

Abstract
Bats are natural reservoirs of several important emerging viruses. Cross‐species transmission appears to be quite common among bats, which may contribute to their unique reservoir potential. Therefore, understanding the importance of bats as reservoirs requires examining them in a community context rather than concentrating on individual species. Here, we use a network approach to identify ecological and biological correlates of cross‐species virus transmission in bats and rodents, another important host group. We show that given our current knowledge the bat viral sharing network is more connected than the rodent network, suggesting viruses may pass more easily between bat species. We identify host traits associated with important reservoir species: gregarious bats are more likely to share more viruses and bats which migrate regionally are important for spreading viruses through the network. We identify multiple communities of viral sharing within bats and rodents and highlight potential species traits that can help guide studies of novel pathogen emergence.
Keywords: Chiroptera, ecological networks, emerging infectious disease, Rodentia, zoonoses
Introduction
Most emerging infectious diseases are zoonotic (passed from animals to humans), with > 70% originating in wildlife (Jones et al. 2008). Cross‐species transmission is one of the most challenging and least studied aspects of disease ecology, yet it is the defining process in zoonotic disease emergence (Lloyd‐Smith et al. 2009).
Bats are the reservoirs of a number of emerging viruses, including Ebolaviruses, SARS coronavirus and Nipah and Hendra paramyxoviruses (Calisher et al. 2006). Viral and malarial parasite diversity in bats is high (Drexler et al. 2012; Luis et al. 2013; Quan et al. 2013; Schaer et al. 2013; Drexler et al. 2014), and bats appear to be major and ancient natural reservoirs of several viral families, including hepaciviruses, pegiviruses, paramyxoviruses, coronaviruses and influenza A viruses ( Drexler, et al. 2012, 2014; Quan et al. 2013; Tong et al. 2013). Since cross‐species transmission can be common among bats (Streicker et al. 2010; Cui et al. 2012), understanding their role as reservoir hosts requires examining them in a community context rather than concentrating on individual species.
Several traits have been hypothesised to make bats particularly suited to hosting and transmitting viruses, including life history traits such as relatively long lifespans for their body size (Munshi‐South & Wilkinson 2010), which may facilitate viral persistence for chronic infections; their reliance on torpor, which can decrease viral replication and immune function (Dempster et al. 1966; Prendergast et al. 2002); and flight, which can facilitate transmission of viruses to new areas. In addition, many bat species are gregarious, living in dense aggregations, and some roosting sites can house diverse assemblages of bat species (Kunz 1982; Kuzmin et al. 2010), which could facilitate transmission of pathogens and sustain acute‐immunising infections. In evolutionary terms, bats are ancient mammals, and it has been hypothesised that viruses that evolved in bats may use conserved cellular receptors enhancing their ability to transmit viruses to other mammals (Calisher et al. 2006). The diet of some species has been hypothesised to facilitate transmission of viruses; after feeding, frugivorous bats often leave partially eaten fruit behind which can be contaminated with viruses (Chua et al. 2002; Dobson 2005). Bats are the second most diverse mammalian order with over 1100 species, with many overlapping species distributions; multiple regions across the globe are home to more than 40 bat species, allowing for species to interact and potentially spread viruses between them (Fig. 1; Calisher et al. 2006; Luis et al. 2013).
Figure 1.
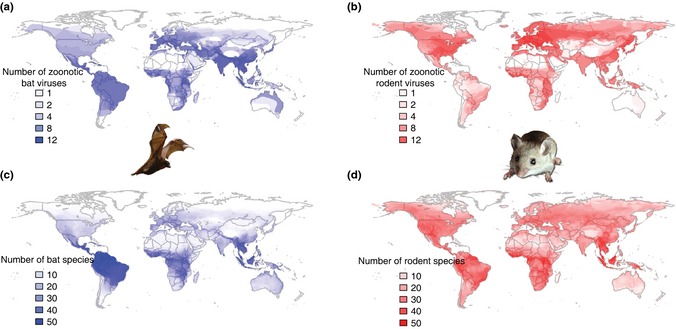
Distributions of zoonotic viral richness for (a) bats and (b) rodents from our data set (as the broadest possible distribution to our current knowledge, for heuristic purposes only); and (c) bat and (d) rodent species richness.
Here, we examine the structure of host–virus communities in bats and rodents and how host traits, such as those above, correlate with propensity to host and transmit viruses. Rodents are a suitable group for comparison because they also host many important zoonotic viruses and share many of the characteristics hypothesised to make bats suitable as viral reservoirs. For example, rodents are also evolutionarily ancient; they are older than bats and more closely related to humans (dos Reis et al. 2012). Rodents are the most diverse mammalian order with approximately twice the number of rodent species as bat species, many of which express torpor, and display a wide range of life history traits, including some long‐lived species.
Networks have been used extensively in disease ecology to model the finite and heterogeneous number of social contacts between individuals. Here, we use networks to describe sharing of viruses between different species, rather than individuals. This is akin to other ecological networks, such as food webs, mutualistic, or host–parasite networks (Montoya et al. 2006). However, we collapse the bipartite network (separate nodes for viruses and host species) to a unipartite projection, weighting contacts between host species by the number of viruses they share. In the same way social networks can highlight the disproportionate importance of specific individuals, such as ‘super‐spreaders’, in population‐level disease dynamics, our networks can highlight the importance of certain species in host–virus communities and disease emergence. In addition, we can use other network methods to look at substructures, or ‘communities’ in the network, highlighting sets of highly interconnected species (Newman 2004).
Here, we use databases of bat and rodent viruses spanning 78 years of publications (Luis et al. 2013) to create networks that connect host species via their shared viruses and examine cross‐species transmission at several scales.
