

Summary
Diamond–Blackfan anaemia (DBA) is a rare congenital disease causing severe anaemia and progressive bone marrow failure. The majority of patients carry mutations in ribosomal proteins, which leads to depletion of erythroid progenitors in the bone marrow. As many as 40% of all DBA patients receive glucocorticoids to alleviate their anaemia. However, despite their use in DBA treatment for more than half a century, the therapeutic mechanisms of glucocorticoids remain largely unknown. Therefore we sought to study disease specific effects of glucocorticoid treatment using a ribosomal protein s19 (Rps19) deficient mouse model of DBA. This study determines for the first time that a mouse model of DBA can respond to glucocorticoid treatment, similar to DBA patients. Our results demonstrate that glucocorticoid treatment reduces apoptosis, rescues erythroid progenitor depletion and premature differentiation of erythroid cells. Furthermore, glucocorticoids prevent Trp53 activation in Rps19‐deficient cells‐ in a disease‐specific manner. Dissecting the therapeutic mechanisms behind glucocorticoid treatment of DBA provides indispensible insight into DBA pathogenesis. Identifying mechanisms important for DBA treatment also enables development of more disease‐specific treatments of DBA.
Keywords: Diamond‐Blackfan anaemia, erythroid differentiation, Trp53, glucocorticoid therapy, erythroid progenitor
Diamond–Blackfan anaemia (DBA) is a rare early‐onset red cell aplasia that is usually manifested during the first year of life (Diamond & Blackfan, 1938; Nathan et al, 1978). At diagnosis, patients present macrocytic anaemia, reticulocytopenia and a normocellular bone marrow where erythroid progenitors are scarce or absent (Vlachos & Muir, 2010). The finding of mutations in ribosomal protein s19 (RPS19) led to the notion that DBA is a ribosomopathy causing functional haploinsufficiency (Draptchinskaia et al, 1999; Farrar & Dahl, 2011). Mutations in RPS19 are found in 25% of all patients and, together with more than 10 other less frequently affected ribosomal protein genes, they cover 60–70% of all DBA cases (Gazda et al, 2004; Landowski et al, 2013; Vlachos et al, 2013). In 2012 the erythroid specific transcription factor GATA1 was found to be mutated in a small cohort of DBA patients, challenging the notion that DBA is a disease of exclusive ribosomal origin (Sankaran et al, 2012).
Diamond–Blackfan anaemia patients usually require lifelong treatment and apart from chronic blood transfusions, glucocorticoid therapy has been standard for more than half a century to alleviate the anaemia (Gasser, 1951; Vlachos et al, 2008). Up to 80% of all DBA patients respond to glucocorticoid therapy and approximately 40% remain steroid responsive over time (Vlachos et al, 2008). Despite being the only available pharmacological therapy for DBA, the therapeutic mechanisms behind glucocorticoid treatment remain elusive. It is well established that glucocorticoid receptor activation is essential for erythroid progenitor expansion during stress erythropoiesis that occurs upon excessive blood loss or chronic anaemia. Mice lacking the ability of glucocorticoid receptor dimerization fail to respond to stress erythropoiesis caused by haemolytic anaemia (Bauer et al, 1999) and self‐renewing colony assay of erythroid progenitors ex vivo requires glucocorticoids (Wessely et al, 1997; Leberbauer et al, 2005; Dolznig et al, 2006; Flygare et al, 2011). Multiple glucocorticoid‐induced genes are important for erythropoiesis, such as the RNA‐binding protein Zfp36l2, which has proved crucial for self‐renewal of erythroid progenitors (Zhang et al, 2013) and the transcription factor Myb, which is important for long‐term erythroid proliferation (Wessely et al, 1997; Lindern von et al, 1999). Glucocorticoids also delayed differentiation in both human and avian erythroid cells, which in turn led to increase in the expansion capacity of erythroid progenitors (Golde et al, 1976; Wessely et al, 1997; Lindern von et al, 1999). Thus, glucocorticoids play an important role in the stressed erythroid system and are important in DBA treatment, but less is known about their therapeutic mechanisms. In order to elucidate this, more knowledge about disease mechanisms in DBA is crucial.
Elevated TP53 activity is brought forward as one of the underlying mechanisms behind DBA pathology, and studies in human cells as well as in mice and zebrafish have shown improvement of the DBA phenotype in the absence of Trp53 (Danilova et al, 2008; Dutt et al, 2011; Jaako et al, 2011; Taylor et al, 2012). In the context of intact ribosomal proteins, erythroid cells with a Trp53 ‐/‐ background proliferate better than wild type counterparts, suggesting that Trp53 has a general inhibitory effect on erythropoiesis (Ganguli et al, 2002). It is not fully known how Trp53 is activated in ribosome protein‐deficient erythroid cells, but it is probably explained in part by stress responses due to impaired ribosomal biogenesis, known as ribosomal stress.
The aim of this study was to identify the therapeutic mechanisms of glucocorticoids in DBA by studying the response in an Rps19‐deficient mouse model and verifying these results in DBA patient cells. Our mouse model proved to be an excellent tool to validate DBA drug treatment and responded to glucocorticoids similarly to DBA patients. We found that glucocorticoid treatment prevented erythroid progenitor depletion and delayed erythroid differentiation, which led to improved expansion of Rps19‐deficient cells. The increased Trp53 activity in Rps19‐deficient cells was reversed by glucocorticoid treatment, indicating a disease specific mechanism of glucocorticoids.
Material and methods
Rps19‐deficient mouse strain, transplantation and shRNA induction
We used a doxycycline‐inducible Rps19‐deficient mouse previously developed by us, carrying a reverse transactivator (rtTA)‐dependent shRNA against Rps19 that develops robust anaemia upon doxycycline induction. For all experiments, we used the shRNA‐D mice (Jaako et al, 2011). The haematopoietic phenotype in these mice is specific to Rps19 down regulation as it can be cured by enforced expression of Rps19 (Jaako et al, 2014). 8‐ to 14‐week‐old WT C57bl/6 males were subjected to lethal irradiation (900 cGy) and transplanted with unfractionated bone marrow from Rps19+/+, rtTA/rtTA (carrying transactivator only) or D/+, rtTA/rtTA recipients (carrying transactivator and heterozygous for Rps19 shRNA). The animals received Ciprofloxacin 125 μg/ml (Arrow Generics, Stevenage, UK) for 2 weeks post‐transplantation. Six to nine weeks after transplantation the mice received doxycycline (2 mg/ml) with sucrose (10 mg/ml) in drinking water ad libitum for 9 days as well as 100 μg/ml of Prednisolone or corresponding volume of solvent in the drinking water (Sigma‐Aldrich, St. Louis, MO, USA). Peripheral blood was analysed fresh on a haematology analyser KX‐21N (Sysmex, Kobe, Japan) after 9 days of doxycycline induction.
Liquid culture and methylcellulose colony assay
Faetal livers from heterozygote D/+ E14·5‐15·5 embryos were dissected and enriched for KIT (CD117) positive cells using magnetic beads and LS columns (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturers’ instructions. The cells were kept in serum‐free expansion medium (SFEM; Stem Cell Technologies, Vancouver, Canada) with 2 u/ml erythropoietin (EPO; LEO Pharma, Ballerup, Denmark), 100 ng/ml murine stem cell factor (mSCF) (PeproTech, Rocky Hill, NJ, USA), with or without 100 nmol/l Dexamethasone (Sigma‐Aldrich), and/or 0·5‐1 μg/ml doxycycline (Sigma‐Aldrich) and kept at 37°C with 5% CO2. Cells were counted manually using trypan blue in a haemocytometer. Colony forming unit‐erythroid (CFU‐E) assays were performed using serum free semisolid MethoCult #3236 (Stem Cell Technologies) supplemented with 5 u/ml EPO and 50 ng/ml mSCF and were cultured at 37°C with 5% CO2 and 4%O2, and the colonies were scored after 4 days.
