

Abstract
Background
Niemann-Pick type C (NPC) disease is a fatal neurodegenerative disorder characterized by the accumulation of free cholesterol in lysosomes. There are currently no effective FDA-approved treatments for NPC, although in the last years the inhibition of Histone Deacetylases (HDACs) has emerged as a potential treatment for this disease. However, the molecular mechanisms that deregulate HDACs activity in NPC disease are unknown. Previously our group had shown that the proapoptotic tyrosine kinase c-Abl signaling is activated in NPC neurons. Here, we demonstrate that c-Abl activity increases HDAC2 levels inducing neuronal gene repression of key synaptic genes in NPC models.
Results
Our data show that: i) HDAC2 levels and activity are increased in NPC neuronal models and in Npc1-/- mice; ii) inhibition of c-Abl or c-Abl deficiency prevents the increase of HDAC2 protein levels and activity in NPC neuronal models; iii) c-Abl inhibition decreases the levels of HDAC2 tyrosine phosphorylation; iv) treatment with methyl-β-cyclodextrin and Vitamin E decrease the activation of the c-Abl/HDAC2 pathway in NPC neurons; v) in vivo treatment with two c-Abl inhibitors prevents the increase of HDAC2 protein levels in the brain of Npc1-/- mice and, vi) c-Abl inhibition prevents HDAC2 recruitment to the promoter of neuronal genes, triggering an increase in their expression.
Conclusion
our data show the involvement of the c-Abl/HDAC2 signaling pathway in the regulation of neuronal gene expression in NPC neuronal models. Thus, inhibition of c-Abl could be a pharmacological target for preventing the deleterious effects of increased HDAC2 levels in NPC disease.
Keywords: Niemann-Pick type C disease, Neuronal genes, Histone deacetylase 2, Tyrosine kinase c-Abl, Imatinib, GNF2
1. Introduction
Neuronal gene repression has been extensively described in several neurodegenerative diseases (1). Specifically, histone deacetylation plays a fundamental role in neuronal gene repression in neurodegenerative processes (2-4). HDACs are a family of enzymes responsible for the regulation of histone deacetylation, which promote greater chromatin compaction making it inaccessible to the transcriptional machinery (5, 6). HDACs are grouped into four classes and have been implicated in diverse biological processes including cellular function, differentiation, development, apoptosis, and synaptogenesis (7). However, the roles of individual HDACs in brain function have only been addressed recently (4, 8, 9). HDAC1, HDAC2, HDAC3 and HDAC8 belong to class I HDACs. Interestingly, HDAC2 is a master regulator of histone deacetylation and gene repression of neuronal genes (1, 3, 4, 10, 11). Surprisingly, HDAC2 is predominantly expressed in neurons the adult brain compared to other HDACs of the same class, such as HDAC1, which is expressed in neural stem cells/progenitors and glia (12). HDAC2 is preferentially recruited on the promoters of neuronal genes triggering neuronal gene repression, cognitive problems and a decrease of synaptic plasticity in the adult brain (4).
Niemann-Pick type C (NPC) disease is a fatal pediatric neurodegenerative lysosomal storage disorder caused by mutations in the NPC1 or NPC2 genes (13, 14). Both genes encode cholesterol transport proteins and their deficiency leads to the pathogenic accumulation of cholesterol and other lipids (lactosylceramide, glucosylceramide, GM2 and GM3 glycosphingolipids and sphingosine) within the late endosomal/lysosomal compartment (15-18). NPC is a systemic disorder characterized primarily by neurodegeneration, especially in the cerebellum and brain cortex, and liver damage (19). Although the etiology of NPC has been already elucidated, there is no curative treatment for this devastating and fatal disorder yet. Nevertheless, Miglustat (N-butyldeoxynojirimycin; NB-DNJ; Zavesca®, Actelion Pharmaceuticals Ltd) is currently approved in the European Union (EU), for the treatment of progressive neurological manifestations in patients with NPC (19, 20). In addition, the cholesterol lowering agent hydroxypropyl-β-cyclodextrin (HP-β-CD) is undergoing a NIH clinical trial in NPC patients (20).
Interestingly, treatment with the class I HDAC inhibitor valproic acid prevents reduction in the expression of neuronal genes in NPC cellular models (21). In addition, treatment with HDACs inhibitors of classes I and II, reduces cholesterol accumulation in NPC human fibroblasts (22). Based on this evidence, HDACs inhibition has emerged as a potential therapeutic treatment for NPC disease (20, 23, 24). Although what HDACs are deregulated in NPC is currently unknown, the evidence suggests that a HDACs class I inhibitor could be a useful treatment to prevent neuronal gene repression and cognitive impairment. As previously mentioned, HDAC2 is the main HDAC I expressed in the adult brain. Moreover, HDAC2-overexpresssing mice show reduced expression of synaptophysin, decreased synaptic plasticity, fewer dendritic spines and cognitive impairments (4). In contrast, Hdac2 knockout mice show increased expression of synaptophysin, augmented synaptic plasticity and a higher number of dendritic spines in the CA-1 region, associated with improvement in associative learning compared to wild-type mice (4, 25). In addition, HDAC2 has been described as a key protein involved in neuronal gene repression in neurodegenerative diseases (3, 4, 10). Indeed, HDAC2 is increased in mouse models of Alzheimer's disease (AD) and samples from AD patients, whereas HDAC1 or HDAC3 are unchanged (3, 10, 26). Therefore, HDAC2 is a good therapeutic candidate for NPC disease.
Interestingly, we recently demonstrated that the activation of the tyrosine kinase c-Abl increases HDAC2 levels in AD models causing neuronal gene repression (10). Our findings show that c-Abl induces HDAC2 phosphorylation on tyrosine 222, increasing its stability and protein levels. This in turn increases histone H3 deacetylation and HDAC2 recruitment to the promoter of key HDAC2 target genes, such as synaptotagmin, NR2a and GluR1, inducing their transcriptional repression in AD models. Our results suggest that c-Abl/HDAC2 signaling is relevant in neurodegeneration (10).
Moreover, recent data show that c-Abl is also involved in HDAC1 regulation by inhibiting its proteasomal degradation (27), suggesting that c-Abl activity controls epigenetic changes of several genes in different cellular contexts.
Interestingly, although AD and NPC have a different etiology, both diseases share some pathological features, including the accumulation of amyloid-β (Aβ), tau pathology and lysosomal dysfunction (28, 29). Moreover, high cholesterol is a risk factor in AD and carrying the ε4 isoform of apolipoprotein E (ApoE) is a risk factor in both disorders (30-32). Accordingly, treatment with HP-β-CD lowered the levels of Aβ42 in a transgenic mouse model of AD, both by reducing Aβ production and enhancing clearance mechanisms (33).
c-Abl has been previously linked to NPC disease pathology. We demonstrated that c-Abl is activated in in vitro and in vivo NPC models, triggering neuronal apoptosis (34). Interestingly, treatment with the c-Abl inhibitor Imatinib decreases apoptosis of Purkinje cerebellar neurons, improving locomotor abilities and increasing the survival rates of Npc1-/- mice (35). In addition, c-Abl activity has been implicated in several neurodegenerative disorders such as AD (36, 37), Parkinson's Disease (38) and Amyotrophic Lateral Sclerosis (39). Finally, c-Abl-overexpressing mice show severe neurodegeneration (40).