Materials and Methods
Data and network formulation
A database of bat and rodent viruses compiled from the literature (Luis et al. 2013) was used to create separate networks of viral sharing for bats and rodents, with each node representing a host species and edges between two nodes representing the presence of virus(es) in both species. Edges were weighted by the number of viruses shared between the two species. See Supplementary Methods in Supporting Information for more details on network formulation.
To account for sampling bias in the analyses, we used the number of citations on Web of Science (http://thomsonreuters.com/products_services/science/science_products/a-z/web_of_science/) with the host species’ Latin species name for the search term (e.g. Altizer et al. 2011). The distribution was highly skewed, therefore we used the log of this number to normalise the distribution. We also performed the analyses using the species name and ‘virus’ as the search terms. All results were qualitatively similar.
We calculated various node and network statistics (Table 1). The node statistics that we calculated were degree (the number of links a node has), weighted degree (incorporating the number of viruses shared) and betweenness (the number of shortest paths that go through a node (weighted)). The network statistics that we calculated included the degree distribution, transitivity (if two nodes are connected, the probability that their adjacent nodes are also connected), degree assortativity (likelihood of high‐degree nodes connecting to other high‐degree degree nodes and low‐degree nodes to other low‐degree nodes) and connectance (links per species2, or the proportion of links that are present out of all possible links). We also calculated quantitative connectance, which is the quantitative linkage density (which takes into account the weights of the edges; Bersier et al. 2002) divided by the number of species in the network.
Table 1.
Network statistics
| Metric | Bat network | Rodent network | P‐valuea |
|---|---|---|---|
| Number of host species | 143 | 196 | |
| Number of viruses | 110 | 185 | |
| Number of viruses per host species | 2.66 | 2.49 | |
| Number of host species per virus | 3.8 | 2.71 | |
| Number of links | 1074 | 1227 | < 0.0001 |
| Mean degree | 15.0 | 12.5 | < 0.0001 |
| Mean weighted degree | 20.1 | 15.2 | < 0.0001 |
| Mean weighted degree adjustedb | 47.4 | 34.0 | < 0.0001 |
| Transitivity | 0.61 | 0.54 | < 0.0001; < 0.0001 |
| Degree assortativity | 0.10 | −0.06 | 0.0002; 0.9766 |
| Connectance | 0.0525 | 0.0319 | < 0.0001 |
| q connectancec | 0.1575 | 0.1147 | < 0.0001 |
| q connectance adjustedd | 0.1639 | 0.1215 | < 0.0001 |
| q connectance Jaccarde | 0.1173 | 0.0741 | < 0.0001 |
P‐values give significance of difference between bat and rodent networks based on random permutations of the networks, except for transitivity and assortativity, which have two P‐values each for the bat and rodent networks, respectively, that indicate significance of difference from a random network (see Supplementary Methods in the Supporting Information).
Mean weighted degree with weights adjusted according to sampling intensity (see Methods).
Quantitative connectance, which takes into account edge weights.
Quantitative connectance, with weights adjusted according to sampling intensity.
Quantitative connectance, where the weights are the proportion of viruses shared rather than the absolute number (see Methods).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
To account for sampling bias, we additionally calculated the quantitative linkage densities and connectance for sampling effort‐corrected networks. See Supporting Information for more details. As another test to account for bias, we calculated the quantitative connectance of the Jaccard matrices (the intersection divided by the union of the viruses for each pair of host species). This gives the proportion of the viruses shared rather than the absolute number. The multiple regression on matrices and GLS analyses described below use the original networks (absolute number of viruses not weighted by sampling effort) because for those, we take sampling effort into account as a covariate.
To calculate P‐values for the network statistics in Table 1, we performed 10 000 permutations of the networks, where we shuffled the edge weights. See Supporting Information for more details.
Species traits were compiled from online databases and the literature as described in Luis et al. (2013). We examined body mass, number of litters per year, litter size, maximum longevity, torpor use, International Union for Conservation of Nature (IUCN) conservation status, geographical distribution area, latitude of the midpoint of the species distribution, number of other species in the same taxonomic order that are sympatric, and for bats only, migratory classification, diet, gregariousness and propensity to roost in caves. See supplemental Figures S4–S7 in Luis et al. (2013) for plots of the raw data. Bat species traits that were new to these analyses were gregariousness, propensity to roost in caves and diet (see Supporting Information for more details). Virus and host trait data and R code are provided online as Supporting Information.
Multiple regression for correlates of viral sharing
Multiple regression on distance matrices (MRM) (Lichstein 2007) was used to detect significant correlations between viral sharing, phylogeny, sympatry and similarity in host traits using ‘MRM’ in the R package ‘ecodist’ (Goslee & Urban 2007; R Core Team 2012). The response variable was the viral sharing matrix (adjacency matrix of the network), with each entry giving the number of viruses shared between each pair of host species.
The ‘ape’ package in R (Paradis et al. 2004) was used to calculate a phylogenetic correlation matrix based on species’ shared branch lengths of the most complete mammalian phylogenetic supertree available containing bats and rodents (Bininda‐Emonds et al. 2007). See Figs S4 and S5 in Supporting Information.
Using the species distribution shape files from the IUCN website (http://www.iucnredlist.org/technical-documents/spatial-data) and the R packages ‘sp’ and ‘rgeos’ (Pebesma & Bivand 2005), a sympatry matrix was created. Each entry of the matrix was either a 1 if the two species’ distributions overlap or a 0 if they do not.
Matrices of sampling effort were created by multiplying the logged number of citations for each species together. Matrices showing similarity in host traits were also calculated. For example for each pair of species, the difference in the number of litters per year was calculated. This matrix was standardised so values ranged between 0 and 1, and the matrix used in the multiple regression models was 1 minus this matrix (so it would be a correlation matrix similar to the phylogenetic and sympatry matrices).