Real time‐polymerase chain reaction (RT‐PCR)For real time (RT)‐PCR analysis, RNA extraction was performed using RNeasy micro kit (Qiagen, Venlo, Netherlands) according to the manufacturers’ instructions and analysis was performed on a 7900 HT fast RT‐PCR (Applied biosystems, Waltham, MA). Primers used for RT‐PCR were from Life Technologies (Carlsbad, CA, USA): Hprt (#01545399_m1), GAPDH (#02758991_g1) Cdkn1a (#04205640_g1 and 00355782_m1), Bax (#00432051_m1 and 00180269_m1), Ccng1 (#00438084_m1), Phlda3 (#00449846_m1), Ptp4a3 (#00477233_m1) and ZFP36L2 (00272828_m1), and Zmat3 (#01292424_m1).
Flow cytometry
A modified version of an erythroid differentiation protocol was used for flow cytometric analysis (Liu et al, 2013). Antibodies used were phycoerythrin‐cyanin 7 (PECy7)‐conjugated TER119 (eBioscience, San Diego, CA, USA), and phycoerythrin (PE)‐conjugated CD44 (BioLegend, San Diego, CA, USA). Allophycocyanin (APC)‐conjugated B220, CD3 and GR‐1 (BioLegend) were used as lineage markers. 7‐aminoactinomycin D (7AAD) was used to monitor viability. The plots were gated on live cells of appropriate size, negative for lineage markers. For flow cytometric detection of apoptosis, PE‐conjugated AnnexinV apoptosis detection kit (BD biosciences, Franklin Lakes, NJ, USA) was used according to the manufacturer's instructions. Data collection was done on a Canto II flow cytometer (BD Biosciences). For cell cycle analysis, the cells were disrupted in ice‐cold ethanol and the DNA was stained with propidium iodide. Cell cycle analysis was done on Aria III flow cytometer (BD Biosciences). All analyses were performed using FlowJo v10 (Tree Star Inc., San Carlos, CA, USA).
Western blot assay
Cells cultured for 72 h were harvested in Laemmli buffer containing 1% mercaptoethanol and proteinase inhibitor cocktail, homogenized and boiled for 5 min. Samples were run on a gradient 4–12% Bis‐Tris gel (Life Technologies), and blotted according to manufacturer's instructions. The anti‐MYB antibody was from Santa Cruz Biotechnology (Dallas, TX, USA) and anti‐Actin antibody from BD Biosciences.
DBA patient cells
A human peripheral blood sample was collected after obtaining informed consent according to ethical guidelines at Universitätsklinikum in Freiburg, Germany. The patient sample was from a 14‐year‐old transfusion‐dependent male DBA patient carrying a heterozygous RPS19 mutation (c.280C>T, p.Arg94X), and healthy control samples were from one male and one female. The blood was enriched for CD34+ fraction using magnetic MicroBead Kit, according to manufacturer's instructions (Miltenyi Biotech). The cells were subsequently expanded in a 2‐phase erythroid culture to promote expansion before being cultured with or without dexamethasone for 24 h. The erythroid expansion protocol was 7 days in serum‐free medium containing 20 ng/ml thrombopoietin, 20 ng/ml Flt3‐ligand, 100 ng/ml human SCF (hSCF), 10 ng/ml interleukin 3 (Peprotech) and 1 nmol/l dexamethasone, followed by 2 days in SFEM containing 30% serum, 2 u/ml EPO and 50 ng/ml hSCF, prior to washing and stimulation in SFEM with 2 u/ml EPO, 50 ng/ml hSCF with or without 100 nmol/l dexamethasone for 24 h. RNA extraction and RT‐PCR analysis were performed as for mouse samples.
Statistical analysis
For statistical analyses the nonparametric unpaired Mann–Whitney U test or paired Wilcoxon test were employed, unless otherwise stated. All statistical calculations were done in the software GraphPad Prism 6·0 (GraphPad Software Inc., San Diego, CA, USA). All graphs display median with interquartile range, unless otherwise stated. P‐values: *≤0·05, **≤0·01, ***≤0·001, ****≤0·0001.
Microarray statistics
The microarray data were Robust Multiarray Average (RMA) normalized. Differentially expressed genes were identified using Significance Analysis of Microarray (SAM), using a ‘multi class’ analysis, where a value of delta was chosen so the resulting differentially expressed genes had a false discovery rate (FDR) of 5%. The differentially expressed genes were then clustered into nine partitions using k‐means. Gene set enrichment analysis (GSEA) was performed using gene sets defined by the following: Trp53 genes were taken from KEGG (http://www.genome.jp/kegg/). TER119 and burst forming unit‐erythroid (BFU‐E) genes were defined from RNA sequencing data http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26086 (Flygare et al, 2011). M/A values were calculated where genes with A > 3 were selected first. Of the remaining, the top and bottom 330 genes were taken from a ranked list of M (log ratios) values to define each list. GSEA was then performed using default settings.
Results
Glucocorticoids reverse anaemia in a mouse model of DBA
In our Rps19‐deficient inducible mouse model 9 days of doxycycline administration caused a robust anaemia with decreased red blood cell (RBC) counts, reduced haematocrit (HCT) and haemoglobin (HGB) levels, as previously reported (Fig 1A–C) (Jaako et al, 2011). The anaemia of Rps19‐deficient mice (D/+) was significantly improved when treated with the glucocorticoid Prednisolone, with a significant increase in RBC counts as well as HGB and HCT compared to untreated Rps19‐deficient animals. Rps19‐deficient mice that received Prednisolone also showed significantly lower MCV compared to untreated Rps19‐deficient mice (Fig 1D). As previously reported, Rps19‐deficiency led to a decrease in white blood cell counts (WBC) and platelet (PLT) counts (Jaako et al, 2011), which were further decreased in prednisolone‐treated animals (Fig S1). These results demonstrate that our Rps19‐deficient mouse model recapitulates the anaemia seen in DBA, and that these mice respond to glucocorticoid treatment similar to DBA patients with improved erythroid blood parameters.
Figure 1.
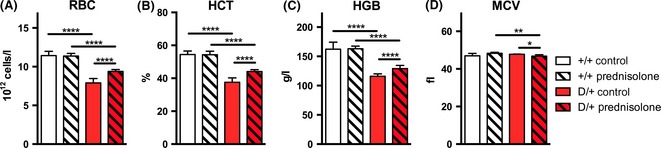
The anaemia of Rps19‐deficient mice is ameliorated by prednisolone treatment. Blood parameters of mice transplanted with +/+ (no Rps19 shRNA) or D/+ (heterozygote for Rps19 shRNA) bone marrow after 9 days of induction with doxycycline and/or prednisolone. A) Red blood cell (RBC) count, B) haemoglobin (HGB) and C) haematocrit (HCT) of D/+ mice receiving prednisolone is significantly improved. D) Mean corpuscular volume (MCV) decreases between Rps19‐deficient animals receiving Prednisolone compared to Rps19‐deficient controls. n = 6–18 for each treatment group.