In this work we analyzed the relevance of the c-Abl/HDAC2 pathway in NPC neuronal models. We found that HDAC2 protein levels and activity are increased in NPC neuronal models and in Npc1-/- mice. Also the pharmacological inhibition of c-Abl or c-Abl deficiency (c-Abl-/-) prevents the increase of HDAC2 levels and activity in NPC neuronal models and decreases the levels of tyrosine phosphorylation of HDAC2. In addition, we found that cholesterol accumulation and oxidative stress are upstream regulators of the c-Abl/HDAC2 pathway in NPC neurons. Furthermore, c-Abl inhibition prevents the recruitment of HDAC2 to the promoter of neuronal genes, triggering an increase in their expression. Our results suggest that c-Abl could be a therapeutic target for preventing HDAC2-mediated neuronal gene repression in NPC disease.
2. Results
2.1. c-Abl mediates the increase in protein levels and repression activity of HDAC2 in NPC neuronal models
To evaluate the relevance of HDAC2 function in the NPC phenotype we first assessed HDAC2 protein levels in NPC neuronal models. We transfected the hippocampal-like cell line HT22 with an shRNA against NPC1 for 48h. As expected, the decrease in NPC1 protein levels was associated with an increase in intracellular free cholesterol levels that was detected by filipin staining (Fig. 1A). Interestingly, HDAC2 protein levels were increased in this neuronal NPC model (Fig. 1B). Although the inhibition of HDACs function has been proposed to have beneficial effects on NPC pathology, this is the first time that HDAC2 levels are reported to be increased in NPC cells.
Figure 1. NPC neuronal models show increased HDAC2 protein levels and activity, effect that is prevented with Imatinib.
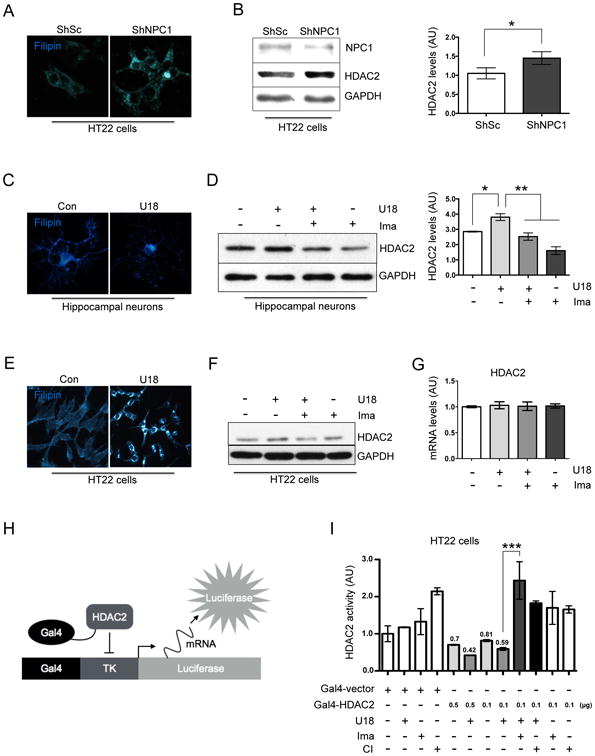
(A) Representative images showing filipin staining of HT22 cells transfected for 48 h with a plasmid expressing ShRNA against NPC1 (ShNPC1) or ShRNA scramble (ShSc) as a control. (B) Western blot analysis and quantification of HDAC2 expression normalized against GAPDH levels, of HT22 cells transfected for 48 h with a plasmid expressing a ShRNA against NPC1 (ShNPC1) or ShRNA scramble (ShSc) as a control (n=3). (C) Representative filipin staining images of hippocampal neurons (7DIV) treated either with vehicle (Ct) or U18 0.5 μg/mL for 24 h. (D) Western blot analysis and quantification of HDAC2 expression normalized against GAPDH levels of hippocampal neurons (7DIV) treated either with vehicle (control), U18 0.5 μg/mL, U18 0.5 μg/mL plus Imatinib (Ima) 5 μM or Ima 5 μM for 24 h (n=3). (E) Representative filipin images of HT22 cells treated with vehicle (Ct) or U18 0.5 μg/mL for 24h. (F) Western blot analysis of HDAC2 and (G) quantitative PCR results of Hdac2 mRNA levels in HT22 cells treated with vehicle (control), U18 0.5 μg/mL, U18 0.5 μg/mL plus Imatinib (Ima) 5 μM or Imatinib 5 μM for 24h (n=3). (H) Diagram of the HDAC2 repression activity assay. (I) HDAC2 repression activity assay were as follows: HT22 cells were co-transfected with the Gal4-TK-Luciferase, Gal4-Vector or decreasing quantities of Gal4-HDAC2. 24h after transfection, the cells were pre-treated with Imatinib 5 μM or CI994 (CI) 1 μM and 1 hour later the cells were treated with U18 0.5 μg/mL for 24 h. Results are from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; values are mean ± SEM.
We confirmed that the increase in HDAC2 protein levels were associated with the NPC phenotype using different NPC cellular models. The Chinese hamster ovary (CHO) cells containing a deletion in the NPC1 locus presented higher HDAC2 levels than wild type cells (Fig. S1A). Moreover, the increase in HDAC2 levels was observed in other NPC neuronal models. 7DIV rat hippocampal neurons or the HT22 cells were treated with the NPC inducer U18666A (U18) for 24h. As expected, U18 promoted intracellular free cholesterol accumulation, in both NPC neuronal models (Fig. 1C & 1E) and a clear increase in HDAC2 protein levels (Fig 1D & 1F). Interestingly, pretreatment with the specific c-Abl inhibitor Imatinib, prevented the increase in HDAC2 protein levels (Fig. 1D & 1F).
Next, we evaluated HDAC2 mRNA levels in NPC neuronal models. We found that HDAC2 mRNA levels remain unchanged under all conditions (Fig. 1G), indicating that the changes in HDAC2 protein levels were not related to changes in its mRNA levels.
To evaluate if the increase in HDAC2 protein levels correlate with an increase in its activity, we evaluated HDAC2 repression activity using a plasmid expressing a Gal4-HDAC2 fusion protein. This construct represses the luciferase expression of a reporter plasmid with a Gal4 binding site upstream of the thymidine kinase promoter (Gal4-TK-Luciferase) (41) (Fig. 1H). HDAC2 repression activity was assessed as the decrease in luciferase activity in HT22 cells transfected with the Gal4-HDAC2 recombinant plasmid versus a Gal4 empty vector. Concordantly with our previous results, treatment of HT22 cells with U18 produced an increase in Gal4-HDAC2-induced repression of luciferase gene in HT22 cells (Fig.1I), whereas Imatinib reduced Gal4-HDAC2-induced repression of the luciferase gene (Fig. 1I). As expected, the class I HDAC inhibitor CI994 also reduced Gal4-HDAC2-induced repression of the luciferase gene (Fig. 1I). Our data confirm an increase in HADC2 protein levels and activity in NPC neuronal models and suggest that c-Abl inhibition decreases the transcriptional repressor activity of HDAC2.
Next, we analyzed the levels of HDAC1, other class I HDAC protein that was previously shown to be upregulated by c-Abl (27). Concordantly, our results suggest that Imatinib treatment decreases HDAC1 levels (Fig. S2). However, we did not find a clear increase in the HDAC1 protein levels in an in vitro NPC model (Fig. S2).
