 |
| Polymicrobial peritonitis (mice) | Imatinib (Abl‐related gene kinase inhibitor) | Reduction of edema in multiple organs | 74 |
| Ventilator‐induced lung injury (mice) | Imatinib (Abl‐related gene kinase inhibitor) | Increased edema and inflammation in lungs | 75 |
| Bacillus anthracis spores (mice) | Pathogen‐directed antimicrobial therapy + nuclear transport modifier (cSN50) | Increased survival | 54 |
| (D) Proof‐of‐concept measures awaiting preclinical testing | |||
| – | Intracellular protein therapy with recombinant cell‐penetrating SOCS‐3 | – |
80
81 |
| – | Intracellular protein therapy with recombinant cell‐penetrating CRADD | – | 35 |
Disclosure of Conflict of Interests
J. Hawiger reports financial support from AGH Therapeutics Inc. outside the submitted work. In addition, R. A. Veach, J. Hawiger and J. Zienkiewicz have multiple issued and pending patents relating to cell‐penetrating NTM peptides and their use for anti‐inflammatory therapy. All rights are assigned to Vanderbilt University.
Acknowledgements
This article is dedicated to the memory of Robert D. Collins, Roger M. Des Prez, M. Glenn Koenig, Grant W. Liddle, David E. Rogers, Samuel I. Rapaport, Roscoe (‘Ike’) Robinson, Daniel Tosteson and Sheldon M. Wolff, our mentors, advisors and friends. We thank Y. Liu, A. Major, D. Liu, M. Hutchens, T. Fletcher, J. Wynn, K. Vickers, O. McGuinness, A. D. Giandomenico, D. Jo, Y.‐Z. Lin, D. Moore, H. Qiao, C. Sethman, T. Torgerson, and L. Wylezinski for their contributions to the ongoing and past projects conducted in the laboratory and cited in this article. We thank N. Brown, Chair of the Department of Medicine at Vanderbilt University, for her steadfast support, and G. Bernard, T. Blackwell, W. Ely, S. Joyce, Z. McGee, S. Peebles, L. Ware and A. Wheeler for inspiring discussions. The authors’ experimental work cited in this article was supported during the past two decades by the National Institutes of Health grants R01HL085833, R01AA15752, R01HL69452, R01HL069452, T32HL069765 (JH), K08DK090146, F32HL099140, F32HL084214 and F32HL087531. Core services were funded in part by scholarships from the Vanderbilt University Medical Center Digestive Disease Research Center, supported by NIH grant P30DK058404, the Vanderbilt Diabetes Research and Training Center, supported by NIH U24DK59637, and the Vanderbilt Ingram Cancer Center, supported by NIH grant P30CA68485. Additional support was provided by the Department of Medicine at Vanderbilt University, the Vanderbilt University Medical Center Immunotherapy Program, and the Vanderbilt Clinical and Translational Science Award UL1TR000445. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors apologize to those whose work was omitted from the reference list because of a limit on the number of cited references.
Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost 2015; 13: 1743–56.
Manuscript handled by: M. Levi
Final decision: P. H. Reitsma, 6 July 2015
References
- 1. Metchnikoff E. Lectures On The Comparative Pathology of Inflammation, Delivered at the Pasteur Instutute in 1891. London: Kegan Paul, 1893. [Google Scholar]
- 2. Hawiger J. Physiology of hemostasis: cellular aspects In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th edn Philadelphia, London, Toronto, Montreal, Sidney, Tokyo: W.B. Saunders Company, 1992: 1495–512. [Google Scholar]
- 3. Skibsted S, Jones AE, Puskarich MA, Arnold R, Sherwin R, Trzeciak S, Schuetz P, Aird WC, Shapiro NI. Biomarkers of endothelial cell activation in early sepsis. Shock. 2013; 39: 427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF‐kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 2008; 205: 1303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4‐dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010; 2:23ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trzeciak S, Cinel I, Phillip Dellinger R, Shapiro NI, Arnold RC, Parrillo JE, Hollenberg SM. Resuscitating the microcirculation in sepsis: the central role of nitric oxide, emerging concepts for novel therapies, and challenges for clinical trials. Academic Emergency Medicine: Official Journal of the Society for Academic Emergency Medicine. 2008;15:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. National Heart Lung and Blood Institute Working Group Report . Blood Systems Response to Sepsis [Internet]. 2010. www.nhlbi.nih.gov/meetings/workshops/bsrts.htm.