Glucocorticoids improve survival and expansion of Rps19‐deficient erythroid cells
Erythroid cells from patients and DBA cell lines have shown to be pro‐apoptotic, which indicates that apoptosis plays an important role in DBA pathogenesis (Perdahl et al, 1994; Miyake et al, 2008). Therefore, we examined apoptosis in Rps19‐deficient erythroid cells when treated with the synthetic glucocorticoid Dexamethasone. As expected, we observed that Rps19‐deficient erythroid cells displayed elevated levels of apoptosis in vitro, shown by an increase in the AnnexinV+/7AAD‐ fraction. Dexamethasone treatment significantly decreased the apoptotic fraction of Rps19‐deficient cells (Fig 2A), and it also reduced apoptotic levels in healthy cells. This indicates that glucocorticoids have a pro‐survival effect on erythroid cells in general, and on Rps19‐deficient cells in particular. In addition to being more pro‐apoptotic, erythroid progenitors from both DBA patients and an Rps19‐deficient mouse model failed to expand in liquid culture (Hamaguchi et al, 2003; Miyake et al, 2005; Jaako et al, 2011). To examine the effect of glucocorticoids on the expansion capacity of Rps19‐deficient cells, erythroid expansion cultures were maintained for 7 days. The expansion of Rps19‐deficient erythroid progenitors was dramatically decreased compared to controls as early as 48 h after doxycycline administration, and did not recover during the 7‐day culture period (Fig 2B). Rps19‐deficient cells receiving Dexamethasone significantly increased their expansion and allowed the culture to be maintained for the full 7 days. As seen before, healthy control cells expanded better in the presence of Dexamethasone, indicating that glucocorticoids promote expansion of erythroid cells in general. Together, these results show that glucocorticoid treatment decreases apoptosis and promotes expansion of Rps19‐deficient erythroid cells and that these effects are more pronounced in Rps19‐deficient cells than in healthy cells.
Figure 2.
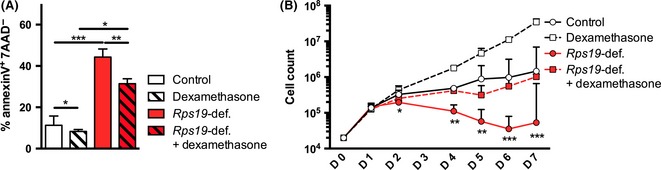
Glucocorticoid treatment improves survival and expansion of Rps19‐deficient faetal liver cells. (A) Rps19‐deficient (Rps19‐def.) erythroid cells show significant increase in apoptosis after 72 h of culture, and this is alleviated by dexamethasone treatment. The apoptotic fraction (AnnexinV+/7AAD −) is determined by flow cytometry. (B) Expansion of erythroid cells increases following dexamethasone treatment both for healthy and Rps19‐deficient cells. Cell density never exceeded 1·5 × 106 cells/ml. For clarity, only statistical significance between Rps19‐deficient and Rps19‐deficient cells treated with dexamethasone is shown.
Glucocorticoids counteract erythroid differentiation and erythroid progenitor depletion caused by Rps19‐deficiency
Glucocorticoids improve the phenotype of Rps19‐deficiency both in vivo and in vitro and we therefore wanted to identify changes in erythroid gene expression caused by glucocorticoid treatment. We performed a microarray analysis on KIT‐enriched faetal liver cells cultured for 24 h in the following conditions: Healthy cells with and without Dexamethasone and Rps19‐deficient cells with and without Dexamethasone. KIT‐enriched faetal liver cell population mostly contains burst forming unit‐erythroid (BFU‐E) and colony forming unit‐erythroid (CFU‐E) progenitors, as well as some immature erythroblasts not yet expressing TER119. Genes significantly altered by either Rps19‐deficiency or Dexamethasone treatment were mapped against previously published RNA sequencing data of healthy cells from BFU‐E or TER119+ cells from murine faetal liver (Flygare et al, 2011). In this previously published data set, the BFU‐E compartment consisted of faetal liver cells highly purified for the early erythroid progenitor BFU‐E by cell sorting. The fraction sorted on positive TER119 expression was composed of more differentiated erythroblasts. In both these fractions, the 330 most highly expressed genes were selected for GSEA (Fig S2A). Genes up regulated by Dexamethasone treatment of healthy cells were much more associated with BFU‐Es than with TER119+ cells (FDR q = 0·032), indicating that glucocorticoids promote maintenance of erythroid progenitors (Fig 3A). Several genes were up regulated by Rps19‐deficiency and these were much more associated with TER119+ cells (FDR q = 0·011) than with BFU‐Es. This was an indication that Rps19‐deficiency caused increased erythroid differentiation by up regulation of genes associated with the more mature erythroblast TER119+ fraction (Fig 3B). Comparison of Rps19‐deficient cells with or without dexamethasone treatment showed that glucocorticoid treatment caused up regulation of genes highly associated with BFU‐Es (FDR q = 0·062) (Fig 3C). Together, this demonstrates that glucocorticoids promote maintenance of erythroid progenitors and delay the increased differentiation and depletion of erythroid progenitors seen in untreated Rps19‐deficient cells. To further identify dexamethasone‐inducible genes that could be responsible for the delayed differentiation and decrease in erythroid progenitor maintenance we compared samples from the microarray analysis cultured under the following three conditions: (i) Rps19‐deficient cells, (ii) Rps19‐deficient cells + dexamethasone and (iii) Wild type controls not receiving dexamethasone. Gene clustering was performed on differentially expressed genes in these conditions. We identified two sets of differentially expressed genes that were normalized by dexamethasone (Cluster 2 and 7 in Fig S2B). Among the genes down regulated by Rps19‐deficiency but reversed by glucocorticoids, we found Kit and Myc, which both are more highly expressed in erythroid progenitors than mature erythroid cells (Suppl Table I). Another transcription factor important for erythropoiesis induced by glucocorticoids is Myb, which is most highly expressed in erythroid progenitors but down regulated with differentiation (Emilia et al, 1986). We observed a significant decrease of Myb gene expression as well as MYB protein levels in Rps19‐deficient cells observed previously (Sieff et al, 2010). At the gene expression level, glucocorticoid treatment significantly increased Myb expression to normal levels (Fig S3). Together, these results indicate that erythroid differentiation is increased in Rps19‐deficient cells and that glucocorticoid treatment counteracts this by maintaining the cells in a more immature erythroid progenitor state. These effects were observed as early as after 24 h of glucocorticoid treatment.
Figure 3.
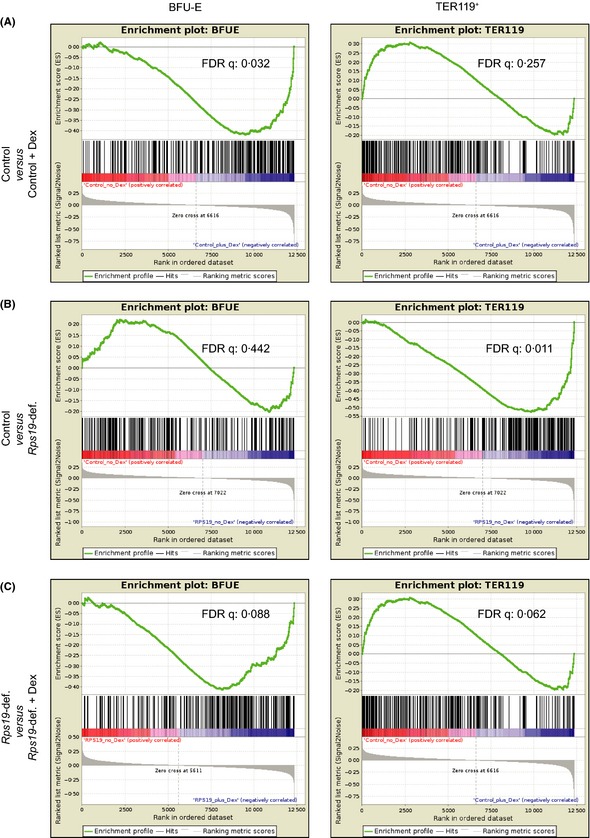
Rps19‐deficiency increases erythroid differentiation and this is counteracted by dexamethasone (Dex). GSEA was performed on differentially expressed genes from microarray analysis mapped against genes most highly expressed in BFU‐Es and TER119+ cells from faetal liver (Flygare et al, 2011). Enrichment of genes associated with BFU‐E or TER119+ erythroid fractions between (A) Control and control + dexamethasone samples, (B) Control and Rps19‐deficient samples, (C) Rps19‐deficient and Rps19‐deficient + dexamethasone samples.