To visualize HDAC2 localization in NPC neuronal models, we performed immunofluorescence experiments in rat hippocampal neurons at 7DIV and treated with U18 for 24 h. We observed an increase in HDAC2 signal and, as expected, HDAC2 was localized in the nucleus. The pre-treatment with the c-Abl inhibitor, Imatinib, prevented this increase (Fig. 2A).
Figure 2. The increase in HDAC2 protein levels in NPC neuronal models is mediated by c-Abl induced tyrosine phosphorylation.
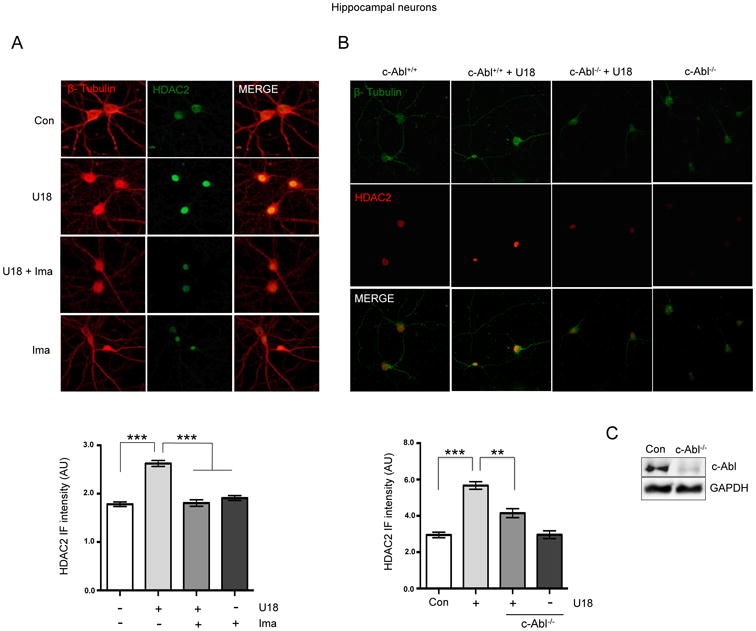
(A) Representative immunofluorescence and quantification of HDAC2 (green) and β-tubulin (red) levels in hippocampal neurons (7DIV) treated with vehicle (Ct), U18 0.5 μg/mL, U18 0.5 μg/mL plus Imatinib (Ima) 5 μM or Ima 5 μM for 24 h (n=3). (B) Representative immunofluorescence and quantification of HDAC2 (red) and β-tubulin (green) levels in hippocampal neurons (7DIV) of c-Abl +/+ and c-Abl -/- mice treated with vehicle (control) or U18 0.5 μg/mL for 24h. (C) Representative western blot of c-Abl expression in hippocampal neurons (7DIV) of c-Abl -/- mice. (D) Representative western blot of HDAC2 tyrosine phosphorylation. HT22 cells were transfected with a Flag-HDAC2 plasmid. After 24h the cells were pre-treated with Imatinib 5 μM, and 1h later cells were treated with U18 0.5 μg/mL for 24h. Flag-HDAC2 was immunoprecipitated and phospho-Tyrosine levels and HDAC2 expression were assessed by western blot analyses. Results are from three independent experiments. **p < 0.01; ***p < 0.001; values are mean ± SEM.
In addition, and to confirm that c-Abl activity mediates the increase in HDAC2 levels in the NPC cells, we analyzed primary cultures of hippocampal neurons (7DIV) from control (c-Abl floxo/floxo) and c-Abl null (c-Abl foxo/floxo Nestin-Cre) P1 mice. Interestingly, the c-Abl null neurons showed no increase in HDAC2 levels in immunofluorescence analysis when they were treated with U18 (Fig. 2B), while control neurons showed a clear increase. As expected, a very weak c-Abl signal was detected in western blot analysis in primary cultures of c-Abl-/-hippocampal neurons, probably due to c-Abl expression in other non-neuronal cells present in the culture (Fig. 2C). In agreement with our previously results in AD models (10), these data strongly suggests that in NPC neuronal models, the increase in HDAC2 levels is mediated by c-Abl.
To confirm the specificity of the Imatinib effect we performed experiments using inhibitors that do not target the c-Abl pathway and evaluated HDAC2 protein levels in HT22 cells (10). We found not change in HDAC2 protein levels using SP600125 (SP), PD 0325901 (PD) and Torin1, that inhibit JNK, MEK1/2 and mTORC1, respectively (Fig. S3).
We have previously demonstrated that c-Abl dependent phosphorylation regulates the stability of HDAC2 preventing its proteosomal degradation in neuronal models (10). We thought that the same mechanism could be involved in the regulation of HDAC2 levels by c-Abl in NPC neurons. To test this idea we transfected HT22 cells with a Flag-HDAC2 expression plasmid and treated the cells with U18. 24h later, we immunoprecipitated HDAC2 from whole cell lysates using anti-Flag beads, and evaluated tyrosine phosphorylation levels. We found that U18 treatment induced tyrosine phosphorylation of Flag-HDAC2, while Imatinib prevented it (Fig. 2D).
Thus, our data suggest that increased HDAC2 protein levels in NPC models is probably mediated by c-Abl tyrosine phosphorylation of HDAC2.
2.2 Cholesterol accumulation and oxidative stress are upstream regulators of the c-Abl/HDAC2 pathway in NPC neurons
Previously, our laboratory reported that cholesterol accumulation mediated by the NPC inducer U18, activates the c-Abl/p73 signaling in NPC neurons (34). Therefore, we evaluated if cholesterol accumulation causes increased HDAC2 protein levels by using the cholesterol reducing agent methyl-β-cyclodextrin (MβCD). U18 treated-HT22 cells were treated with increasing concentrations of MβCD and 24h later we evaluated cholesterol accumulation by filipin staining. Also, we measured the activation of c-Abl with a phopsho-c-Abl antibody and HDAC2 protein levels by western blot analysis. Interestingly, we found a decrease in both phospho-c-Abl and HDAC2 protein levels that correlated with the decrease in cholesterol filipin staining caused by MβCD treatment (Fig. 3A-D). Our previous results demonstrated that cholesterol accumulation in NPC neurons triggers oxidative stress and the activation of c-Abl/p73 signaling pathway. Accordingly, the use of antioxidants reduces c-Abl/p73 activation (34, 42). Therefore, we evaluated the effect of antioxidant treatment on HDAC2 protein levels in U18-treated HT22 cells. As expected, we found that the antioxidant Vitamin E (VitE) significantly decreased HDAC2 protein levels (Fig. 3E).
Figure 3. Cholesterol accumulation and oxidative stress are upstream regulators of the c-Abl/HDAC2 pathway in NPC neurons.