Community detection
Host species were grouped into ‘communities’, partitions of highly interconnected nodes, by using the community detection algorithm described in Blondel et al. (2008), which maximises the modularity between groups of the weighted networks. See Supporting Information for more details.
As an alternative to using modularity to assign nodes to communities, we also used clique percolation theory (Palla et al. 2005). In this method, nodes can belong to multiple communities. See Supporting Information for more details.
Generalised least squares – traits associated with important hosts
Generalised least squares (GLS) were used to examine correlates of viral richness per host species, degree and betweenness, while allowing for phylogenetic correlation to be incorporated into the error structure as described below.
Because many life history traits are correlated, we performed principal components analyses (PCA) on the life history traits: logged body mass, maximum longevity, number of litters per year and litter size (separately for bats and rodents). The variables were rescaled to have unit variance before analysis in R using the ‘prcomp’ function (R Core Team 2012), and these principal components were then used in subsequent analyses. See Fig. S6 for PCA plots and Table S1 for loading values and variance explained by each principal component.
Because closely related species share traits, we tested for phylogenetic dependence by setting the error term for the GLS to the phylogenetic correlation matrix multiplied by an additional parameter, Pagel's λ, that was estimated to determine the strength of phylogenetic dependence (Pagel 1999; Freckleton et al. 2002). A λ estimate of 1 indicates that the error structure of the model was directly proportional to the species shared branch lengths. A λ of 0 indicates the error structure of the model was not related to the species shared branch lengths (i.e., phylogeny does not explain any additional variation). Note that phylogeny is not a covariate in this analysis, and may not be significant if the model covariates themselves have phylogenetic dependence. Models were ranked by their AICc (Akaike information criterion adjusting for finite sample sizes) values. Correlation coefficients (R) were Pearson's product moment correlation comparing observed values to model predictions.
We also ran these models using MCMC generalised linear mixed effects models (Hadfield, 2010). See the Supporting Information for more details.
Maps
Maps were made using the R packages ‘maptools’, ‘maps’, ‘sp’ and ‘PBSmapping’ (Pebesma & Bivand 2005; Becker et al., 2012; Schnute et al. 2012; Bivand & Lewin‐Koh 2013). Viruses were mapped by the union of their hosts’ distributions using the command ‘gUnionCascaded’ from the ‘rgeos’ package (Pebesma & Bivand 2005). The assumption was made that the virus exists in the entire distribution of its host(s), analogous to the fundamental niche concept (Hutchinson 1957; Harris & Dunn 2010). To visually assess whether sampling effort had a strong effect on the virus distributions, we also plotted these distributions discounting by sampling effort of the host species (Figs S7 and S8).
Results
The bat network had 143 host species connected by 110 viruses, and the rodent network had 196 host species connected by 185 viruses (Figs 3a, 4a, S1 and S2). A species’ degree is the number of other host species it is connected to by shared viruses. Degree in bats ranged from 0 to 53 with a mean of 15.0 and in rodents ranged from 0 to 78 with a mean of 12.5 (Table 1; See Fig. S3 for degree distributions). The mean degree and mean weighted degree were significantly higher in bats than in rodents. There was significant transitivity (the probability that the adjacent nodes of a node are also connected) in both bats and rodents. Assortativity in bats was significant, suggesting that bat species with high degree tend to interact with other bat species of high degree (and low degree with low degree), but there is no evidence of this in rodents (Table 1).
The bat network was 1.64 times more connected (connectance = links/species2) than the rodent network (Table 1). This was statistically significant – not once in 10 000 permutations was the difference between connectance for the bat and rodent networks at least the observed difference. The quantitative connectance adjusted for sampling effort and the quantitative connectance on the Jaccard matrices were also higher in bats (Table 1).
Multiple regression on matrices for correlates of viral sharing
Next, we examined the connections between species, using multiple regression on matrices (Lichstein 2007) to determine the correlates of the number of viruses that two host species share. Both phylogeny (relatedness) and sympatry (geographic range overlap) were important predictors of viral sharing (Table 2). Species with overlapping distributions and closer phylogenetic relationships shared more viruses. Sympatry was more important than phylogeny for viral sharing in both orders, but both explained more variation in bats. Sampling effort (by number of citations on Web of Science) was also important. Well‐studied pairs of species were found to have more viruses shared between them. The amount of variation explained by sympatry, phylogeny and citations together was 21% in bats and 14.3% in rodents. Similarity in life history traits explained a small amount of additional variation (Table S2).
Table 2.
Multiple regression of matrices for correlates of the number of viruses shared between two species
| Model | R 2 | P |
|---|---|---|
| Bats | ||
| ∼ phylogeny + sympatry + citations | 0.210 | 0.0001 |
| ∼ sympatry + citations | 0.186 | 0.0001 |
| ∼ phylogeny + sympatry | 0.175 | 0.0001 |
| ∼ sympatry | 0.148 | 0.0001 |
| ∼ phylogeny + citations | 0.102 | 0.0001 |
| ∼ phylogeny | 0.058 | 0.0001 |
| ∼ citations | 0.050 | 0.0001 |
| Rodents | ||
| ∼ phylogeny + sympatry + citations | 0.143 | 0.0001 |
| ∼ sympatry + citations | 0.138 | 0.0001 |
| ∼ phylogeny + sympatry | 0.104 | 0.0001 |
| ∼ sympatry | 0.102 | 0.0001 |
| ∼ phylogeny + citations | 0.073 | 0.0001 |
| ∼ citations | 0.060 | 0.0001 |
| ∼ phylogeny | 0.006 | 0.0001 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Traits associated with important hosts
Although bats generally have received attention as disease reservoirs, some species are more important for disease emergence. We determined which species have the most viruses, the highest degree (the most connections) and the highest betweenness in the network (a measure of network centrality – the number of shortest paths between any two nodes that go through the node of interest) and host traits associated with these network metrics. For all analyses of the bat and rodent networks, sampling effort was important – the more a species was studied, the more viruses, higher degree and higher betweenness. Consequently, we adjusted for sampling effort by number of citations on Web of Science.