- 8. Hawiger J, Musser JM. How to approach genome wars in sepsis? Crit Care 2011; 15: 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992; 101: 1644–55. [DOI] [PubMed] [Google Scholar]
- 10. Vincent JL. Dear SIRS, I'm sorry to say that I don't like you. Crit Care Med 1997; 25: 372–4. [DOI] [PubMed] [Google Scholar]
- 11. Rhee C, Gohil S, Klompas M. Regulatory mandates for sepsis care–reasons for caution. The New England Journal of Medicine. 2014; 370: 1673–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International sepsis definitions conference. Intensive Care Med 2003; 29: 530–8. [DOI] [PubMed] [Google Scholar]
- 13. Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun‐Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, et al Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 2008; 36: 296–327. [DOI] [PubMed] [Google Scholar]
- 14. Yang ZY, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med 2000; 6: 886–9. [DOI] [PubMed] [Google Scholar]
- 15. Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, Pappu RV, Leung DW, Basler CF, Amarasinghe GK. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 2014; 16: 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong G, Kobinger GP. Backs against the wall: novel and existing strategies used during the 2014‐2015 Ebola virus outbreak. Clin Microbiol Rev 2015; 28: 593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 2004; 4: 487–98. [DOI] [PubMed] [Google Scholar]
- 18. Pauli NT, Kim HK, Falugi F, Huang M, Dulac J, Henry Dunand C, Zheng NY, Kaur K, Andrews SF, Huang Y, DeDent A, Frank KM, Charnot‐Katsikas A, Schneewind O, Wilson PC. Staphylococcus aureus infection induces protein A‐mediated immune evasion in humans. J Exp Med. 2014;211:2331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flick MJ, Du X, Prasad JM, Raghu H, Palumbo JS, Smeds E, Hook M, Degen JL. Genetic elimination of the binding motif on fibrinogen for the S. aureus virulence factor ClfA improves host survival in septicemia. Blood. 2013;121:1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 2004; 305: 1283–6. [DOI] [PubMed] [Google Scholar]
- 21. DiGiandomenico A, Veach RA, Zienkiewicz J, Moore DJ, Wylezinski LS, Hutchens MA, Hawiger J. The, “genomic storm” induced by bacterial endotoxin is calmed by a nuclear transport modifier that attenuates localized and systemic inflammation. PLoS ONE 2014; 9: e110183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. International study of the prevalence and outcomes of infection in intensive care units. JAMA: The Journal of the American Medical Association. 2009; 302: 2323–9. [DOI] [PubMed] [Google Scholar]
- 23. Beutler BA. TLRs and innate immunity. Blood 2009; 113: 1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamzik M, Hamburger T, Petrat F, Peters J, de Groot H, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care 2012; 16: R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Angus DC, Yang L, Kong L, Kellum JA, Delude RL, Tracey KJ, Weissfeld L. Circulating high‐mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med 2007; 35: 1061–7. [DOI] [PubMed] [Google Scholar]
- 26. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med 2009; 15: 1318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012; 12: 324–33. [DOI] [PubMed] [Google Scholar]
- 28. Woywodt A, Blann AD, Kirsch T, Erdbruegger U, Banzet N, Haubitz M, Dignat‐George F. Isolation and enumeration of circulating endothelial cells by immunomagnetic isolation: proposal of a definition and a consensus protocol. J Thromb Haemost 2006; 4: 671–7. [DOI] [PubMed] [Google Scholar]
- 29. Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Cheang M. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006; 34: 1589–96. [DOI] [PubMed] [Google Scholar]
- 30. Gaieski DF, Mikkelsen ME, Band RA, Pines JM, Massone R, Furia FF, Shofer FS, Goyal M. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal‐directed therapy was initiated in the emergency department. Crit Care Med 2010; 38: 1045–53. [DOI] [PubMed] [Google Scholar]
- 31. Sharawy N. Vasoplegia in septic shock: do we really fight the right enemy? J Crit Care 2014; 29: 83–7. [DOI] [PubMed] [Google Scholar]
- 32. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med 2014; 20: 195–203. [DOI] [PubMed] [Google Scholar]
- 33. Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med. 2011; 3: 88 ps25. [DOI] [PubMed] [Google Scholar]
- 34. Rauch S, Gough P, Kim HK, Schneewind O, Missiakas D. Vaccine protection of leukopenic mice against Staphylococcus aureus bloodstream infection. Infect Immun 2014; 82: 4889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiao H, Liu Y, Veach RA, Wylezinski L, Hawiger J. The adaptor CRADD/RAIDD controls activation of endothelial cells by proinflammatory stimuli. J Biol Chem 2014; 289: 21973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanchez A, Wagoner KE, Rollin PE. Sequence‐based human leukocyte antigen‐B typing of patients infected with Ebola virus in Uganda in 2000: identification of alleles associated with fatal and nonfatal disease outcomes. J Infect Dis 2007; 196(Suppl. 2): S329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK‐4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev 2011; 24: 490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Conley ME, Casanova JL. Discovery of single‐gene inborn errors of immunity by next generation sequencing. Curr Opin Immunol 2014; 30: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Collins FS, Varmus H. A new initiative on precision medicine. The New England Journal of Medicine. 2015; 372: 793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanapathipillai R, Henao Restrepo AM, Fast P, Wood D, Dye C, Kieny MP, Moorthy V. Ebola vaccine–an urgent international priority. The New England Journal of Medicine. 2014; 371: 2249–51. [DOI] [PubMed] [Google Scholar]
- 41. Griffin MR, Zhu Y, Moore MR, Whitney CG, Grijalva CG. U.S. hospitalizations for pneumonia after a decade of pneumococcal vaccination. The New England Journal of Medicine. 2013; 369: 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Help Protect Your Preteen and Teen Against Meningococcal Disease . National Center for Immunizations and Respiratory Diseases; 2014. www.cdc.gov/features/meningococcal. Accessed 21 April 2014.