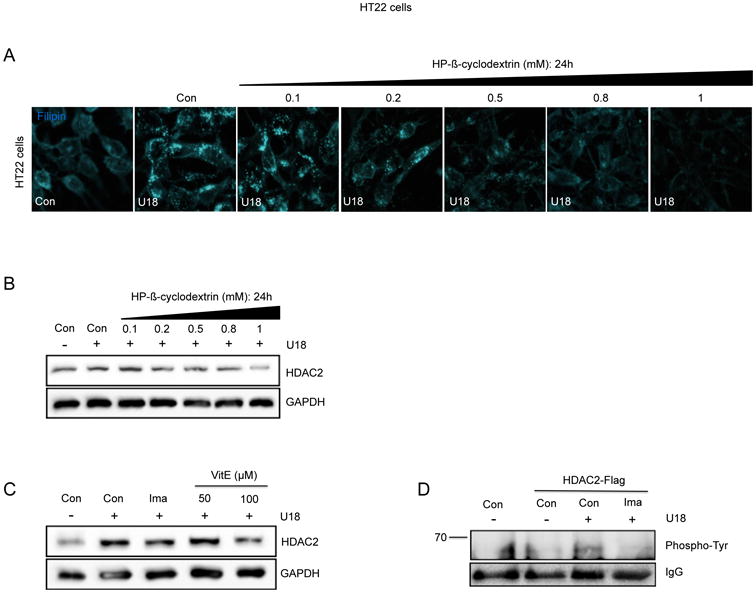
(A) Filipin stained HT22 cells treated with vehicle (Ct), U18 0.5 μg/mL for 24h, or U18 0.5 μg/mL for 24h plus increasing concentrations of methyl-β-cyclodextrin (MβCD) (0.1, 0.2, 0.5, 0.8 and 1 mM). (B-D) Representative western blot of HDAC2 expression in HT22 cells treated with vehicle (Ct), U18 0.5 μg/mL for 24h or U18 0.5 μg/mL for 24h plus increasing concentrations of methyl-β-cyclodextrin (0.1, 0.2, 0.5, 0.8 and 1 mM) (n=3). (E) Western blot of HDAC2 expression in HT22 cells treated with vehicle (Ct), U18 0.5 μg/mL for 24h, U18 0.5 μg/mL for 24h plus Imatinib (Ima) 10 μM and U18 0.5 μg/mL for 24h plus Vitamin E (VitE) at 50 and 100 μM.
Our results suggest that both, lipid accumulation and oxidative stress induce impairment of c-Abl/HDAC2 signaling in NPC neuronal models.
2.3. Imatinib and GNF2 treatments prevent the increase of HDAC2 protein levels in Npc1-/- mice
Previously, our laboratory showed that treatment of Npc1-/- mice with Imatinib increases locomotor function and survival (35). Therefore, we evaluated whether HDAC2 protein levels are upregulated in vivo in the NPC murine model, and if c-Abl activation is involved in this increase. We decided to analyze brain and cerebellum, two of the most affected areas in the CNS of NPC mice. To this end, four week-old Npc1-/- mice underwent daily treatment with Imatinib by intraperitoneal injection for 1 month. Then, we analyzed HDAC2 protein levels in the brain and in the cerebellum by western blot analysis. In concordance with the in vitro NPC models, we observed that NPC mice show higher levels of HDAC2 than wild type mice in brain and cerebellum (Fig 4A). Moreover, we observed that Npc1-/- mice treated with Imatinib presented HDAC2 levels similar to those of wild type mice (Fig. 4A). This result indicates that there is an increase in HDAC2 levels, which is prevented by c-Abl inhibition, in brain and cerebellum of Npc1-/-mice.
Figure 4. NPC in vivo models show increased HDAC2 levels, which are prevented by Imatinib and GNF2 treatments.
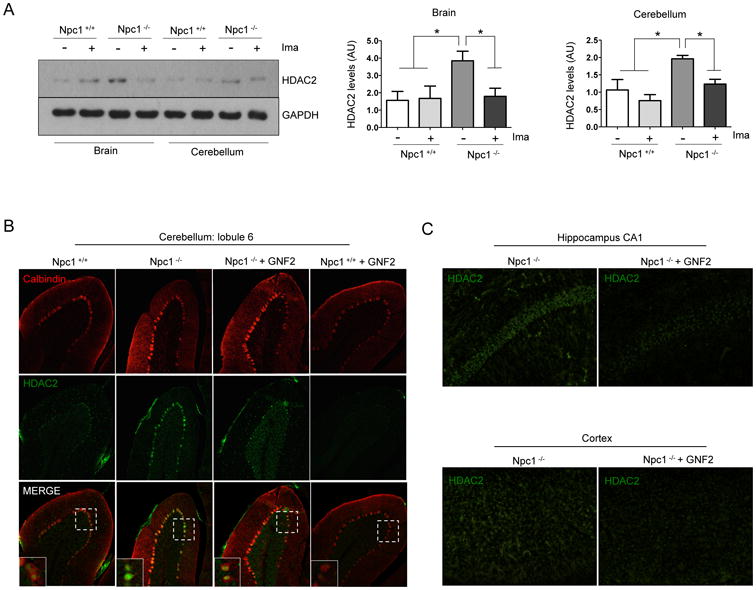
(A) Representative western blot and quantification of HDAC2 levels in brain and cerebellum extracts from 8-month-old wild-type (Npc1 +/+) and Npc1 -/- mice treated with Imatinib (Ima) 12.5 mg/Kg or vehicle, every day for four weeks by intraperitoneal injection (n=3). (B) Representative immunofluorescence micrographs showing HDAC2 levels in the lobule 6 of cerebellums of Npc1 +/+ and Npc1 -/- mice treated with vehicle or GNF2 5 mg/Kg. Results are from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; values are mean ± SEM.
To confirm our in vivo results we used a different c-Abl allosteric inhibitor, GNF2, and evaluated HDAC2 levels in the brain by immunofluorescence. We treated four weeks-old Npc1-/- animals for three weeks with GNF2. The immunofluorescence assays of Npc1-/- mice cerebellum clearly showed higher HDAC2 levels than wild type animals; while the cerebellum of Npc1-/- mice treated with GNF2 presented a fainter HDAC2 staining, similar to the wild type (Fig. 4B and Fig. S4). Moreover, HDAC2 was almost undetectable in wild type animals treated with GNF2. In addition, we analyzed HDAC2 levels in the CA1 area of the hippocampus and in the cortex. We also observed higher levels of HDAC2 in Npc1-/- mice. GNF2 treatment prevented the increase of HDAC2 levels in these same brain structures in Npc1-/- mice (Fig. 4C). Altogether, these results indicate that the increased HDAC2 levels in brain and cerebellum of Npc1-/- mice is mediated by c-Abl activity.
2.4. c-Abl inhibition prevents increased HDAC2 recruitment on neuronal genes and decreased mRNA levels of neuronal genes
After establishing that there is an increase of HDAC2 protein levels mediated by c-Abl in NPC neuronal models, we investigated whether this increment was associated with the repression of well-known HDAC2 neuronal target genes. First, we assessed the recruitment of HDAC2 to the promoter of the neuronal genes GluR1, NR2A, Synaptophysin and Synaptotagmin by chromatin immunoprecipitation (ChIP). We treated HT22 cells with U18 and observed that HDAC2 recruitment on the neuronal genes promoters was increased when compared with vehicle-treated cells (Fig. 5A). In agreement with our previous results, the recruitment of HDAC2 on the promoter of these four genes was prevented by the c-Abl inhibitor Imatinib (Fig. 5A). In the same way, U18 treatment of HT22 cells induced a significant decrease of acetylated histone H3 (H3ac) levels at the promoter of these genes (Fig. 5B), which correlates with increased recruitment of HDAC2 in HT22 cells treated with U18 (Fig. 5A). In addition, Imatinib pre-treatment significantly increased H3ac levels at the promoter of these neuronal genes (Fig. 5B), which also correlates with a decrease in the recruitment of HDAC2 to these promoters (Fig. 5A). Finally, we evaluated mRNA levels of these four neuronal genes by q-PCR. We observed a decrease in mRNA expression of GluR1, NR2A, Synaptophysin and Synaptotagmin in HT22 cells treated with U18 for 24h (Fig. 5C). Consistently, this repression of neuronal genes was prevented by the class I HDAC inhibitor, MS275 (Fig. 5C). More importantly, pretreatment with Imatinib, also prevented the reduction of the mRNA levels of the four neuronal genes evaluated (Fig. 5C). As a control, we analyzed the expression of the α-tocopherol transport protein gene (TtpA), which we had previously shown was not regulated by U18 treatment (43). As expected, the expression of this gene did not change with Imatinib or CI994 treatments (Fig. S5).