The most important variable associated with hosting more viruses in bats was diet (Fig. 2a, Table S3 and S4). Host traits that correlated with the highest degree within the bat network (the most connections or viruses shared), in order of importance, were gregariousness and sympatry; diet was marginally important (Fig. 2b, Table S6 and S7). Migration was the most important host trait associated with betweenness in bats (Fig. 2c, Table S9 and S10).
Figure 2.
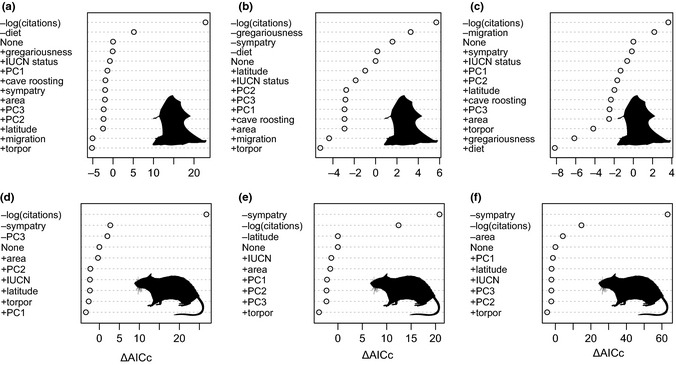
Ranking of host trait variables from the GLS models by ΔAICc: the change in AICc values when each variable is individually added (+) or removed (−) from the best model for (a) the number of viruses identified in a bat species (best model: number of viruses ∼ log(citations) + diet), (b) the degree of a bat species in the viral sharing network (best model: degree ∼ log(citations) + gregariousness + sympatry + diet), (c) the betweenness of a bat species in the viral sharing network (best model: betweenness ∼ log(citations) + migration), (d) the number of viruses identified in a rodent species (best model: number of viruses ∼ log(citations) + sympatry + PC3), (e) the degree of a rodent species in the viral sharing network (best model: degree ∼ log(citations) + sympatry + latitude) and (f) the betweenness of a rodent species in the viral sharing network (best model: betweenness ∼ log(citations) + sympatry + area).
In these analyses, phylogeny is not a covariate, but incorporated into the error structure, and an additional phylogenetic correlation parameter (λ; Pagel 1999) is estimated, to determine if phylogeny can explain variation unexplained by the model covariates. For bats, λ, was 0 for the best models for number of viruses and degree (Table S3 and S6), indicating that phylogeny did not explain any additional variation. A likely explanation is that the best models included diet, which is strongly related to phylogeny. However, λ for the best model for betweenness was 0.79 (Table S9), indicating that phylogeny explained additional variation.
For rodents, sympatry was the most important host trait; species whose distributions overlapped with a greater number of other rodent species had more viruses and higher degree and betweenness (Fig. 2d–f and Table S11–S19). In addition, rodent species that are larger, longer lived and with higher reproductive rates (e.g. Sigmodon hispidus, Rattus norvegicus) had more viruses (PC3; Table S1, S11–S13). Species at higher latitudes had marginally higher degree (after controlling for sympatry: Table S14–S16).
The results from the MCMC generalised linear mixed effects models were largely similar to these results (See Tables S5, S8, S13, S16 and S19). The only significant difference was the best model for the number of viruses in bats included only citations and phylogeny (Table S5). See the Supporting Information for more details.
Community detection
Although 88 and 69% of all possible pair‐wise combinations of species are connected by < 8 degrees of separation in the bat and rodent networks, respectively, the influence of a single species may be more localised. We detected distinct ‘communities’ of viral sharing within orders (Figs 3a and 4a), by maximising the modularity (Blondel et al. 2008). Modularity can attain values in the range from −0.5 to 1. If positive, there are more within‐group connections than would be expected at random, and in practice, values > 0.3 indicate significant community structure (Newman 2004). Modularity scores were 0.54 and 0.55 for the bat and rodent networks respectively.
Figure 3.
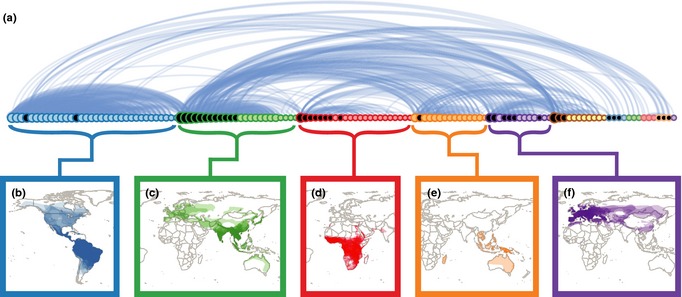
(a) The viral sharing network in bats, organised by communities. Each circle represents a bat species. Lines and their thickness represent number of viruses shared between species. Circle size represents the host's degree and is coloured by community. Species with a black centre were classified as part of multiple communities in an alternative method for community identification. (See Fig. S1 for diagram with species names) Panels (b) through (f) show the distributions of the species (with darker areas representing more host species) that make up the five largest communities. Important viruses include (b) Dengue, Eastern equine encephalitis and Yellow fever viruses in the blue community, (c) Japanese encephalitis virus and SARS coronavirus in the green community, (d) Ebola‐Zaire and Lake Victoria Marburg viruses in the red community, (e) Hendra and Nipah viruses in the orange community, (f) European bat lyssavirus‐1 and ‐2, and Issyk‐kul virus in the purple community.
Figure 4.