- 43. O'Neill LA, Bowie AG. The family of five: TIR‐domain‐containing adaptors in Toll‐like receptor signalling. Nat Rev Immunol 2007; 7: 353–64. [DOI] [PubMed] [Google Scholar]
- 44. Hawiger J. Innate immunity and inflammation: a transcriptional paradigm. Immunol Res 2001; 23: 99–109. [DOI] [PubMed] [Google Scholar]
- 45. Liu Y, Major AS, Zienkiewicz J, Gabriel CL, Veach RA, Moore DJ, Collins RD, Hawiger J. Nuclear transport modulation reduces hypercholesterolemia, atherosclerosis, and fatty liver. J Am Heart Assoc 2013; 2: e000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Brownstein BH, Mason PH, Baker HV, Finnerty CC, et al A genomic storm in critically injured humans. J Exp Med 2011; 208: 2581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Janz DR, Bastarache JA, Rice TW, Bernard GR, Warren MA, Wickersham N, Sills G, Oates JA, Roberts LJ 2nd, Ware LB. Randomized, placebo‐controlled trial of acetaminophen for the reduction of oxidative injury in severe sepsis: the acetaminophen for the reduction of oxidative injury in severe sepsis trial. Crit Care Med 2015; 43: 534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol 2011; 73: 503–14. [DOI] [PubMed] [Google Scholar]
- 49. Angus DC, van der Poll T. Severe sepsis and septic shock. The New England Journal of Medicine. 2013; 369: 2063. [DOI] [PubMed] [Google Scholar]
- 50. Singer M. The role of mitochondrial dysfunction in sepsis‐induced multi‐organ failure. Virulence 2014; 5: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado‐Lourenco L, Levin MG, Thacker S, Sethupathy P, Barter PJ, Remaley AT, Rye KA.HDL‐transferred microRNA‐223 regulates ICAM‐1 expression in endothelial cells. Nat Commun. 2014; 5: 3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW. MicroRNA‐181b regulates NF‐kappaB‐mediated vascular inflammation. J Clin Investig 2012; 122: 1973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deck MK, Anderson ES, Buckner RJ, Colasante G, Coull JM, Crystal B, Della Latta P, Fuchs M, Fuller D, Harris W, Hazen K, Klimas LL, Lindao D, Meltzer MC, Morgan M, Shepard J, Stevens S, Wu F, Fiandaca MJ. Multicenter evaluation of the Staphylococcus QuickFISH method for simultaneous identification of Staphylococcus aureus and coagulase‐negative staphylococci directly from blood culture bottles in less than 30 minutes. J Clin Microbiol 2012; 50: 1994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Veach RA, Zienkiewicz J, Collins RD, Hawiger J. Lethality in a murine model of pulmonary anthrax is reduced by combining nuclear transport modifier with antimicrobial therapy. PLoS ONE 2012; 7: e30527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Levin J, Poore TE, Young NS, Margolis S, Zauber NP, Townes AS, Bell WR. Gram‐negative sepsis: detection of endotoxemia with the limulus test. With studies of associated changes in blood coagulation, serum lipids, and complement. Ann Intern Med 1972; 76: 1–7. [DOI] [PubMed] [Google Scholar]
- 56. Sun H, Xu Y, Sitkiewicz I, Ma Y, Wang X, Yestrepsky BD, Huang Y, Lapadatescu MC, Larsen MJ, Larsen SD, Musser JM, Ginsburg D. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc Natl Acad Sci USA 2012; 109: 3469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Andreasen AS, Krabbe KS, Krogh‐Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem 2008; 15: 1697–705. [DOI] [PubMed] [Google Scholar]
- 58. Zienkiewicz J, Armitage A, Hawiger J. Targeting nuclear import shuttles, importins/karyopherins alpha by a peptide mimicking the NFkappaB1/p50 nuclear localization sequence. J Am Heart Assoc 2013; 2: e000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu D, Zienkiewicz J, DiGiandomenico A, Hawiger J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol Ther 2009; 17: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu D, Li C, Chen Y, Burnett C, Liu XY, Downs S, Collins RD, Hawiger J. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem 2004; 279: 48434–42. [DOI] [PubMed] [Google Scholar]