Glucocorticoids maintain erythroid progenitors and delay differentiation of Rps19‐deficient cells
During the course of in vitro erythroid differentiation in liquid culture there is a progressive loss of cells with early erythroid progenitor potential, such as CFU‐E cells, and an accumulation of more mature erythroid cells expressing TER119. Our gene expression analysis indicated that glucocorticoid treatment of Rps19‐deficient cells maintained erythroid progenitors and delayed erythroid differentiation (Fig 3). To validate these findings, we examined CFU‐E colony forming potential of KIT‐enriched Rps19‐deficient and healthy faetal liver cells in liquid culture. We hypothesized that CFU‐E potential would decrease in Rps19‐deficient cultures compared to controls over time, and that glucocorticoid treatment could rescue this depletion. As expected, Rps19‐deficient cells showed decreased CFU‐E colony forming potential, starting as early as 24 h after induction with doxycycline (Fig 4A). Dexamethasone treatment significantly retained cells with CFU‐E potential in both Rps19‐deficient and healthy cultures after 72 h. This trend was observed as early as 24 h of culture. CFU‐E potential for glucocorticoid treated Rps19‐deficient cells was equal to that of healthy controls, indicating that glucocorticoids can reverse erythroid progenitor depletion significantly (Fig 4A).
Figure 4.
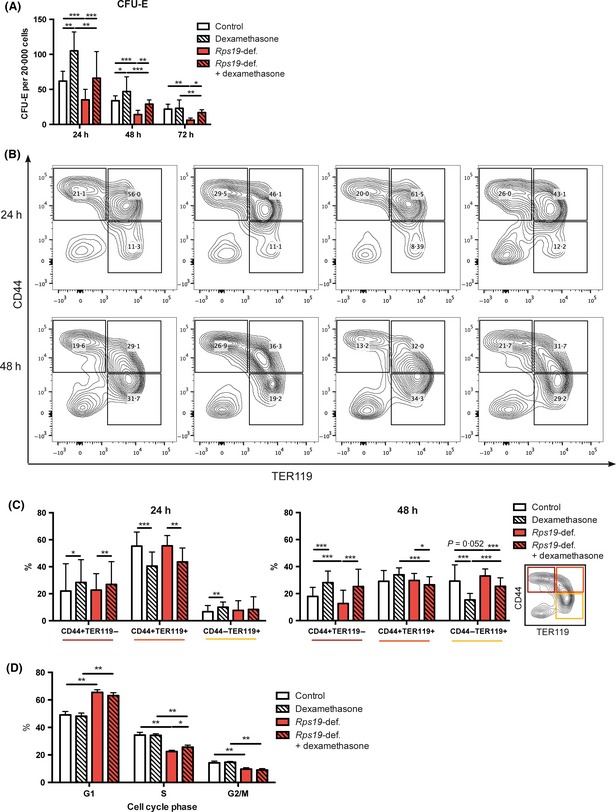
Glucocorticoids prevent erythroid progenitor loss and decrease differentiation in Rps19‐deficiency. (A) Healthy and Rps19‐deficient KIT‐enriched faetal liver cells cultured with and without dexamethasone for 24, 48 or 72 h and then plated for colony assay, in three separate experiments. (B) Flow cytometric analysis of the erythroid differentiation markers CD44 and TER119 in Rps19‐deficient and healthy cultures with or without Dexamethasone treatment. Shown here are representative plots for each condition and time point from three separate experiments. (C) Quantification of data from the flow cytometric experiment in (B). (D) Cell cycle distribution analysis of cells cultured for 72 h. Displayed here is mean with standard deviation and paired student's t‐test was used for statistical analysis.
To further examine the maintenance of early erythroid progenitors and identify changes in differentiation between Rps19‐deficient cells and controls, we employed flow cytometric analysis of the well‐established markers of erythroid differentiation CD44 and TER119 (Fig 4B) (Liu et al, 2013). Rps19‐deficient erythroid cells showed increased erythroid differentiation compared to controls, with less cells in the immature CD44+TER119− fraction and more cells in the differentiated CD44−TER119+ fraction after 48 h of culture. Dexamethasone treatment significantly reversed this and retained more cells in the immature CD44+TER119− fraction (Fig 4C). These findings were in line with data from the microarray analysis, indicating that increased differentiation of Rps19‐deficient cells and depletion of erythroid progenitors could be reversed by glucocorticoid treatment.
During differentiation, erythroid cells undergo a set of cell divisions before they become mature red cells and exit the cell cycle. Cell cycle analysis of Rps19‐deficient cells revealed that these cells to a greater extent reside in the G1 phase and less in S and M phase compared to healthy controls (Fig 4D). This cell cycle difference was not significantly reversed by glucocorticoid treatment. Together, these results demonstrate that Rps19‐deficiency increases erythroid differentiation and depletes erythroid progenitors. Importantly, glucocorticoid administration significantly reverses both these effects, which allows expansion of erythroid progenitors, despite Rps19‐deficiency.
Glucocorticoids reverse up regulation of Trp53 targets caused by Rps19‐deficiency
The tumour suppressor Trp53 is known to be highly involved in the DBA phenotype, as shown in animal models of DBA (Danilova et al, 2008; Taylor et al, 2012). In our mouse model, Rps19‐deficient erythroid cells demonstrated a dramatic up regulation of Trp53 downstream target genes, including Cdkn1a (p21) and Bax that regulate cell cycle and apoptosis (Jaako et al, 2011). Activation of Trp53 in the Rps19‐deficient mice involves 5S ribonucleoprotein particle (5S RNP)‐mediated inhibition of MDM2. Deletion of the Trp53 gene or disruption of the 5S RNP‐MDM2 interaction in the Rps19‐deficient DBA mouse ameliorates the anaemia in vivo (Jaako et al, 2015). We therefore wanted to examine the effect of glucocorticoid treatment on Trp53 activity in Rps19‐deficient erythroid cells. The expression of multiple Trp53 target genes was significantly elevated in Rps19‐deficient erythroid cells, shown by GSEA of microarray data mapped against the Trp53 KEGG pathway (Fig 5A). The same thing was seen for multiple Trp53 target genes including Cdkn1a and Bax by RT‐PCR (Fig 5B–C). Dexamethasone treatment significantly blocked the up regulation of multiple Trp53 target genes, demonstrating that glucocorticoids have a potent dampening effect on Trp53 activity in Rps19‐deficient cells (Fig 5C). Glucocorticoids clearly displayed this effect after 72 h, but a trend was also seen after 24 h of treatment (compare Fig 5B and 5C). Importantly, glucocorticoids did not down regulate expression of the Trp53 target genes in healthy cells, indicating that glucocorticoids had this effect specifically in cells with elevated Trp53 activity. Both Rps19‐deficient and control cells responded to glucocorticoid stimulation by up regulation of the glucocorticoid receptor target gene Per1 at both 24 and 72 h (Fig S4A–B).