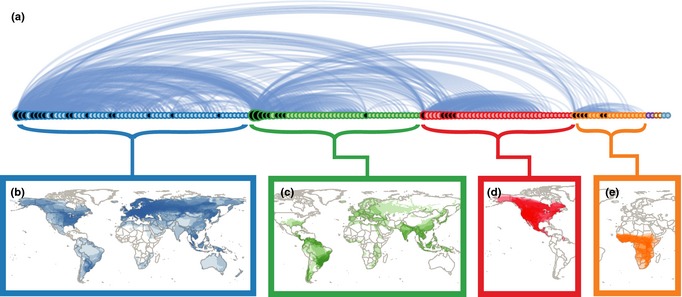
(a) The viral sharing network in rodents. See Fig. 2 caption for description. Panels (b) through (e) show the distributions of the species that make up the four largest communities of viral sharing in rodents. (b) Important viruses in the blue community include California encephalitis, Cowpox and Lymphocytic choriomeningitis viruses, among others. (c) Important viruses in the green community include Andes and Encephalomyocarditis viruses. (d) Important viruses in the red community include Colorado Tick fever and Sin Nombre viruses. (e) Important viruses in the orange community include Bunyamwera and Rift Valley fever viruses.
In the bat network, there were 10 viral sharing communities. Each species had distributions that on average overlapped with 85% of the species within its community vs. 18% of species outside (t = 8.82, d.f. = 12.54, P = 9.8 × 10−7; Fig. 3b–f). The largest and most densely connected community (Fig. 3b) consisted of 34 bat species in the Americas, largely connected by vector‐borne viruses. The remaining communities were connected by a mixture of vector‐borne and non‐vector‐borne viruses. The green community (Fig. 3c) was broadly distributed from Europe to Australia, and approximately half of the species in this community also shared viruses with species in other communities that overlapped spatially.
There were two globally distributed communities of rodents with 61 and 44 species (Fig. 4b,c) and two communities that were more spatially restricted. Each rodent species on average overlapped with 68% of the species within its community vs. 7% outside of its community (t = 4.66, d.f. = 6.12, P = 0.003; Fig. 4b–e). The house mouse, Mus musculus, has spread across the globe along with its viruses (Fig. 4b). Removing this species results in largely the same community structure, indicating this species has already facilitated the transmission of viruses to multiple sympatric species across the globe. The black rat, Rattus rattus, and the brown rat, R. norvegicus, are also widely distributed and are the main species holding the green community together (Fig. 4c). When either Rattus species is removed from this community, it splits into several smaller communities, suggesting less secondary viral spread to sympatric rodent species.
Discussion
Overall properties of the bat and rodent networks provide a global view of viral sharing within each order, to our current knowledge. The number of viruses per host species, the number of hosts per virus, the mean degree and connectance were higher in the bat network than the rodent network. This suggests that viruses may pass more easily between bat species than between rodent species. Our analyses indicate that characteristics unique to bats, such as gregariousness and migration, may facilitate this increased transmission.
Previous studies hypothesised frugivory as an important viral transmission mechanism among bats and from bats to other species, because virus has been isolated from partially eaten fruit, which is often shared and dropped to the ground (Chua et al. 2002; Dobson 2005). Our study is consistent with that hypothesis, with frugivores hosting more viruses than nectivores, insectivores and sanguivores. However, diet is strongly correlated with phylogeny, and the best model using MCMC glmm only included phylogeny. Therefore, these results could indicate the importance of diet or, alternatively, some other factor correlated with phylogeny, such as immunological functioning, for example.
The most important host characteristics associated with degree in bats were gregariousness and sympatry. These characteristics have been hypothesised as important mechanisms for viral maintenance and spread (Calisher et al. 2006; Luis et al. 2013) – bat species with distributions that overlap with a greater number of other bat species, and particularly those which are gregarious, will have greater interspecific contacts and chance for cross‐species transmission. At some roosting sites, bat densities can be as high as 3000 bats per square metre, and some roosts house diverse assemblages of species (Constantine 1967; Kunz 1982). High densities can lead to high contact rates, facilitating pathogen transmission and persistence. These dense roosting sites are often caves, therefore we had hypothesised that propensity to roost in caves would be an important predictor of viral sharing in bats. However, colony size (gregariousness) was more important than where the colony roosts. Large colony sizes do not occur on the same scale in rodents and may have led to the higher mean degree and connectance in bats compared to rodents.
Species with high betweenness might not have or share the most viruses, but could play a key role in disseminating viruses because they can connect disparate regions of the network. Migration was the most important host trait associated with betweenness in bats. Long‐distance animal movement has been hypothesised to enhance geographic spread of infectious disease, however, evidence in most cases is lacking (Altizer et al. 2011). In the bat network, many of the shortest network paths went through regional migrants (moving 100–500 km annually), which were more important than long‐distance migrants (> 500 km). This is consistent with the idea of ‘migratory culling’ (Bradley & Altizer 2005) in which the physiological stress of long‐distance migration can increase mortality of infected individuals.
For rodents, sympatry was the most important covariate, again emphasising the importance of spatial overlap in cross‐species transmission. We found that species that had higher reproductive rates and body mass had more viruses (PC3 of the PCA), which is in accordance with the findings of Han et al. for all rodent pathogens and parasites (2015).
This is the first analysis we are aware of that attempts to identify communities of pathogen sharing at a global scale. These communities reveal which species are most highly connected, providing insight into virus maintenance and spread. They may also be useful in guiding public health strategies. For example if a new zoonotic virus is identified in Hipposideros speoris, it may be useful to look for it in other species of the green community (Fig. 3c) as well.
The links in our networks represent real connections, the sharing of viruses, and may be evidence of cross‐species transmission, coevolution of host and virus lineages, or indirect links of cross‐species transmission through an intermediate host or vector. The importance of phylogeny for viral sharing could indicate the virus originated in a common ancestor and was maintained or coevolved in separate lineages, or the greater ability of viruses to transmit between closely related species. However, the greater importance of sympatry in viral sharing indicates that cross‐species transmission is at least as important if not more important than coevolution in establishing the network.