Figure 5. HDAC2 regulates synaptic gene expression in NPC neuronal models.

(A) Quantitative PCR results of the promoters of Glur1, NR2a, Synaptophysin and Synaptotagmin from HDAC2-immunoprecipitated chromatin and (B) histone H3-acetylated immmunoprecipitated chromatin from HT22 cells treated with vehicle (control), U18 0.5 μg/mL and U18 0.5 μg/mL plus Imatinib (Ima) 5 μM for 24h (n=3). (C) Quantitative PCR results of Glur1, NR2a, Synaptophysin and Synaptotagmin mRNA expression in HT22 cells treated with vehicle (control), U18 0.5 μg/mL, U18 0.5 μg/mL plus Imatinib (Ima) 5 μM and U18 0.5 μg/mL plus MS275 0.1 μM (MS) for 24h (n=3). Results are from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; values are mean ± SEM
These results suggest that, in NPC neuronal models, c-Abl inhibition prevents the recruitment of HDAC2 on the promoter of some key genes involved in synaptic plasticity resulting in an increase in their expression.
3. Discussion
In this work we report the participation of the c-Abl/HDAC2 signaling pathway in epigenetic alterations in NPC neurons. We found that HDAC2 levels and repression activity are increased in NPC neuronal models and also in Npc1-/-mice, and that the pharmacological inhibition of c-Abl or c-Abl deficiency prevents the increase of HDAC2 levels. In addition, we show that cholesterol accumulation and oxidative stress are upstream regulators of the c-Abl/HDAC2 pathway. Furthermore, c-Abl inhibition prevents the recruitment of HDAC2 to the promoter of neuronal genes, allowing an increase in their expression.
To our knowledge, this is the first report showing an increase in HDAC2 protein levels in different NPC neuronal models, including HT22 cells and hippocampal neurons treated with U18, and HT22 cells transfected with an shRNA against NPC1. Interestingly, although we did not find a clear increase in HDAC1 protein levels after U18 treatment of HT22 cells, Imatinib did decrease HDAC1 levels. These results suggest that in NPC neuronal models HDAC1 levels are regulated by other signaling pathways in addition to c-Abl. We also found increased HDAC2 protein levels in NPC mice brain and cerebellum, two CNS areas that are damaged in NPC patients. Increased expression of HDAC2 has also been reported in other neurodegenerative disease models. In AD, the increase in HDAC2 levels and activity has been linked to the deterioration of neuronal and synaptic function. Indeed, postmortem samples from AD patients show increased HDAC2 levels (3). Thus, the evidence suggests that HDAC2 inhibition, or the restraint of HDAC2 activity could be an effective strategy to ameliorate the progression of the disease and to improve the cognitive and motors skills in NPC patients, as has been proposed for AD.
Treatment with HDAC inhibitors was demonstrated to be effective against cognitive decline and neuronal gene repression in AD mouse models (2, 44-47), reducing cholesterol accumulation in mutant human fibroblasts and preventing the decrease in neuronal gene expression in NPC models (21, 22, 48). Indeed, HDACs inhibition was proposed as a therapeutic approach for NPC pathology (23, 24). Considering our results, HDAC2 should be considered a good therapeutic candidate for NPC. However, there are currently no available selective inhibitors of HDAC2 activity (49). Furthermore, the development of a specific HDAC2 inhibitor is challenging because the members of class I HDACs have a highly conserved catalytic domain (50). Thus, an alternative strategy could be to modulate the pathways involved in regulating HDAC2 levels and activity in NPC neurons.
Previously, our laboratory demonstrated that c-Abl is mainly activated in NPC neuronal models due to increased oxidative stress, and that treatment with antioxidants such as N-acetylcysteine and vitamin E can prevent c-Abl activation (34, 42). We also showed that treatment of Npc1-/- mice with Imatinib, a specific c-Abl inhibitor, improves locomotor abilities and increases survival rates (35). This effect correlated with decreases in c-Abl/p73 proapoptotic signaling and Purkinje neuron death and apoptosis (35). Here, we show that cholesterol accumulation and oxidative stress are upstream activators of the c-Abl/HDCA2 pathway and accordingly, treatment with MβCD and Vit E decrease phospho-c-Abl and HDAC2 levels in NPC neuronal models. In addition, our results suggest that tyrosine phosphorylation of HDAC2 by c-Abl, increases HDAC2 protein levels in NPC models. These results are in agreement with our previous findings in neuronal models in which we found that specific phosphorylation on tyrosine 222 promotes HDAC2 repression activity and prevents poly-ubiquitination and proteasomal degradation (10). However, further studies are required to establish if this specific HDAC2 tyrosine is phosphorylated by c-Abl, preventing its degradation in NPC neuronal models.
To demonstrate the connection between c-Abl and the increase in HDAC2, we used different NPC models and approaches. We found that the specific inhibitor Imatinib, prevents the increase in HDAC2 levels and activity in HT22 cells and hippocampal neurons treated with U18. Moreover, treatment with two different and specific c-Abl inhibitors prevents the increase in HDAC2 levels in the brain and cerebellum of NPC mice. In addition, we demonstrated that c-Abl activity is responsible for the increase of HDAC2 levels in NPC neuronal models using c-Abl-/- hippocampal neurons.
Interestingly, our results show that the change in HDAC2 protein levels and activity does not correlate with changes in HDAC2 mRNA levels, suggesting that c-Abl activity increases the stability of the HDAC2 protein, probably through c-Abl mediated HDAC2 tyrosine phosphorylation, a similar mechanism to that occurring in AD models (10).
HDAC2 has been described as an epigenetic repressor. Therefore, we focused our attention on known HDAC2 target genes that are relevant in the context of synaptic plasticity, including GluR1, NR2a, synaptophysin and synaptotagmin. These genes are down regulated in neurodegenerative diseases, such as in the human AD brain (3). In addition, several studies have shown that GluR1 and NR2a KO mice display learning and memory deficits (51, 52).
We found increased recruitment of HDAC2 on the promoter of GluR1, NR2a, synaptophysin and synaptotagmin genes and that this increase was prevented by Imatinib. Concordantly, U18 treatment promoted a reduction in acetylated histone H3 levels on the promoter of these synaptic genes, effect that was prevented by Imatinib. As a consequence of these chromatin changes, we expected a change in the mRNA expression of these genes. Accordingly, we found a reduction in their mRNA levels that was prevented by pre-treatments with the HDAC inhibitor MS-275 and with Imatinib.
We have previously described the reduction in neuronal gene expression in NPC models. Indeed, our laboratory demonstrated a change in the expression profile of NPC mice, especially in genes related with synaptic function and axogenesis (53). Although the molecular mechanisms involved are unknown, in this work we show evidence supporting the role of HDAC2 in repression of neuronal genes in NPC models.