- 61. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, Tiglao E, Figueiredo J‐L, Iwamoto Y, Theurl I, Gorbatov R, Waring MT, Chicoine AT, Mouded M, Pittet MJ, Nahrendorf M, Weissleder R, Swirski FK. Innate response activator B cells protect against microbial sepsis. Science (New York, Ny) 2012; 335: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, Brenner T, Uhle F, Iwamoto Y, Robbins CS, Noiret L, Maier SL, Zonnchen T, Rahbari NN, Scholch S, Klotzsche‐von Ameln A, Chavakis T, Weitz J, Hofer S, Weigand MA, et al Interleukin‐3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 2015; 347: 1260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, Chen B, Carin L, Suarez A, Mohney RP, Freeman DH, Wang M, You J, Wulff J, Thompson JW, Moseley MA, Reisinger S, Edmonds BT, Grinnell B, Nelson DR, et al An integrated clinico‐metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013; 5: 195ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tomaiuolo M, Stalker TJ, Welsh JD, Diamond SL, Sinno T, Brass LF. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood. 2014; 124: 1816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 2008; 8: 776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Venkata C, Kashyap R, Farmer JC, Afessa B. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. Journal of Intensive Care. 2013; 1: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Voves C, Wuillemin WA, Zeerleder S. International Society on Thrombosis and Haemostasis score for overt disseminated intravascular coagulation predicts organ dysfunction and fatality in sepsis patients. Blood Coagulation & Fibrinolysis: An International Journal in Haemostasis and Thrombosis. 2006; 17: 445–51. [DOI] [PubMed] [Google Scholar]
- 68. Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood 2014; 124: 1705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Camerer E, Cornelissen I, Kataoka H, Duong DN, Zheng YW, Coughlin SR. Roles of protease‐activated receptors in a mouse model of endotoxemia. Blood 2006; 107: 3912–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Spiel AO, Firbas C, Mayr FB, Leitner JM, Schmidt B, Knobl P, Varadi K, Jilma B. The effects of supra‐normal protein C levels on markers of coagulation, fibrinolysis and inflammation in a human model of endotoxemia. Thromb Haemost 2005; 94: 1148–55. [DOI] [PubMed] [Google Scholar]
- 71. Zarychanski R, Abou‐Setta AM, Kanji S, Turgeon AF, Kumar A, Houston DS, Rimmer E, Houston BL, McIntyre L, Fox‐Robichaud AE, Hebert P, Cook DJ, Fergusson DA. The efficacy and safety of heparin in patients with sepsis: a systematic review and metaanalysis. Crit Care Med 2015; 43: 511–8. [DOI] [PubMed] [Google Scholar]
- 72. Yu OM, Heller Brown J. GPCR and RhoA‐stimulated transcriptional responses mediating inflammation, differentiation and cell proliferation. Mol Pharmacol 2015; 88: 171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis‐induced vascular damage. Nat Immunol 2007; 8: 1303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, Groeneveld ABJ, Vonk Noordegraaf A, van Hinsbergh VWM, van Nieuw Amerongen GP. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 2012; 126: 2728–38. [DOI] [PubMed] [Google Scholar]
- 75. Letsiou E, Rizzo AN, Sammani S, Naureckas P, Jacobson JR, Garcia JG, Dudek SM. Differential and opposing effects of imatinib on LPS‐ and ventilator‐induced lung injury. Am J Physiol Lung Cell Mol Physiol 2015; 308: L259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013; 13: 260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez‐Jimenez E, Toledano V, Cubillos‐Zapata C, Rapisarda A, Chen J, Duan K, Yang H, Poidinger M, Melillo G, Nizet V, Arnalich F, Lopez‐Collazo E, Biswas SK. Human monocytes undergo functional re‐programming during sepsis mediated by hypoxia‐inducible factor‐1alpha. Immunity 2015; 42: 484–98. [DOI] [PubMed] [Google Scholar]
- 78. Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, Shifrin N, Malynn BA, Ma A. Homeostatic MyD88‐dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 2008; 205: 451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carow B, Rottenberg ME. SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol 2014; 5: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med 2005; 11: 892–8. [DOI] [PubMed] [Google Scholar]
- 81. Fletcher TC, DiGiandomenico A, Hawiger J. Extended anti‐inflammatory action of a degradation‐resistant mutant of cell‐penetrating suppressor of cytokine signaling 3. The Journal of Biological Chemistry. 2010; 285: 18727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ridder DA, Lang MF, Salinin S, Roderer JP, Struss M, Maser‐Gluth C, Schwaninger M. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med 2011; 208: 2615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hughes CG, Morandi A, Girard TD, Riedel B, Thompson JL, Shintani AK, Pun BT, Ely EW, Pandharipande PP. Association between endothelial dysfunction and acute brain dysfunction during critical illness. Anesthesiology. 2013;118:631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Morandi A, Rogers BP, Gunther ML, Merkle K, Pandharipande P, Girard TD, Jackson JC, Thompson J, Shintani AK, Geevarghese S, Miller RR 3rd, Canonico A, Cannistraci CJ, Gore JC, Ely EW, Hopkins RO. The relationship between delirium duration, white matter integrity, and cognitive impairment in intensive care unit survivors as determined by diffusion tensor imaging: the VISIONS prospective cohort magnetic resonance imaging study*. Crit Care Med 2012; 40: 2182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iwashyna TJ, Ely EW, Smith DM, Langa KM.Long‐term cognitive impairment and functional disability among survivors of severe sepsis. JAMA: The Journal of the American Medical Association. 2010;304:1787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hou JW. Natural course of neonatal progeroid syndrome. Pediatrics and Neonatology. 2009; 50: 102–9. [DOI] [PubMed] [Google Scholar]
- 87. Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010; 121: 2200–10. [DOI] [PubMed] [Google Scholar]
- 88. Sethman CR, Hawiger J. The innate immunity adaptor SARM translocates to the nucleus to stabilize lamins and prevent DNA fragmentation in response to pro‐apoptotic signaling. PLoS One 2013; 8: e70994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014; 40: 315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jung TW, Hwang HJ, Hong HC, Choi HY, Yoo HJ, Baik SH, Choi KM. Resolvin D1 reduces ER stress‐induced apoptosis and triglyceride accumulation through JNK pathway in HepG2 cells. Mol Cell Endocrinol 2014; 391: 30–40. [DOI] [PubMed] [Google Scholar]
- 91. Wynn JL, Scumpia PO, Delano MJ, O'Malley KA, Ungaro R, Abouhamze A, Moldawer KL. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007; 28: 675–83. [DOI] [PubMed] [Google Scholar]
- 92. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald‐Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, et al Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 2013; 110: 3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Osuchowski MF, Remick DG, Lederer JA, Lang CH, Aasen AO, Aibiki M, Azevedo LC, Bahrami S, Boros M, Cooney R, Cuzzocrea S, Jiang Y, Junger WG, Hirasawa H, Hotchkiss RS, Li XA, Radermacher P, Redl H, Salomao R, Soebandrio A, et al Abandon the mouse research ship? Not just yet!. Shock. 2014; 41: 463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2015. (published ahead of print August 4, 2014); 112: 1167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Levi M, ten Cate H, Bauer KA, van der Poll T, Edgington TS, Buller HR, van Deventer SJ, Hack CE, ten Cate JW, Rosenberg RD. Inhibition of endotoxin‐induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti‐tissue factor antibody in chimpanzees. J Clin Investig 1994; 93: 114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Marshall JC, Deitch E, Moldawer LL, Opal S, Redl H, van der Poll T. Preclinical models of shock and sepsis: what can they tell us? Shock. 2005; 24(Suppl 1): 1–6. [DOI] [PubMed] [Google Scholar]
- 97. Torio CM, Andrews RM. National inpatient hospital costs: the most expensive conditions by payer, 2011: statistical brief #160 In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs [Internet], Rockville, MD: Agency for Health Care Policy and Research (US), 2013. Aug. www.ncbi.nlm.nih.gov/books/NBK169005/. Accessed August 2013. [Google Scholar]
- 98. Angus DC, Linde‐Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29: 1303–10. [DOI] [PubMed] [Google Scholar]
- 99. Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sanchez PJ, O'Shea TM, Goldberg RN, van Meurs KP, Faix RG, et al Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 2010; 126: 443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2013; 41: 1167–74. [DOI] [PubMed] [Google Scholar]