For cross‐species transmission to take place, there must be contact, direct or indirect, between two host species, and therefore, it is not surprising that sympatry is important in the sharing of viruses. The importance of sympatry is reflected in similarities between viral and host species richness maps (Fig. 1). Locations where host species diversity is highest generally have more viruses. However, bats in Europe and India have greater viral richness than predicted by the host species richness map (Fig. 1a,c), and this does not appear to be due to sampling bias (Fig. S7). The community context may provide additional insight. There are two host–virus communities connecting Europe to East Asia where species richness is high (Fig. 3c,f), and these communities may provide a bridge. Higher viral richness than expected from host species richness in India may also reflect connections with areas of high species richness in S.E. Asia (Fig. 3c). For the viral richness maps, we assumed that the virus exists throughout the distribution of its host(s) analogous to the fundamental niche concept (Hutchinson 1957; Harris & Dunn 2010) and representing the broadest possible distribution. However, there may be barriers that restrict the realised niche of a virus. For example Rhinolophus ferrumequinum hosts SARS‐like coronavirus and Japanese encephalitis virus, and its range extends from E. Asia to W. Europe, but the populations in E. Asia and Europe do not appear to mix (Flanders et al. 2009).
Thirteen viruses were present in both bats and rodents. Ten of these were vector transmitted. Therefore, direct transmission between bats and rodents appears rare. Of the three directly transmitted viruses, only Ebola could have been transmitted from bats via fruit. The other viruses shared between bats and rodents in this study, Hantaan and Puumala, were found in insectivorous bats, and transmission may have occurred from rodents, as they are currently thought to be the reservoirs of these hantaviruses. However, a recent study indicates bats may be important and ancient reservoirs of hantaviruses more generally (Guo et al. 2013).
A recent study examining the sharing of parasites in primates using similar network methods used an overall metric for network centrality which included weighted degree, betweenness and several other network measures of the importance of a node (Gómez et al. 2013). In this study, the authors found that hosts with denser populations living in larger groups and having broad distributions had higher centrality. Although overall, degree and betweenness were correlated in our study, the metrics were different enough to be best predicted by different covariates in bats – gregariousness for degree, and migration for betweenness. However, in rodents, the most important factor was the same for the different metrics – sympatry best predicted the number of viruses, degree and betweenness.
For our analysis we used a database covering 78 years of publications (1934–2011). Subsequent analyses, particularly focusing on bats as reservoirs of viruses, have been published (e.g. Drexler et al. 2012; Quan et al. 2013). Studies such as Drexler et al.'s (2012) report short genome fragments that are yet to be categorised by the ICTV, which makes it difficult to discern what sequences should be considered the same virus for classification of viral sharing, and thus we have not included them in our analyses. However, molecular studies increase our ability to distinguish viruses and understand virus–host relationships. As interest in reservoirs of emerging infectious diseases increases, especially in bats, we predict that results such as these will further strengthen our findings and allow for more refined analyses at the community level. Indeed, several recent studies support our conclusions, showing a preponderance of host switching between bats and from bats to other mammals (Drexler et al. 2012; Guo et al. 2013; Drexler et al. 2014).
This study is based on a large literature search, and therefore there are necessarily constraints on inference, given different motivations for, and methods used during studies of both rodent and bat viruses through time. We were concerned that the recent attention on bats as reservoirs may have biased the sampling and artificially increased the apparent connectance of the bat network; therefore as a secondary analysis, we also created networks using only viral accounts before the year 2000. We found that although both the bat and rodent networks were smaller (the bat network was reduced to 120 species and 744 links; and the rodent network to 145 species and 738 links), the connectance remained relatively steady (5.2% in bats and 3.5% in rodents), suggesting that the apparent greater viral sharing in bats compared to rodents is not merely a function of recent attention on bats. However, the importance of citations in our analyses for both orders highlights the importance of sampling effort overall.
Although we were able to account for sampling effort in the analyses examining species traits associated with viral sharing, it is difficult to account for sampling effort in creating the networks themselves. One approach which we implemented was creating networks using weighted edges based on sampling effort; this gave us confidence that among the species in our database, the connectance is higher in bats than in rodents. Although our database only has 9% of extant rodent species and 13% of extant bat species, the rodents in our database have been more studied overall and more studied for viruses than bats even when removing the most highly studied, lab rodents – Mus musculus and Rattus norvegicus (significant by t‐test on log number of citations for the species name, means = 4.90 & 4.44, P = 0.002; and the species name + virus, means = 1.99 & 1.43, P < 0.001). We would expect more of the total viral community to have been identified in the more highly studied group of species (rodents), thus the sampling bias should increase the apparent connectance in rodents compared to bats. However, this does not account for species not in our database. If the rodent species that have not been sampled share more viruses than the bat species that have not been sampled, then these results may not hold. Similarly, the species trait data was only available for roughly 37% of the bat and rodent species in our networks, which corresponds to 5 and 3% of known extant bat and rodent species respectively; therefore, the species traits associated with important hosts may also change as our knowledge increases. New viruses are being discovered frequently, suggesting that there are many more viruses yet undetected in both bats and rodents. Therefore, our conclusions and the maps of viral diversity for both orders (Fig. 1) may change significantly as our knowledge increases. We hope that this first, broad examination of viral sharing will help motivate future efforts including more directed data collection.
Another potential source of bias may be reports of spillover or incidental hosts that are not important reservoirs but are treated with equal weight in these analyses. We took care to distinguish the hantaviruses and arenaviruses, for which there is relatively good knowledge of host–virus relationships. However, we found that this did not impact the qualitative conclusions – when using ICTV classifications the rodent network still had lower mean degree (13.8), mean weighted degree (19.01), connectance (3.51%) and transitivity (0.57) than the bat network. However, for most of the reported viruses in bats and rodents, there is little knowledge of the ecology and epidemiology in these hosts. This again highlights the importance of further study of both bats and rodents as reservoirs of viruses.