4. Conclusions
Our data show that HDAC2 levels and repression activity are increased in NPC neuronal models. The pharmacological inhibition of c-Abl or the deficiency of c-Abl prevents the increase of HDAC2 proteins levels and activity and the recruitment of HDAC2 to the promoter of neuronal genes, triggering an increase in their expression, in NPC neuronal models.
The results of this work suggest that the c-Abl/HDAC2 signaling pathway could also contribute to the pathogenesis of NPC disease, decreasing neuronal gene expression and worsening neuronal function. Consequently, treatment with c-Abl inhibitors would also decrease c-Abl/HDAC2 signaling and therefore rescue neuronal gene expression, contributing to the improvement of NPC disease.
Finally, our results reveal a new mechanism of damage in NPC through the activation of the c-Abl/HDAC2 signaling pathway and the repression of neuronal genes expression. Thus, inhibition of c-Abl could be a pharmacological target for the prevention of the deleterious effects caused by increased HDAC2 levels in NPC.
5. Material and methods
5.1. Antibodies and reagents
Mouse anti-HDAC2 3F3 ChIP grade (ab51832), rabbit anti-HDAC2 ChIP grade (ab7029) and rabbit anti-HDAC1 (ab184651) were purchased from Abcam (Cambridge, United Kingdom). Rabbit anti-acetyl-Histone H3 (06-599) were purchased from Millipore (Billerica, USA). Mouse anti-c-Abl (sc-23), rabbit anti-B-tubulin (sc-9104), mouse anti-GAPDH (sc-32233) were purchased from Santa Cruz biotechnology (Dallas, United States of America). Rabbit anti-phospho-c-Abl (C4240), Filipin, The ANTI-FLAG M2 affinity gel (A2220), methyl-β-cyclodextrin and Vitamin E ((+)-α-tocopherol acetate semisynthetic from natural α-tocopherol) was purchased from Sigma-aldrich. Rabbit anti-Calbindin D-28K (AB1778) was purchased from Chemicon International (Temecula, CA, USA). The anti-NPC1 antibody was kindly donated by Dr. William Garver (University of Arizona, Tucson, AZ). Imatinib mesylate (13139), MS275 and CI994 was purchased from Cayman Chemical Company (Ann Arbor, United States of America). Torin1 (4247) was purchased from Tocris.
GNF2 was obtained from National Center for Advancing Tranlational Sciences (Center at the National Institutes of Health (NIH). U18666A (U18) drug was purchased from Enzo Life Sciences Inc. (Farmingdale, NY, USA). The mouse anti-phospho-tyrosine (4G10) was purchased from Millipore.
5.2. Cell Culture
HT22 cells were kindly donated by Elena Pasquale (Sanford-Burnham Medical Research Institute, La Jolla, California, USA). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 μg/ml streptomycin. CHO cells were maintained in Ham's F12 nutrient mixture medium (Gibco) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 μg/ml streptomycin.
5.3. Rat and mice hippocampal neuron cultures
Rat hippocampal cultures were prepared as described previously with some modifications (36, 54). Hippocampi from Sprague Dawley rats at embryonic day 18 or Hippocampi from mouse at postnatal day 1 were removed (the genotype of the animals (c-Abl+/+ or c-Abl-/-) was determined 1 day later), dissected in Ca2+/Mg2+-free Hank's balanced salt solution (HBSS) and rinsed twice with HBSS. Then, the tissue was resuspended in HBSS containing 0.25% trypsin and incubated for 15 min at 37°C. After three rinses with HBSS, the tissue was mechanically dissociated in DMEM, supplemented with 10% horse serum (Invitrogen), 100 U/mL penicillin, and 100 μg/mL streptomycin. Dissociated hippocampal cells were seeded onto polyLysine coated coverslips. Cultures were maintained at 37°C in 5% CO2 for 2 h before the plating medium was replaced with Neurobasal growth medium (Invitrogen) supplemented with B27 (Invitrogen), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. At day 2, cultured neurons were treated with AraC 2μM for 24 h; this method resulted in cultures highly enriched in neurons (approximately 5% glia).
5.4. Imatinib, GNF2, Vitamin E, MS275, U18666A (U18), SP600125 (SP), Torin, PD 0325901 (PD) treatments
Rat hippocampal neurons and HT22 cells (neuronal hippocampal cell line) were pre-treated with Imatinib at 5 μM or MS275 0.1 μM or SP 10 μM or PD 25 μM or Torin1 0.25 μM and Vitamin E (α-tocopherol) at 50 or 100 μM. Later, cells were treated with U18 at 0.5 μg/mL for 24 h. c-Abl-/- hippocampal neurons were treated with U18 at 0.5 μg/mL for 24 h. Mice were treated with Imatinib 12.5 mg/kg or GNF2 5 mg/kg by intraperitoneal injection every day, starting at p28 for 1 month or 3 weeks respectively. Control groups received daily injections of 0.9% NaCl.
5.5. Plasmids
pM18S Flag-HDAC2, pGal4-HDAC2, p-Gal4E1B-TK-Luciferase were previously described (41). ShNPC1 were purchased from Open Biosystem Inc. (Colorado, USA). ShNPC1 were transfected into HT22 cells using Lipofectamine 2000 reagent (Life Technologies, Carlsbad, US). After 48 hours of transfection, the cells were lysed in RIPA buffer and performed immunoblot analysis. We use a plasmid scramble ShRNA as a control.
5.5. HDAC2 repression activity assay
Plasmids Gal4-HDAC2 and luciferase reporters were transfected into HT22 cells. 48h later, cells were collected and luciferase activity was determined with the Dual Luciferase Reporter Assay System (Promega, E1910).
5.6. Methyl-β-cyclodextrin (MβCD) treatments
HT22 cells were treated with vehicle or U18 at 0.5 μg/mL. 4 h later cells were co-treated with different concentrations of MβCD: 0.1, 0.2, 0.5, 0.8 and 1 mM for 20 h. Cells were then fixed for filipin staining or collected to perform immunoblot analyses.
5.7 HDAC2 tyrosine phosphorylation assay
HT22 cells were lysed in RIPA buffer plus protease and phosphatase inhibitors. We used Anti-FLAG M2 affinity Gel beads for Flag-HDAC2 immunoprecipitations. HDAC2 tyrosine phosphorylation was evaluated with an anti-phospho-Tyr antibody.
5.8. Immunofluorescence procedures
Hippocampal neurons were seeded onto poly-Lysine coated coverslips in 24-well culture plates at a density of 3.0 × 104 cells per well. After treatment, cells were washed twice with PBS, fixed in 4% paraformaldehyde in PBS for 20 min, and permeabilized for 10 min in 0.2% Triton X-100 in PBS. After 2 washes with PBS, the cells were incubated in 3% BSA in PBS for 30 min at room temperature, followed by an overnight incubation at 4°C with primary antibodies against HDAC2 and β-Tubulin. The cells were washed four times with PBS and then incubated with anti-mouse Alexa 488 and anti-rabbit Alexa 555 antibodies (Life Technologies, Carlsbad, United States of America) for 1h at room temperature. The fluorescence images were captured with an Olympus BX51 microscope and analyzed and quantified with the ImageJ software.