Figure 5.
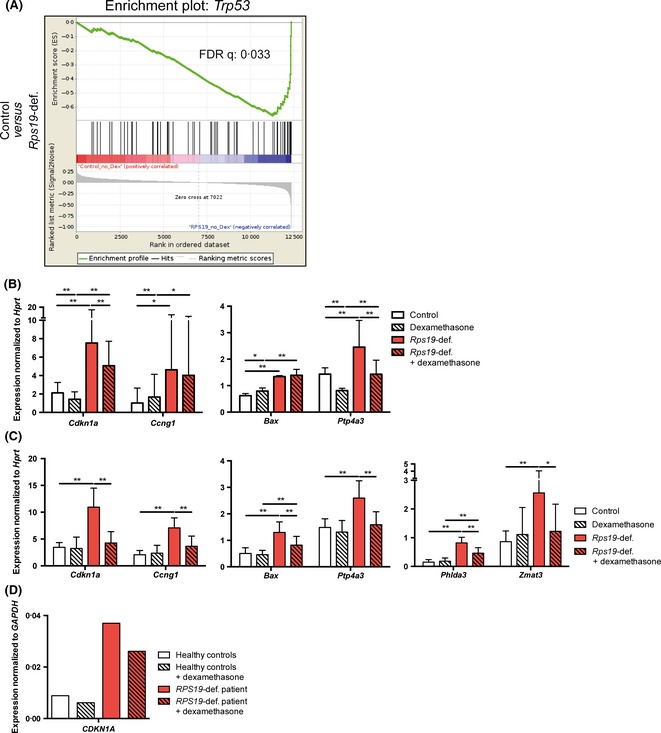
Trp53 target genes are up regulated in Rps19‐deficient cells, but not in the presence glucocorticoids. (A) GSEA of genes significantly up regulated by Rps19‐deficiency mapped to the Trp53 KEGG pathway. (B) Trp53 responsive gene expression after dexamethasone and doxycycline treatment for 24 h and (C) 72 h. Results from three separate experiments. (D) Gene expression of CDKN1A in blood cells from peripheral blood of one DBA patient carrying an RPS19 mutation and two healthy control subjects.
In order to determine if glucocorticoid receptor activation reduces nuclear TP53 levels we performed a mini screen with selected steroid‐associated compounds including 30 glucocorticoid receptor agonists. The readout of the screen was average nuclear TP53 staining intensity in RPS19‐deficient human A549 (TP53 wild‐type) cells (Fig S5). While RPS19‐deficiency caused a dramatic increase in nuclear TP53 protein levels, this increase was moderately but significantly reduced when cells were treated with the group of glucocorticoid compounds. Neither androgens nor oestrogens had this effect. The results suggest that glucocorticoids are able to reduce nuclear TP53 protein accumulation caused by ribosomal stress.
To validate the clinical relevance of this effect, we examined gene expression of Cdkn1a in erythroid cells from one DBA patient carrying an RPS19 mutation. Indeed, the expression of CDKN1A was highly elevated in this patient compared to healthy donors, and CDKN1A expression was dampened after only 24 h of glucocorticoid treatment (Fig 5D). The patient cells were responsive to glucocorticoids, shown by up regulation of ZFP36L2, a direct target of the glucocorticoid receptor important for erythroid progenitor self‐renewal (Fig S4C). Overall, these results demonstrate that glucocorticoids counteract up regulation of multiple Trp53 target genes in Rps19‐deficient erythroid cells. This effect is not seen in healthy controls, which indicates that it is specific to DBA pathology.
Discussion
Glucocorticoids are often chosen as first line treatment for DBA, and they have now been used to treat DBA patients for more than half a century (Gasser, 1951). It is well known that glucocorticoids are crucial for stress erythropoiesis and self‐renewal of erythroid progenitors in vitro, but little is known about their therapeutic mechanisms in DBA treatment. One previous attempt has been made to study glucocorticoid responsiveness in a mouse model of Rps6‐deficiency, but these mice did not respond to glucocorticoid treatment (Keel et al, 2012). Here, for the first time, we show glucocorticoid responsiveness in a mouse model of DBA, demonstrating that it is a relevant and useful model to study therapeutic mechanisms of DBA treatment. We also demonstrate that glucocorticoids prevent the depletion of erythroid progenitors and the increase in erythroid differentiation observed in Rps19‐deficient cells. Furthermore, we demonstrate that glucocorticoids inhibit up regulation of multiple Trp53 target genes, an effect not seen in healthy cells and thus specific to the DBA phenotype.
Erythropoiesis requires a tight regulation between self‐renewal and differentiation of erythroid progenitors to generate sufficient numbers of mature red blood cells. Steady state erythropoiesis involves a set number of differentiation divisions and is regulated by EPO and SCF (Muta et al, 1995). Glucocorticoids are not required for steady state erythropoiesis, but are vital for situations of stress erythropoiesis, such as excessive bleeding or severe anaemia. This is best illustrated by lack of recovery from haemolytic anaemia of mice with disrupted glucocorticoid receptor function (Bauer et al, 1999). In steady state erythropoiesis, cultures treated with glucocorticoids give rise to more BFU‐E colonies than untreated controls, indicating that they are important for erythroid progenitor self‐renewal and maintenance (Flygare et al, 2011; Narla et al, 2011). We observe that Rps19‐deficient erythroid cells have decreased capacity of CFU‐E formation, which leads to decreased erythroid output and thus a decreased number of cells able to reach erythroid maturation. This is in line with other studies showing decreased CFU‐E and BFU‐E colony‐forming abilities in erythroid cells from DBA patients (Nathan et al, 1978; Lipton et al, 1986; Giri et al, 2000). We show that glucocorticoid‐treated Rps19‐deficient cultures retain more CFU‐E colony‐forming capacity compared to their untreated counterparts, indicating that glucocorticoid treatment can significantly rescue erythroid progenitor depletion. This is in line with a study observing an increase in the erythroid progenitor population of steroid responsive DBA patients compared to transfusion‐dependent patients (Iskander et al, 2015). In human bone marrow the erythroid output was decreased when RPS19 was knocked down and, similar to our findings, this was rescued by glucocorticoids (Ebert et al, 2005).
In our study, gene expression analysis demonstrated a signature of increased differentiation of Rps19‐deficient cells. The gene expression results further suggested that glucocorticoid treatment of Rps19‐deficient cells decreased differentiation and increased maintenance of erythroid progenitors. This explanation to the therapeutic effect of glucocorticoids was confirmed by colony forming, cell expansion and flow cytometry analyses, demonstrating that glucocorticoid treatment reversed both increase in differentiation and depletion of erythroid progenitors in Rps19‐deficient cells. It could seem counterproductive for glucocorticoids to delay differentiation in an Rps19‐deficient erythroid system, but we postulate that this, together with increased maintenance of erythroid progenitors, prevents the system from being prematurely differentiated, which would result in exhaustion of self‐renewing cells. Counteracting this exhaustion could allow erythroid expansion to occur despite Rps19‐deficiency. This would be in line with the significant improvement of anaemia we observed in our mouse model of DBA.