Our networks connect hosts by their shared viruses. For a given pair of connected species, we do not know in which direction the cross‐species transmission may have taken place, or if transmission even occurred between these species. There may have been a third species that transmitted the virus to both species. As more viral sequences become available, molecular methods, such as those used by Streicker et al. (2010), may be used to infer directionality in cross‐species transmission, and help resolve these issues.
Our study highlights the interconnectedness of species with respect to viruses and shows the benefits of examining pathogens in a community ecology context at several scales. Our analyses suggest that unique bat characteristics figure importantly in sharing and spreading viruses, lending quantitative support for bats’ special status in zoonotic virus emergence and demonstrating which characteristics affect reservoir potential. Practically, our analyses may help guide future surveillance for optimal prevention of emerging zoonoses. This could help campaigns aimed at preventing spillover between bats and humans to benefit human health while conserving bats’ important roles in ecosystem services such as pest management, plant pollination and seed dispersal (Boyles et al. 2011).
Author Contributions
ADL, CTW, JLNW and AAC designed study. ADL, TJO, DTSH, ATG and JNM compiled data. ADL performed analyses and wrote the manuscript, and all other authors contributed substantially to editing.
Supporting information
Acknowledgements
This work was supported by the Research and Policy for Infectious Disease Dynamics (RAPIDD) program of the Science and Technology Directorate (US Department of Homeland Security) and the Fogarty International Center (National Institutes of Health). D.T.S.H. acknowledges funding from a David H. Smith post‐doctoral fellowship. A.A.C. is partially funded by a Royal Society Wolfson Research Merit award, and J.L.N.W. is supported by the Alborada Trust. Thanks to Paul Cryan and Michael O'Donnell of the USGS Fort Collins Science Center for help with species distribution analyses.
References
- Altizer, S. , Bartel, R. & Han, B.A. (2011). Animal migration and infectious disease risk. Science, 331, 296–302. [DOI] [PubMed] [Google Scholar]
- Becker, R.A. , Wilks, A.R. & R version by Ray Brownrigg. Enhancements by Thomas P Minka . (2012). maps: Draw Geographical Maps. R package version 2.3‐0. http://CRAN.R-project.org/package&=maps. Last accessed 30 September 2014. [Google Scholar]
- Bersier, L.F. , Banašek‐Richter, C. & Cattin, M.F. (2002). Quantitative descriptors of food‐web matrices. Ecology, 83, 2394–2407. [Google Scholar]
- Bininda‐Emonds, O.R. , Cardillo, M. , Jones, K.E. , MacPhee, R.D. , Beck, R.M. , Grenyer, R. et al (2007). The delayed rise of present‐day mammals. Nature, 446, 507. [DOI] [PubMed] [Google Scholar]
- Bivand, R. & Lewin‐Koh, N. (2013). maptools: Tools for Reading and Handling Spatial Objects. R package version 0.8‐23. [Google Scholar]
- Blondel, V.D. , Guillaume, J.L. , Lambiotte, R. & Lefebvre, E. (2008). Fast unfolding of communities in large networks. J. Stat. Mech: Theory Exp., 2008, P10008. [Google Scholar]
- Boyles, J.G. , Cryan, P.M. , McCracken, G.F. & Kunz, T.H. (2011). Economic importance of bats in agriculture. Science, 332, 41–42. [DOI] [PubMed] [Google Scholar]
- Bradley, C.A. & Altizer, S. (2005). Parasites hinder monarch butterfly flight: implications for disease spread in migratory hosts. Ecol. Lett., 8, 290–300. [Google Scholar]
- Calisher, C.H. , Childs, J.E. , Field, H.E. , Holmes, K.V. & Schountz, T. (2006). Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev., 19, 531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K.B. , Lek Koh, C. , Hooi, P.S. , Wee, K.F. , Khong, J.H. , Chua, B.H. et al (2002). Isolation of Nipah virus from Malaysian island flying‐foxes. Microbes Infect., 4, 145–151. [DOI] [PubMed] [Google Scholar]
- Constantine, D.G. (1967). Activity patterns of the Mexican free‐tailed bat. University of New Mexico Publications in Biology no. 7.