5.9. Filipin staining
HT22 cells or hippocampal neurons were fixed in 4% paraformaldehyde/4% sucrose in PBS for 30 min. Then, cells were washed in PBS and treated with 1.5 mg/mL glycine for 20 min. Finally cells were treated with 25 μg/mL Filipin (Sigma) for 30 min, washed with PBS and covered with Fluoromount-G (Southern Biotech, Birmingham, AL, USA). Images were captured with an Olympus BX51 microscope (Olympus) and analyzed with the ImageJ software.
5.10. Immunoblot analysis
Cells and brain homogenates were lysed in RIPA buffer (25mM Tris-HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors cocktail (Roche), Na3VO4, NaF, PMSF and Aprotinina. Proteins were resolved in SDS-PAGE, transferred to Nitrocellulose membranes (Thermo Scientific), and detected with primary antibodies against HDAC1, HDAC2, c-Abl, GAPDH and NPC1. The reactions were followed by incubation with HRP labeled secondary antibodies and developed using the ECL technique (Thermo Scientific).
5.11. Histological analysis
Mice treated with GNF2 5 mg/kg were perfused with 4% paraformaldehyde in PBS. Brains were removed and post-fixed overnight at 4°C, placed in 20% sucrose in PBS at 4°C overnight, and then cut in 25 μm thick sagittal sections using a cryostat (Leika) at -20°C. Slices were permeabilized with 0.1% Triton X-100, blocked in 5% BSA in PBS and incubated overnight with the anti-Calbindin D-28K (AB1778) and rabbit anti-HDAC2 antibody (ab7029) or the mouse anti-HDAC2 3F3 ChIP grade antibody (ab51832) in 5% BSA in PBS. The primary antibody was visualized with anti-rabbit Alexa-Fluor 555, anti-rabbit Alexa-Fluor 488 or ari-mouse Alexa Fluor 488.
5.12. Quantitative Real-Time PCR
Total RNA from HT22 cells was extracted using Trizol (LifeTechnologies, 15596), and reverse-transcribed into cDNA using iScript RT Supermix (Bio-Rad 1708840). Real-time quantitative PCR assays were performed in triplicate using iQ SYBR Green supermix (Bio-Rad, 170-8882) and exon specific primers (primers are listed in the Supplemental experimental procedures table I) in a CFX96 real-time PCR Detection system (Bio-Rad). The relative quantities of cDNA were calculated using the comparative CT method. Data were derived from three independent amplifications.
5.13. Chromatin Immunoprecipitation
ChIP was performed with HT22 cells. The cells were rinsed with PBS, fixed in 4% paraformaldehyde in PBS for 10 min and washed with 0.125M Glycine in PBS. Then, they were lysed in Lysis Buffer I (50mM HEPES, 3mM MgCl2, 20mM KCl and 0.1% NP-40) and disrupted with a Dounce Homogenizer. Then, the samples were sonicated using a Bioruptor (Diagenode B01010002), using a cycle of eight 30-second pulses (30s between pulses) at a frequency of 20 KHz, 8 times at 4oC. The resulting whole cell extract was diluted in 50mM HEPES, 140 mM NaCl, 1mM EDTA and 1% Triton and incubated in Protein A 25 μL (sc-2001, Santa Cruz Biotechnology) or G agarose (sc-2002, Santa Cruz Biotechnology), depending on the primary antibody isotype, and then incubated for 4 h at 4oC. The samples were then centrifuged and the supernatant was incubated overnight at 4oC in 4 μg of mouse anti-HDAC2 3F3 ChIP grade antibodies (ab51832) or rabbit anti-acetyl-Histone H3 (06-599) antibodies. Later, 50 μL of Protein A or G, depending on the antibody isotype, was added and incubated for another 4 h at 4°C in a rotator. The pellet was then washed four times in 1M HEPES, 140 mM NaCl, 1mM EDTA and 1% Triton. The immunocomplex was eluted by incubation at 65 oC for 15 min in 50mM Tris pH = 8.0, 1mM EDTA, 1% SDS and NaCl 200 mM. The immunocomplex and whole-cell extracts were treated with RNAseA, (Qiagen 19101) proteinase K (Life Technologies 25530) and the DNA was then purified using the Qiagen DNA purification kit (Cat# 28106). The enrichment of DNA fragments was evaluated by Real Time PCR analysis, which was performed using iQ SYBR Green supermix (Bio-Rad, 170-8882) and primers designed to amplify the promoters of the genes analyzed (primers are described in the Supplemental experimental procedures table II), in a CFX96 realtime PCR Detection system (Bio-Rad). The relative quantities of immunoprecipitated DNA fragments were calculated using Pfaffl method (55).
5.14. Statistical analysis
Mean and s.e.m values and the number of experiments are indicated in each figure. One-way ANOVA tests were performed followed by Bonferroni post-test using the Prisma Software.
5.15. Statement of ethics
All protocols were approved and followed local guidance documents generated by the ad hoc committee of Chile (CONICYT) and were approved by the Bioethics Committee of the School of Medicine from Pontificia Universidad Católica de Chile (CEBA Protocol # 14-038). They were in agreement with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals recommended by the Institute for Laboratory Animal Research in its Guide for Care and Use of Laboratory Animals.
Supplementary Material
Acknowledgments
This study was supported by grants from the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) (grant numbers 1120512 to A.R.A, and 1110310 and 1150186 to S.Z); Fondo de Fomento al Desarrollo Científico y Tecnológico FONDEF D10I1077 (to A.R.A and S.Z.); Fondo Nacional de Desarrollo de Areas Prioritarias, FONDAP, Project no. 15090007, Center for Genome Regulation (CGR) to S.Z and Projecto Basal PFB12/2007, 2013-2017 to A.R.A. M.G-Z. acknowledges support from CONICYT, VRI and MECESUP. L.G-H acknowledges support from VRI and P.S.C and M.J. Y. acknowledges support from CONICYT.
Abbreviations
- NPC
Niemann-Pick type C
- HDAC2
Histone deacetylase 2
- U18
U18666A
- Ima
Imatinib
- MS
MS275
Footnotes
Competing interests: The authors declare that there are no conflicts of interest.
Author Contributions: Conceived and designed the experiments: P.S.C., M.G-Z., A.R.A., S.Z.; performed the experiments: P.S.C., M.G-Z., L.G-H. M.J.Y.; Analyzed the data: P.S.C., M.G-Z., J. M., E. S., A.R.A., S.Z.; contributed reagents/materials/analysis tools: A.D., J.M., E.S.; wrote the paper: P.S.C., M.G-Z., A.R.A., S.Z.
References
- 1.Penney J, Tsai LH. Histone deacetylases in memory and cognition. Science signaling. 2014;7(355):re12. doi: 10.1126/scisignal.aaa0069. [DOI] [PubMed] [Google Scholar]
- 2.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447(7141):178–82. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 3.Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–6. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Current opinion in genetics & development. 2003;13(2):143–53. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- 6.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Molecular cell. 2008;31(4):449–61. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews Genetics. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. The International journal of developmental biology. 2009;53(2-3):275–89. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- 9.Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):7876–81. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez-Zuniga M, Contreras PS, Estrada LD, Chamorro D, Villagra A, Zanlungo S, et al. c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer's disease. Molecular cell. 2014;56(1):163–73. doi: 10.1016/j.molcel.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Jawerka M, Colak D, Dimou L, Spiller C, Lagger S, Montgomery RL, et al. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron glia biology. 2010;6(2):93–107. doi: 10.1017/S1740925X10000049. [DOI] [PubMed] [Google Scholar]
- 12.MacDonald JL, Roskams AJ. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237(8):2256–67. doi: 10.1002/dvdy.21626. [DOI] [PubMed] [Google Scholar]
- 13.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 14.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 15.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–24. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature medicine. 2008;14(11):1247–55. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 17.Pentchev PG, Boothe AD, Kruth HS, Weintroub H, Stivers J, Brady RO. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. The Journal of biological chemistry. 1984;259(9):5784–91. [PubMed] [Google Scholar]
- 18.Sturley SL, Patterson MC, Balch W, Liscum L. The pathophysiology and mechanisms of NP-C disease. Biochimica et biophysica acta. 2004;1685(1-3):83–7. doi: 10.1016/j.bbalip.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Molecular genetics and metabolism. 2012;106(3):330–44. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Klein AD, Alvarez A, Zanlungo S. The unique case of the Niemann-Pick type C cholesterol storage disorder. Pediatric endocrinology reviews : PER. 2014;12(1):166–75. [PubMed] [Google Scholar]
- 21.Kim SJ, Lee BH, Lee YS, Kang KS. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochemical and biophysical research communications. 2007;360(3):593–9. doi: 10.1016/j.bbrc.2007.06.116. [DOI] [PubMed] [Google Scholar]
- 22.Pipalia NH, Cosner CC, Huang A, Chatterjee A, Bourbon P, Farley N, et al. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5620–5. doi: 10.1073/pnas.1014890108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helquist P, Maxfield FR, Wiech NL, Wiest O. Treatment of Niemann--pick type C disease by histone deacetylase inhibitors. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2013;10(4):688–97. doi: 10.1007/s13311-013-0217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maceyka M, Milstien S, Spiegel S. The potential of histone deacetylase inhibitors in Niemann - Pick type C disease. The FEBS journal. 2013;280(24):6367–72. doi: 10.1111/febs.12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(15):6401–11. doi: 10.1523/JNEUROSCI.1001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bie B, Wu J, Yang H, Xu JJ, Brown DL, Naguib M. Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nature neuroscience. 2014;17(2):223–31. doi: 10.1038/nn.3618. [DOI] [PubMed] [Google Scholar]
- 27.Aoyama K, Yamaguchi N, Yuki R, Morii M, Kubota S, Hirata K, et al. c-Abl induces stabilization of histone deacetylase 1 (HDAC1) in a kinase activity-dependent manner. Cell biology international. 2015;39(4):446–56. doi: 10.1002/cbin.10413. [DOI] [PubMed] [Google Scholar]
- 28.Malnar M, Hecimovic S, Mattsson N, Zetterberg H. Bidirectional links between Alzheimer's disease and Niemann-Pick type C disease. Neurobiology of disease. 2014;72 Pt A:37–47. doi: 10.1016/j.nbd.2014.05.033. [DOI] [PubMed] [Google Scholar]
- 29.Nixon RA. Niemann-Pick Type C disease and Alzheimer's disease: the APP-endosome connection fattens up. The American journal of pathology. 2004;164(3):757–61. doi: 10.1016/S0002-9440(10)63163-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu R, Yanjanin NM, Elrick MJ, Ware C, Lieberman AP, Porter FD. Apolipoprotein E genotype and neurological disease onset in Niemann-Pick disease, type C1. American journal of medical genetics Part A. 2012;158A(11):2775–80. doi: 10.1002/ajmg.a.35395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Molecular psychiatry. 2011;16(9):903–7. doi: 10.1038/mp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poirier J. Apolipoprotein E and cholesterol metabolism in the pathogenesis and treatment of Alzheimer's disease. Trends in molecular medicine. 2003;9(3):94–101. doi: 10.1016/s1471-4914(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 33.Yao J, Ho D, Calingasan NY, Pipalia NH, Lin MT, Beal MF. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. The Journal of experimental medicine. 2012;209(13):2501–13. doi: 10.1084/jem.20121239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein A, Maldonado C, Vargas LM, Gonzalez M, Robledo F, Perez de Arce K, et al. Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiology of disease. 2011;41(1):209–18. doi: 10.1016/j.nbd.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Alvarez AR, Klein A, Castro J, Cancino GI, Amigo J, Mosqueira M, et al. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22(10):3617–27. doi: 10.1096/fj.07-102715. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiology of disease. 2004;17(2):326–36. doi: 10.1016/j.nbd.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Cancino GI, Toledo EM, Leal NR, Hernandez DE, Yevenes LF, Inestrosa NC, et al. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer's beta-amyloid deposits. Brain : a journal of neurology. 2008;131(Pt 9):2425–42. doi: 10.1093/brain/awn125. [DOI] [PubMed] [Google Scholar]
- 38.Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(38):16691–6. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsumata R, Ishigaki S, Katsuno M, Kawai K, Sone J, Huang Z, et al. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PloS one. 2012;7(9):e46185. doi: 10.1371/journal.pone.0046185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlatterer SD, Tremblay MA, Acker CM, Davies P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. Journal of Alzheimer's disease : JAD. 2011;25(1):119–33. doi: 10.3233/JAD-2011-102025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. The Journal of biological chemistry. 2002;277(35):31826–33. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 42.Marin T, Contreras P, Castro JF, Chamorro D, Balboa E, Bosch-Morato M, et al. Vitamin E dietary supplementation improves neurological symptoms and decreases c-Abl/p73 activation in Niemann-Pick C mice. Nutrients. 2014;6(8):3000–17. doi: 10.3390/nu6083000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yevenes LF, Klein A, Castro JF, Marin T, Leal N, Leighton F, et al. Lysosomal vitamin E accumulation in Niemann-Pick type C disease. Biochimica et biophysica acta. 2012;1822(2):150–60. doi: 10.1016/j.bbadis.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2009;18(1):131–9. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer's disease mouse model when administered at an advanced stage of disease progression. Journal of Alzheimer's disease : JAD. 2011;26(1):187–97. doi: 10.3233/JAD-2011-110080. [DOI] [PubMed] [Google Scholar]
- 46.Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010;35(4):870–80. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2009;34(7):1721–32. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 48.Nunes MJ, Moutinho M, Gama MJ, Rodrigues CM, Rodrigues E. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PloS one. 2013;8(1):e53394. doi: 10.1371/journal.pone.0053394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annual review of pharmacology and toxicology. 2013;53:311–30. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- 50.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nature reviews Molecular cell biology. 2008;9(3):206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A. Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learning & memory. 2008;15(2):50–4. doi: 10.1101/lm.777308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou R, Holmes A, Du J, Malkesman O, Yuan P, Wang Y, et al. Genome-wide gene expression profiling in GluR1 knockout mice: key role of the calcium signaling pathway in glutamatergically mediated hippocampal transmission. The European journal of neuroscience. 2009;30(12):2318–26. doi: 10.1111/j.1460-9568.2009.07022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vazquez MC, del Pozo T, Robledo FA, Carrasco G, Pavez L, Olivares F, et al. Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PloS one. 2011;6(12):e28777. doi: 10.1371/journal.pone.0028777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaech S, Banker G. Culturing hippocampal neurons. Nature protocols. 2006;1(5):2406–15. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 55.Pfaffl MW. A new mathematical model for relative quantification in realtime RT-PCR. Nucleic acids research. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.