TP53 has been identified a key player in the pathogenesis of DBA in both fish and human model systems (Danilova et al, 2008; Dutt et al, 2011; Taylor et al, 2012). Similarly to others, we have also shown that the anaemia of our Rps19‐deficient mouse model could be significantly improved in a Trp53 null background (Jaako et al, 2011). Likewise, the phenotype of a mouse model of the ribosomal disease Treacher Collins syndrome is reversed in a Trp53 null background, demonstrating an important role for Trp53 in the pathology of ribosomopathies (Jones et al, 2008). The mechanism behind Trp53 activation in DBA is not fully understood, but probably involves activation of ribosomal stress pathways. In situations where one ribosomal component is not available in sufficient amounts, ribosome biogenesis fails and the ribosomal proteins RPL5, RPL11 and 5S rRNA diffuse from the nucleolus and act as sensors of ribosomal stress by forming a complex that inhibits MDM2. As MDM2 is a E3 ubiquitin ligase that promotes degradation of TRP53, formation of this complex leads to TRP53 stabilization (Lohrum et al, 2003; Zhang et al, 2003; Dai & Lu, 2004; Donati et al, 2013). Some studies have reported that erythroid defects in mouse cells and zebrafish models of ribosomal deficiency are not always corrected by Trp53 depletion (Torihara et al, 2011; Jia et al, 2013; Singh et al, 2014). Although TP53 seems to be important for the disease, it is plausible that there is more to the pathology of DBA than increased TP53 activity alone. Interestingly, both RPL5 and RPL11 are found mutated in DBA, but in these cases it is unclear if TP53 activation has a role in the pathogenic mechanism (Singh et al, 2014) and more studies are needed to elucidate if ribosomal stress and TP53 activation is a central pathogenic mechanism in DBA. In this study, we found both a significant up regulation of TP53 protein and multiple TP53 responsive genes in both human and murine Rps19‐deficient cells. Also, our microarray analysis reveals a strong Trp53 activation signature in Rps19‐deficient erythroid cells. Glucocorticoids reverse the increased expression of multiple Trp53 target genes involved in both cell cycle (Cdkn1a and Ccng1) and apoptosis (Bax). Interestingly, no change in Trp53 target gene expression was seen in healthy cells regardless of glucocorticoid administration, indicating that this mechanism is specific to Rps19‐deficient cells and thus disease‐specific for DBA. Neither treatment with androgen‐ nor oestrogen‐associated compounds resulted in any decrease of TP53 protein levels, indicating that this effect is specific to glucocorticoids. This effect was seen with treatment of multiple glucocorticoids, leading us to conclude that glucocorticoid receptor activation can have a TP53 dampening effect. One previous study reported negative cross talk between TP53 and the glucocorticoid receptor in a neuroblastoma cell line (Sengupta et al, 2000), but because glucocorticoids have very cell type‐specific effects, it remained highly unclear if this was also true in a DBA setting. Here, we show, for the first time, that glucocorticoids dampen the elevated Trp53 activity seen in DBA, in a disease‐specific manner.
Together, the results from this study demonstrate that glucocorticoid treatment dampens the elevated Trp53 activity seen in erythroid cells from DBA patients and murine cells, by decreasing the expression of multiple Trp53 responsive genes, coupled with decreased levels of apoptosis in these cells. To our knowledge, this is the first time it has been shown that glucocorticoid treatment reverses Trp53 activity and affects erythroid maintenance in the context of DBA. This study also demonstrates that glucocorticoid treatment of Rps19‐deficient cells reverse the depletion of erythroid progenitors as well as counteracts the increase in erythroid differentiation, leading to increased expansion and CFU‐E colony forming capacity (Fig 6).
Figure 6.
Glucocorticoids act by rescuing depletion of erythroid progenitors and dampen Trp53 activity. Glucocorticoid treatment thereby allows expansion and replenishment of the Rps19‐deficient erythroid system.
Glucocorticoid treatment of DBA patients has been frequently used for decades, but comes at the cost of severe side effects. In the effort to develop more disease‐specific treatments for DBA it is important to identify the specific mechanisms responsible for increased erythroid output of glucocorticoid treatment. Based on our results, it is likely that other glucocorticoid receptor ligands with dissociated functions have a more potent effect on the mechanisms relevant for DBA therapy and at the same time induce fewer side effects. This has been seen in other diseases of chronic glucocorticoid treatment, such as Duchenne muscular dystrophy (Biggar et al, 2006). The present study identifies the desired effects for a better DBA drug and is therefore a step forward toward development of more potent and safer glucocorticoid receptor agonists for DBA therapy.
Author contributions
SES and JF designed the research study, SES, KS and MW performed the research, SES analysed the data and performed statistical analyses, SS performed statistical analysis of the microarray data, MW provided DBA patient samples, AJG and RDH performed research on the human cell line, PJ and SK gave intellectual input to the study and provided the mouse model, and SES and JF wrote the manuscript.
Competing interests
The authors have no competing interests.
Supporting information
Data S1. Materials and methods.
Fig S1. Blood parameters of Rps19‐deficient and healthy mice.
Fig S2. Gene expression analyses of microarray and RNA sequencing data.
Fig S3. Decreased Myb expression is normalized by glucocorticoids in Rps19‐deficient cells.
Fig S4. Murine and patient cells respond to glucocorticoid stimulation.
Fig S5. TP53 protein is less upregulated in RPS19‐deficient human cells treated with glucocorticoids.
Table SI. List of genes significantly altered by Dexamethasone treatment of Rps19‐deficient cells. Log2 gene expression values of genes from cluster 2 and 7 from Fig S2.
Acknowledgements
We thank the staff of the BMC animal facility for excellent animal care and Abdul Ghani Alattar for cDNA preparations. We also thank Carolina Guibentif and Alexander Mattebo for critical revision of this manuscript. We would also like to acknowledge the Australian Diamond Blackfan Anaemia (ADBA) programme members, consisting of Sheren Al‐Obaidi, Sarah CE Bray, Richard J D'Andrea, Jianmin Ding, Amee J George, Thomas J Gonda, S Peter Klinken, Piyush Madhamshettiwar, Lorena Núñez Villacis, Richard B Pearson, Ben Saxon, Hamish S Scott, Kaylene J Simpson, Adam Stephenson, Amilia Wee, Louise N Winteringham, Mei S Wong, and Ross D Hannan for their contribution to this work. This work was supported by the Söderberg Foundation, The Diamond–Blackfan Anemia Foundation, DBA Canada, Swedish Research Council, Ellen Bacharach's Foundation, Åke Wiberg's Foundation, Marie Curie Integration Grant and an Australian Diamond Blackfan Anaemia (ADBA) programme grant from the Captain Courageous Foundation (www.captaincourageousfoundation.com).
References
- Bauer, A. , Tronche, F. , Wessely, O. , Kellendonk, C. , Reichardt, H.M. , Steinlein, P. , Schütz, G. & Beug, H. (1999) The glucocorticoid receptor is required for stress erythropoiesis. Genes & Development, 13, 2996–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar, W.D. , Harris, V.A. , Eliasoph, L. & Alman, B. (2006) Long‐term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscular Disorders: NMD, 16, 249–255. [DOI] [PubMed] [Google Scholar]
- Dai, M.‐S. & Lu, H. (2004) Inhibition of MDM2‐mediated p53 ubiquitination and degradation by ribosomal protein L5. The Journal of Biological Chemistry, 279, 44475–44482. [DOI] [PubMed] [Google Scholar]
- Danilova, N. , Sakamoto, K.M. & Lin, S. (2008) Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood, 112, 5228–5237. [DOI] [PubMed] [Google Scholar]
- Diamond, L.K. & Blackfan, K.D. (1938) Hypoplastic anemia. American Journal of Diseases of Children, 56, 464–467. [Google Scholar]
- Dolznig, H. , Grebien, F. , Deiner, E.M. , Stangl, K. , Kolbus, A. , Habermann, B. , Kerenyi, M.A. , Kieslinger, M. , Moriggl, R. , Beug, H. & Müllner, E.W. (2006) Erythroid progenitor renewal versus differentiation: genetic evidence for cell autonomous, essential functions of EpoR, Stat5 and the GR. Oncogene, 25, 2890–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati, G. , Peddigari, S. , Mercer, C.A. & Thomas, G. (2013) 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2‐p53 checkpoint. Cell Reports, 4, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia, N. , Gustavsson, P. , Andersson, B. , Pettersson, M. , Willig, T.N. , Dianzani, I. , Ball, S. , Tchernia, G. , Klar, J. , Matsson, H. , Tentler, D. , Mohandas, N. , Carlsson, B. & Dahl, N. (1999) The gene encoding ribosomal protein S19 is mutated in Diamond‐Blackfan anaemia. Nature Genetics, 21, 169–175. [DOI] [PubMed] [Google Scholar]
- Dutt, S. , Narla, A. , Lin, K. , Mullally, A. , Abayasekara, N. , Megerdichian, C. , Wilson, F.H. , Currie, T. , Khanna‐Gupta, A. , Berliner, N. , Kutok, J.L. & Ebert, B.L. (2011) Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood, 117, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, B.L. , Lee, M.M. , Pretz, J.L. , Subramanian, A. , Mak, R. , Golub, T.R. & Sieff, C.A. (2005) An RNA interference model of RPS19 deficiency in Diamond‐Blackfan anemia recapitulates defective hematopoiesis and rescue by dexamethasone: identification of dexamethasone‐responsive genes by microarray. Blood, 105, 4620–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilia, G. , Donelli, A. , Ferrari, S. , Selleri, L. , Zucchini, P. , Moretti, L. , Venturelli, D. , Ceccherelli, G. & Torelli, G. (1986) Cellular‐levels of messenger‐Rna From C‐Myc, C‐Myb and C‐Fes Onc‐Genes in normal myeloid and erythroid precursors of human‐bone marrow ‐ an Insitu Hybridization Study. British Journal of Haematology, 62, 287–292. [DOI] [PubMed] [Google Scholar]
- Farrar, J.E. & Dahl, N. (2011) Untangling the phenotypic heterogeneity of Diamond Blackfan Anemia. Seminars in Hematology, 48, 124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flygare, J. , Rayon Estrada, V. , Shin, C. , Gupta, S. & Lodish, H.F. (2011) HIF1alpha synergizes with glucocorticoids to promote BFU‐E progenitor self‐renewal. Blood, 117, 3435–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguli, G. , Back, J. , Sengupta, S. & Wasylyk, B. (2002) The p53 tumour suppressor inhibits glucocorticoid‐induced proliferation of erythroid progenitors. EMBO Reports, 3, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser, C. (1951) Aplastic anemia (chronic erythroblastophthisis) and cortisone. Schweizerische Medizinische Wochenschrift, 81, 1241–1242. [PubMed] [Google Scholar]
- Gazda, H.T. , Zhong, R. , Long, L. , Niewiadomska, E. , Lipton, J.M. , Ploszynska, A. , Zaucha, J.M. , Vlachos, A. , Atsidaftos, E. , Viskochil, D.H. , Niemeyer, C.M. , Meerpohl, J.J. , Rokicka‐Milewska, R. , Pospisilova, D. , Wiktor‐Jedrzejczak, W. , Nathan, D.G. , Beggs, A.H. & Sieff, C.A. (2004) RNA and protein evidence for haplo‐insufficiency in Diamond‐Blackfan anaemia patients with RPS19 mutations. British Journal of Haematology, 127, 105–113. [DOI] [PubMed] [Google Scholar]
- Giri, N. , Kang, E. , Tisdale, J.F. , Follman, D. , Rivera, M. , Schwartz, G.N. , Kim, S. , Young, N.S. , Rick, M.E. & Dunbar, C.E. (2000) Clinical and laboratory evidence for a trilineage haematopoietic defect in patients with refractory Diamond‐Blackfan anaemia. British Journal of Haematology, 108, 167–175. [DOI] [PubMed] [Google Scholar]
- Golde, D.W. , Bersch, N. & Cline, M.J. (1976) Potentiation of erythropoiesis in vitro by dexamethasone. The Journal of Clinical Investigation, 57, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi, I. , Flygare, J. , Nishiura, H. , Brun, A.C.M. , Ooka, A. , Kiefer, T. , Ma, Z. , Dahl, N. , Richter, J. & Karlsson, S. (2003) Proliferation deficiency of multipotent hematopoietic progenitors in ribosomal protein S19 (RPS19)‐deficient diamond‐Blackfan anemia improves following RPS19 gene transfer. Molecular Therapy: The Journal of the American Society of Gene Therapy, 7, 613–622. [DOI] [PubMed] [Google Scholar]
- Iskander, D. , Psaila, B. , Gerrard, G. , Chaidos, A. , En Foong, H. , Harrington, Y. , Karnik, L.C. , Roberts, I. , la Fuente de, J. & Karadimitris, A. (2015) Elucidation of the erythroid progenitor defect in Diamond‐Blackfan Anemia by characterization and prospective isolation of human erythroid progenitors. Blood, 125, 2553–2557. [DOI] [PubMed] [Google Scholar]
- Jaako, P. , Flygare, J. , Olsson, K. , Quere, R. , Ehinger, M. , Henson, A. , Ellis, S. , Schambach, A. , Baum, C. , Richter, J. , Larsson, J. , Bryder, D. & Karlsson, S. (2011) Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond‐Blackfan anemia. Blood, 118, 6087–6096. [DOI] [PubMed] [Google Scholar]
- Jaako, P. , Debnath, S. , Olsson, K. , Modlich, U. , Rothe, M. , Schambach, A. , Flygare, J. & Karlsson, S. (2014) Gene therapy cures the anemia and lethal bone marrow failure in mouse model for RPS19‐deficient Diamond‐Blackfan anemia. Haematologica, 12, 1792–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaako, P. , Debnath, S. , Olsson, K. , Zhang, Y. , Flygare, J. , Lindstrom, M.S. , Bryder, D. & Karlsson, S. (2015) Disruption of the 5S RNP‐Mdm2 interaction significantly improves the erythroid defect in a mouse model for Diamond‐Blackfan anemia. Leukemia. doi:10.1038/leu.2015.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, Q. , Zhang, Q. , Zhang, Z. , Wang, Y. , Zhang, W. , Zhou, Y. , Wan, Y. , Cheng, T. , Zhu, X. , Fang, X. , Yuan, W. & Jia, H. (2013) Transcriptome analysis of the zebrafish model of Diamond‐Blackfan anemia from RPS19 deficiency via p53‐dependent and ‐independent pathways. PLoS ONE, 8, e71782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, N.C. , Lynn, M.L. , Gaudenz, K. , Sakai, D. , Aoto, K. , Rey, J.‐P. , Glynn, E.F. , Ellington, L. , Du, C. , Dixon, J. , Dixon, M.J. & Trainor, P.A. (2008) Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nature Medicine, 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keel, S.B. , Phelps, S. , Sabo, K.M. , O'Leary, M.N. , Kirn‐Safran, C.B. & Abkowitz, J.L. (2012) Establishing Rps6 hemizygous mice as a model for studying how ribosomal protein haploinsufficiency impairs erythropoiesis. Experimental Hematology, 40, 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landowski, M. , O'Donohue, M.‐F. , Buros, C. , Ghazvinian, R. , Montel‐Lehry, N. , Vlachos, A. , Sieff, C.A. , Newburger, P.E. , Niewiadomska, E. , Matysiak, M. , Glader, B. , Atsidaftos, E. , Lipton, J.M. , Beggs, A.H. , Gleizes, P.‐E. & Gazda, H.T. (2013) Novel deletion of RPL15 identified by array‐comparative genomic hybridization in Diamond‐Blackfan anemia. Human Genetics, 132, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindern von, M. , Zauner, W. , Mellitzer, G. , Steinlein, P. , Fritsch, G. , Huber, K. , Löwenberg, B. & Beug, H. (1999) The glucocorticoid receptor cooperates with the erythropoietin receptor and c‐Kit to enhance and sustain proliferation of erythroid progenitors in vitro . Blood, 94, 550–559. [PubMed] [Google Scholar]
- Lipton, J.M. , Kudisch, M. , Gross, R. & Nathan, D.G. (1986) Defective erythroid progenitor differentiation system in congenital hypoplastic (Diamond‐Blackfan) anemia. Blood, 67, 962–968. [PubMed] [Google Scholar]
- Liu, J. , Zhang, J. , Ginzburg, Y. , Li, H. , Xue, F. , De Franceschi, L. , Chasis, J.A. , Mohandas, N. & An, X. (2013) Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood, 121, e43–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrum, M.A.E. , Ludwig, R.L. , Kubbutat, M.H.G. , Hanlon, M. & Vousden, K.H. (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell, 3, 577–587. [DOI] [PubMed] [Google Scholar]
- Miyake, K. , Flygare, J. , Kiefer, T. , Utsugisawa, T. , Richter, J. , Ma, Z. , Wiznerowicz, M. , Trono, D. & Karlsson, S. (2005) Development of cellular models for ribosomal protein S19 (RPS19)‐deficient diamond‐blackfan anemia using inducible expression of siRNA against RPS19. Molecular Therapy: The Journal of the American Society of Gene Therapy, 11, 627–637. [DOI] [PubMed] [Google Scholar]
- Miyake, K. , Utsugisawa, T. , Flygare, J. , Kiefer, T. , Hamaguchi, I. , Richter, J. & Karlsson, S. (2008) Ribosomal protein S19 deficiency leads to reduced proliferation and increased apoptosis but does not affect terminal erythroid differentiation in a cell line model of Diamond‐Blackfan anemia. Stem Cells (Dayton, Ohio), 26, 323–329. [DOI] [PubMed] [Google Scholar]
- Muta, K. , Krantz, S.B. , Bondurant, M.C. & Dai, C.H. (1995) Stem cell factor retards differentiation of normal human erythroid progenitor cells while stimulating proliferation. Blood, 86, 572–580. [PubMed] [Google Scholar]
- Narla, A. , Dutt, S. , McAuley, J.R. , Al‐Shahrour, F. , Hurst, S. , McConkey, M. , Neuberg, D. & Ebert, B.L. (2011) Dexamethasone and lenalidomide have distinct functional effects on erythropoiesis. Blood, 118, 2296–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, D.G. , Clarke, B.J. , Hillman, D.G. , Alter, B.P. & Housman, D.E. (1978) Erythroid precursors in congenital hypoplastic (Diamond‐Blackfan) anemia. The Journal of Clinical Investigation, 61, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdahl, E.B. , Naprstek, B.L. , Wallace, W.C. & Lipton, J.M. (1994) Erythroid failure in Diamond‐Blackfan anemia is characterized by apoptosis. Blood, 83, 645–650. [PubMed] [Google Scholar]
- Sankaran, V.G. , Ghazvinian, R. , Do, R. , Thiru, P. , Vergilio, J.‐A. , Beggs, A.H. , Sieff, C.A. , Orkin, S.H. , Nathan, D.G. , Lander, E.S. & Gazda, H.T. (2012) Exome sequencing identifies GATA1 mutations resulting in Diamond‐Blackfan anemia. The Journal of Clinical Investigation, 122, 2439–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta, S. , Vonesch, J.L. , Waltzinger, C. , Zheng, H. & Wasylyk, B. (2000) Negative cross‐talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. The EMBO Journal, 19, 6051–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieff, C.A. , Yang, J. , Merida‐Long, L.B. & Lodish, H.F. (2010) Pathogenesis of the erythroid failure in Diamond Blackfan anaemia. British Journal of Haematology, 148, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S.A. , Goldberg, T.A. , Henson, A.L. , Husain‐Krautter, S. , Nihrane, A. , Blanc, L. , Ellis, S.R. , Lipton, J.M. & Liu, J.M. (2014) p53‐independent cell cycle and erythroid differentiation defects in murine embryonic stem cells haploinsufficient for Diamond Blackfan Anemia‐Proteins: RPS19 versus RPL5. PLoS ONE, 9, e89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, A.M. , Humphries, J.M. , White, R.M. , Murphey, R.D. , Burns, C.E. & Zon, L.I. (2012) Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Experimental Hematology, 40, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torihara, H. , Uechi, T. , Chakraborty, A. , Shinya, M. , Sakai, N. & Kenmochi, N. (2011) Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond‐Blackfan anaemia. British Journal of Haematology, 152, 648–654. [DOI] [PubMed] [Google Scholar]
- Vlachos, A. & Muir, E. (2010) How I treat Diamond‐Blackfan anemia. Blood, 116, 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos, A. , Ball, S. , Dahl, N. , Alter, B.P. , Sheth, S. , Ramenghi, U. , Meerpohl, J. , Karlsson, S. , Liu, J.M. , Leblanc, T. , Paley, C. , Kang, E.M. , Leder, E.J. , Atsidaftos, E. , Shimamura, A. , Bessler, M. , Glader, B. & Lipton, J.M. (2008) Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. British Journal of Haematology, 142, 859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos, A. , Dahl, N. , Dianzani, I. & Lipton, J.M. (2013) Clinical utility gene card for: Diamond ‐ Blackfan Anemia ‐ update 2013. European Journal of Human Genetics: EJHG, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessely, O. , Deiner, E.M. , Beug, H. & Lindern von, M. (1997) The glucocorticoid receptor is a key regulator of the decision between self‐renewal and differentiation in erythroid progenitors. The EMBO Journal, 16, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wolf, G.W. , Bhat, K. , Jin, A. , Allio, T. , Burkhart, W.A. & Xiong, Y. (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53‐dependent ribosomal‐stress checkpoint pathway. Molecular and Cellular Biology, 23, 8902–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Prak, L. , Rayon Estrada, V. , Thiru, P. , Flygare, J. , Lim, B. & Lodish, H.F. (2013) ZFP36L2 is required for self‐renewal of early burst‐forming unit erythroid progenitors. Nature, 499, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Fig S1. Blood parameters of Rps19‐deficient and healthy mice.
Fig S2. Gene expression analyses of microarray and RNA sequencing data.
Fig S3. Decreased Myb expression is normalized by glucocorticoids in Rps19‐deficient cells.
Fig S4. Murine and patient cells respond to glucocorticoid stimulation.
Fig S5. TP53 protein is less upregulated in RPS19‐deficient human cells treated with glucocorticoids.
Table SI. List of genes significantly altered by Dexamethasone treatment of Rps19‐deficient cells. Log2 gene expression values of genes from cluster 2 and 7 from Fig S2.