- Cui, J. , Tachedjian, M. , Wang, L. , Tachedjian, G. , Wang, L.F. & Zhang, S. (2012). Discovery of retroviral homologs in bats: implications for the origin of mammalian gammaretroviruses. J. Virol., 86, 4288–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, G. , Grodums, E.I. & Spencer, W.A. (1966). Experimental Coxsackie B‐3 virus infection in Citellus lateralis. J. Cell. Physiol., 67, 443–454. [DOI] [PubMed] [Google Scholar]
- Dobson, A.P. (2005). What links bats to emerging infectious diseases? Science, 310, 628. [DOI] [PubMed] [Google Scholar]
- Drexler, J.F. , Corman, V.M. , Müller, M.A. , Maganga, G.D. , Vallo, P. , Binger, T. et al (2012). Bats host major mammalian paramyxoviruses. Nat. Commun., 3, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler, J.F. , Corman, V.M. & Drosten, C. (2014). Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res., 101, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders, J. , Jones, G. , Benda, P. , Dietz, C. , Zhang, S. , Li, G. et al (2009). Phylogeography of the greater horseshoe bat, Rhinolophus ferrumequinum: contrasting results from mitochondrial and microsatellite data. Mol. Ecol., 18, 306–318. [DOI] [PubMed] [Google Scholar]
- Freckleton, R. , Harvey, P. & Pagel, M. (2002). Phylogenetic analysis and comparative data: a test and review of evidence. Am. Nat., 160, 712–726. [DOI] [PubMed] [Google Scholar]
- Gómez, J.M. , Nunn, C.L. & Verdú, M. (2013). Centrality in primate–parasite networks reveals the potential for the transmission of emerging infectious diseases to humans. Proc. Natl Acad. Sci., 110, 7738–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslee, S.C. & Urban, D.L. (2007). The ecodist package for dissimilarity‐based analysis of ecological data. J. Stat. Softw., 22, 1–19. [Google Scholar]
- Guo, W.P. , Lin, X.D. , Wang, W. , Tian, J.H. , Cong, M.L. , Zhang, H.L. et al (2013). Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog., 9, e1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield, J.D. (2010). MCMC methods for multi‐response generalized linear mixed models: the MCMCglmm R package. J. Stat. Softw., 33, 1–22.20808728 [Google Scholar]
- Han, B.A. , Schmidt, J.P. , Bowden, S.E. , Drake, J.M. (2015). Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci., 112, 7039–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, N.C. & Dunn, R.R. (2010). Using host associations to predict spatial patterns in the species richness of the parasites of North American carnivores. Ecol. Lett., 13, 1411–1418. [DOI] [PubMed] [Google Scholar]
- Hutchinson, G.E. (1957). Concluding remarks. Cold Spring Harbor Symp. Quant. Biol., 22, 415–427. [Google Scholar]
- IUCN (2010). IUCN red list of threatened species. version 2010.4. Available at: http://www.iucnredlist.org. Last accessed 20 October 2010.
- Jones, K.E. , Patel, N.G. , Levy, M.A. , Storeygard, A. , Balk, D. , Gittleman, J.L. & Daszak, P. (2008). Global trends in emerging infectious diseases. Nature, 451, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz, T. (1982). Ecology of Bats. Plenum Press, New York. [Google Scholar]
- Kuzmin, I. , Mayer, A. , Niezgoda, M. , Markotter, W. , Agwanda, B. , Breiman, R. & Rupprecht, C. (2010). Shimoni bat virus, a new representative of the lyssavirus genus. Virus Res., 149, 197–210. [DOI] [PubMed] [Google Scholar]
- Lichstein, J.W. (2007). Multiple regression on distance matrices: a multivariate spatial analysis tool. Plant Ecol., 188, 117–131. [Google Scholar]
- Lloyd‐Smith, J.O. , George, D. , Pepin, K.M. , Pitzer, V.E. , Pulliam, J.R.C. , Dobson, A.P. et al (2009). Epidemic dynamics at the human‐animal interface. Science, 326, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis, A.D. , Hayman, D.T. , O'Shea, T.J. , Cryan, P.M. , Gilbert, A.T. , Pulliam, J.R. et al (2013). A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. Biol. Sci., 280, 20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya, J.M. , Pimm, S.L. & Solé, R.V. (2006). Ecological networks and their fragility. Nature, 442, 259–264. [DOI] [PubMed] [Google Scholar]
- Munshi‐South, J. & Wilkinson, G.S. (2010). Bats and birds: Exceptional longevity despite high metabolic rates. Ageing Res. Rev., 9, 12–19. [DOI] [PubMed] [Google Scholar]
- Newman, M.E. (2004). Fast algorithm for detecting community structure in networks. Phys. Rev. E, 69, 066133. [DOI] [PubMed] [Google Scholar]
- Pagel, M. (1999). Inferring the historical patterns of biological evolution. Nature, 401, 877–884. [DOI] [PubMed] [Google Scholar]
- Palla, G. , Derényi, I. , Farkas, I. & Vicsek, T. (2005). Uncovering the overlapping community structure of complex networks in nature and society. Nature, 435, 814–818. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , Claude, J. & Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics, 20, 289. [DOI] [PubMed] [Google Scholar]
- Pebesma, E.J. & Bivand, R.S. (2005). Classes and methods for spatial data in R. R. News, 5, 9–13. [Google Scholar]
- Prendergast, B.J. , Freeman, D.A. , Zucker, I. & Nelson, R.J. (2002). Periodic arousal from hibernation is necessary for initiation of immune responses in ground squirrels. Am. J. Physiol. Regul. Integr. Comp. Physiol., 282, R1054–R1062. [DOI] [PubMed] [Google Scholar]
- Quan, P.L. , Firth, C. , Conte, J.M. , Williams, S.H. , Zambrana‐Torrelio, C.M. , Anthony, S.J. et al (2013). Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl Acad. Sci., 110, 8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2012). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3‐900051‐07‐0. [Google Scholar]
- dos Reis, M. , Inoue, J. , Hasegawa, M. , Asher, R. , Donoghue, P. & Yang, Z. (2012). Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. Biol. Sci., 279, 3491–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer, J. , Perkins, S.L. , Decher, J. , Leendertz, F.H. , Fahr, J. , Weber, N. & Matuschewski, K. (2013). High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc. Natl Acad. Sci., 110, 17415–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute, J.T. , Boers, N. & library by Alan Murta, R.H.G. ( (2012). PBSmapping: Mapping Fisheries Data and Spatial Analysis Tools. R package version 2.65.40. http://CRAN.R-project.org/package=PBSmapping. Last accessed 30 September 2014. [Google Scholar]
- Streicker, D.G. , Turmelle, A.S. , Vonhof, M.J. , Kuzmin, I.V. , McCracken, G.F. & Rupprecht, C.E. (2010). Host phylogeny constrains cross‐species emergence and establishment of rabies virus in bats. Science, 329, 676. [DOI] [PubMed] [Google Scholar]
- Tong, S. , Zhu, X. , Li, Y. , Shi, M. , Zhang, J. , Bourgeois, M. et al (2013). New world bats harbor diverse Influenza A viruses. PLoS Pathog., 9, e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials