

Abstract
No vaccine exists against group A Streptococcus (GAS), a leading cause of worldwide morbidity and mortality. A severe hurdle is the hypervariability of its major antigen, the M protein, with >200 different M types known. Neutralizing antibodies typically recognize M protein hypervariable regions (HVRs) and confer narrow protection. In stark contrast, human C4b-binding protein (C4BP), which is recruited to the GAS surface to block phagocytic killing, interacts with a remarkably large number of M protein HVRs (apparently ~90%). Such broad recognition is rare, and we discovered a unique mechanism for this through structure determination of four sequence-diverse M proteins in complex with C4BP. The structures revealed a uniform and tolerant ‘reading head’ in C4BP, which detected conserved sequence patterns hidden within hypervariability. Our results open up possibilities for rational therapies targeting the M-C4BP interaction, and also inform a path towards vaccine design.
Introduction
Group A Streptococcus (GAS, S. pyogenes) is a major cause of worldwide morbidity and mortality1. This bacterial pathogen is responsible for mucosal infections (e.g. pharyngitis), acute invasive diseases (e.g., necrotizing fasciitis) and autoimmune sequelae (e.g., rheumatic heart disease)2. Currently, no vaccine against GAS exists3,4. A major impediment to immunization is the hypervariability of the antigenic M protein, a surface-anchored virulence factor5,6 that is also the target of neutralizing antibodies. These antibodies typically recognize the hypervariable region7,8,9 (HVR, N-terminal ~50 amino acids) of M proteins, which are dimeric α-helical coiled coils, and thus confer M type-specific immunity. One approach to overcoming hypervariability is to include multiple M protein HVRs in a vaccine, and indeed a vaccine candidate that includes 30 HVRs10 has advanced into early clinical testing. However, with >200 distinct M protein HVRs11 and the complexity of global GAS epidemiology1, even the most extensive multivalent vaccine is unlikely to offer universal protection. Here, we offer structural details that have implications for overcoming M protein hypervariability for vaccine design. This approach is based on the finding that human C4b-binding protein (C4BP) recognizes M protein HVRs with broad specificity12, in stark contrast to the narrow type-specificity displayed by antibodies. In one study, a remarkable ~90% of GAS strains of differing M types bound C4BP12. While this study examined binding to whole bacteria, no protein other than M protein (or an M-like protein, such as Protein H) has been described to bind C4BP, and no region other than the M protein HVR has been described to bind C4BP. C4BP13 is a negative regulator of the complement system that binds the complement protein C4b, and thereby disables the C3 convertase of the classical and lectin pathways. GAS recruits C4BP to its surface, like a number of others pathogens14,15, to evade opsonophagocytic killing16,17.
Broad specificity in recognition is rare, having been observed only in a few cases. A prominent example is the interaction between major histocompatibility complex (MHC) glycoproteins and peptides18,19. The breadth of this particular interaction is explained by MHC glycoproteins primarily making contact to the peptide main chain. To understand the basis for broad specificity in the case of M protein and C4BP, co-crystal structures of four M protein HVRs (M2, M22, M28, and M49) bound to the first two domains of the C4BP α chain were determined (Fig. 1a, Supplementary Fig. S1, and Supplementary Table S1). C4BP consists of 7 α chains disulfide-bonded to a single β chain, with each of these chains being composed of multiple ~60-residue complement control protein (CCP) domains20. The first two CCP domains of the α chain (C4BPα1-2) are sufficient to bind M protein HVRs21 and C4b21,22 (Fig. 1a). Overlapping but non-identical sites on C4BP are engaged by M protein HVRs and C4b22.
Figure 1. Structures of M-C4BP complexes.
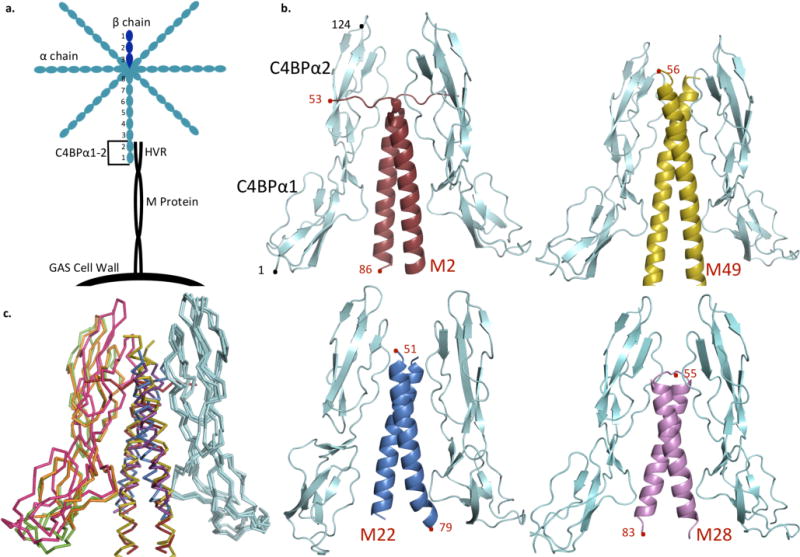
a. Schematic of C4BP (blue) bound to surface-associated M protein (black), highlighting the M HVR-C4BPα1-2 interaction.
b. C4BPα1-2 (cyan) in complex with the HVR of M2 (red), M49 (yellow), M22 (blue), and M28 (magenta). Terminal residues are numbered.
c. Superposition of M-C4BP complexes, based on the bound C4BPα1-2 molecule shown at right in cyan. M2 is red and its second bound C4BPα1-2 molecule green; M49 is yellow and its second bound C4BPα1-2 orange; M22 is blue and its second bound C4BPα1-2 is omitted (because a crystal contact restricts its orientation into an artifactual conformation); and M28 is magenta and its second bound C4BPα1-2 pink.
Results
Structural similarity
The structures of the four M protein HVR-C4BPα1-2 complexes (determined between 2.54–3.02 Å resolution limits) were astonishingly similar, given the lack of sequence relationship among the M proteins (Supplementary Fig. S1). The M protein-C4BP interface was in well defined electron density and unambiguously modeled (Supplementary Fig. S2), whereas portions of C4BPα1-2 distal to the interface were ill defined, consistent with the inherent flexibility of these domains20. The M protein HVRs form parallel, dimeric α-helical coiled coils, with two C4BPα1-2 molecules bound to each M protein dimer, as prior reports suggested20,23 (Figs. 1b–c; detailed view of M2 shown later, and detailed views of M22, M28, and M49 are in Supplementary Figs. S3–S5). The portions of the M proteins that contact C4BPα1-2 are in canonical coiled-coil conformation, except for M2, which is underwound (Supplementary Fig. S6). C4BPα1 is proximal to the C-terminal portion of the M protein HVR and C4BPα2 to the N-terminal portion, in agreement with the approach of intact C4BP to the streptococcal surface (Fig 1a). The C4BPα1 and α2 domains are relatively unchanged from their unbound NMR structures20 (average RMSD ~1.5 and ~1.0 Å for domains 1 and 2, respectively), except that domain 1 is rotated 180° with respect to domain 2 (Supplementary Figs. S7 and S8). This rotation is consistent with evidence from mutagenic22 and structural20 studies, and is discussed further below. The M-C4BP interface is extensive, with a total of ~1450–1690 Å2 of surface area being buried (in the 2:2 complex). Most of this surface area is polar, and the fit is far from hand-in-glove (surface complementarities 0.56–0.66)24, except for M22 which has a better fit (0.72). These observations suggest a modest binding affinity, consistent with the 0.5 μM Kd20 for the interaction between C4BPα1-2 and the M4 HVR. A much tighter association of picomolar Kd25 results from avidity between C4BP, which has multiple, bundled arms26, and surface-localized M protein.
Uniform ‘reading head’
Most significantly, the four structures revealed a uniform set of amino acids in C4BP that act as a ‘reading head’ for recognizing M protein HVRs. Most of this ‘reading head’ resides in C4BPα2 (Fig. 2a) and takes the form of a quadrilateral that is composed of: (1) a hydrophobic pocket that contains C4BP H67, I78, and L82; (2) a hydrogen bonding group in the form of the main chain nitrogen of C4BP H67; and two positively charged residues, C4BP (3) R64 and (4) R66. The segment that holds this quadrilateral is structurally invariant, being stabilized by a disulfide bond at C65 and limited in conformation by P68 (not depicted). The M proteins supply amino acid side chains that interact with these C4BP residues to form complementary quadrilaterals (Fig. 2b). In all four M-C4BP structures, a hydrophobic M protein residue (usually an aromatic) fits into the (1) hydrophobic pocket, and a polar M protein residue immediately following in sequence hydrogen bonds to the (2) main chain nitrogen of H67. The contacts to C4BP (3) R64 and (4) R66 are predominantly electrostatic (usually salt bridges), but in the case of M49, a polar residue is absent and R64 instead makes hydrophobic contacts, extending its alkyl chains across several M49 residues. These data are compatible with a report that substitution of C4BP residues R64, R66, or H67 with Gln affects binding to M4 and M2222. Decreased affinity results in the case of R64Q and H67Q, but increased affinity occurs for R66Q (likely through a gain-of-function).
Figure 2. C4BP Binding Mode.
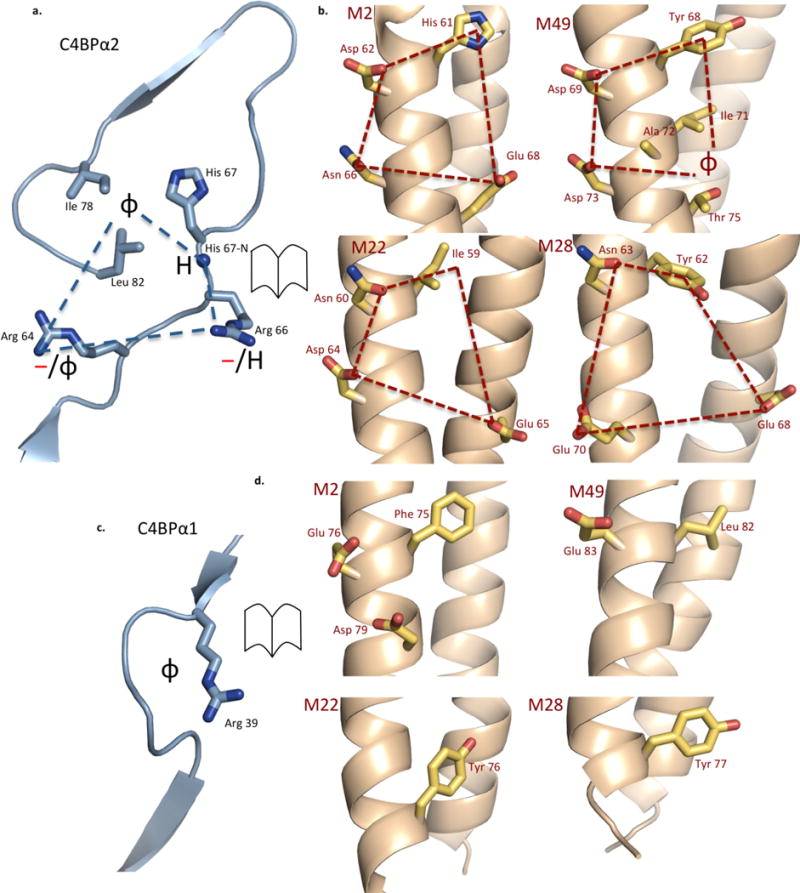
a. The C4BPα2 quadrilateral (blue dashed lines), with the C4BPα2 backbone shown in ribbon representation and key side chains shown as bonds, in which carbons are cyan and nitrogens blue (here and in following panels). The chemical character of M protein residues that interact with the quadrilateral is depicted: φ, hydrophobic; —, negative; H, hydrogen bond forming.
b. M2, M49, M22, and M28 residues that interact with the C4BPα2 quadrilateral and form a complementary quadrilateral (red dashed lines), shown in open-book representation with respect to C4BPα2. The M protein backbone is in ribbon representation and key side chains shown as bonds, in which carbons are yellow, oxygens red, and nitrogens blue. The numbering of M proteins is such that the initiator Met is residue 1.
c. The C4BPα1 Arg39 nook. The depiction and symbols are as for panel a.
d. M2, M49, M22, and M28 residues that interact with the C4BPα1 Arg39 nook shown in open-book representation. The depiction is as for panel b.
Uniform ‘reading head’ contacts from C4BPα1 were far fewer. The key C4BPα1 residue was R39, which formed electrostatic contacts through its guanidinium group as well as hydrophobic contacts through its alkyl chain, creating a ‘hydrophobic nook’ in conjunction with main chain atoms of C4BPα1 (Fig 2c). Thus, out of the six C4BP residues that form uniform contacts, three are arginines. This high proportion is likely significant, as the combination of polar and apolar atoms in Arg along with its chain length increase the possibilities for interactions with variable residues. Substitution of C4BP R39 with Gln results in decreased binding to M4 but increased binding to M2222 (again, likely a gain-of-function). All four M proteins have hydrophobic residues that insert into the C4BPα1 ‘hydrophobic nook’. M2 and M49 also have negatively charged residues that interact with C4BP R39, whereas neither M22 nor M28 do. The importance of C4BP R39 provided an explanation for the aforementioned 180° rotation of C4BPα1 (around a hinge at K63, Supplementary Fig. S7). In free C4BP, the C4BPα1 R39 nook and the C4BPα2 quadrilateral are on opposite sides, and require a 180° reorientation to interact simultaneously with M protein. This 180° rotation was seen in all four structures. However, in one of the two C4BPα1-2 molecules bound to M22, the 180° rotation was prevented due to a crystal contact (Supplementary Figs. S7c, d and S8). A similar 180° rotation appears necessary for the interaction of C4BP with C4b, as it has been demonstrated that R39 and the set of residues in C4BPα2 that interact with M protein HVRs also interact with C4b22. The purpose of requiring a 180° rotation in C4BPα1 to transition between free and bound forms is unclear.
Sequence conservation hidden within hypervariability
The evidence gathered from these structures proved powerful in bringing to light weak sequence conservation in M protein HVRs. Comparison of the heptad position of M protein residues that interacted with C4BP made it clear that there were two binding patterns observed in these structures, with chemically similar residues contacting C4BP (Fig. 3a): One for M2 and M49, and a separate one for M22 and M28. These two patterns were also evident in the way the coiled coils interacted with C4BP. In the case of M2 and M49, the coiled coils ran roughly parallel to C4BPα1-2, such that each C4BPα1-2 molecule contacted a single α-helix (Figs. 1b and 2b). But in the case of M22 and M28, the coiled coils lay crosswise across C4BPα1-2 such that each C4BPα1-2 molecule contacted both α-helices. Remarkably, these same two patterns were evident in a larger number of M proteins (Fig. 3b, and Supplementary Figs. S9, and S10), which had chemically similar amino acids to the ones in M2/M49 or M22/M28 that had been visualized to contact C4BP. We were able to assign 13 M proteins to the M2/M49 pattern and 32 (including the M-like Protein H) to the M22/M28 pattern. Thus, these two patterns may explain the interaction of nearly half of the M strains that were studied for C4BP binding12. A further 46 M proteins from this study12 could not be assigned to either pattern (Supplementary Fig. S11). In these cases, it is possible that there are proteins besides M that recruit C4BP to the GAS surface or other regions of the M proteins that do. However since, for GAS, only M proteins and M protein HVRs have been documented to bind C4BP, we think it more likely that there are still other arrangements by which M protein HVRs interact with C4BP.
Figure 3. C4BP-binding modes of M proteins.
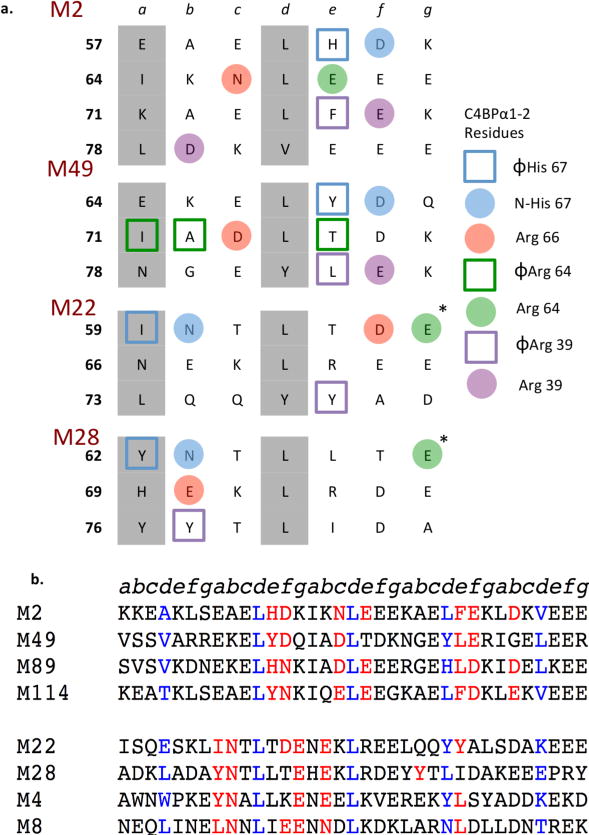
a. Heptad registers of M2, M49, M22, and M28 HVRs (a and d residues in grey). M protein residues interacting with C4BPα1-2 residues are highlighted according to their corresponding C4BPα1-2 interaction.
b. Sequence alignment of M protein HVRs that belong to the M2/M49 group (top) or the M22/M28 group (bottom). Residues that contact or are predicted to contact C4BPα1-2 are in red. Residues observed or predicted to be at core d positions of the heptad register are highlighted in blue for visual reference. Protein H (PrtH) is an M-like protein expressed by certain M1 strains.
Tolerance to hypervariability
We next sought to understand tolerance in C4BP to sequence variation in M protein, as single amino acid changes have been shown to alter recognition by antibodies but not C4BP12. Alanine substitutions were created in the M2 residues mentioned above that make contact with the uniform ‘reading head’. In addition, substitutions were made in two M2 residues, K65 and E83 (numbering of M proteins such that initiator Met is residue 1), which make contacts observed only in M2 (Fig. 4). The M2-C4BP interaction was evaluated by a Ni2+-NTA agarose coprecipitation assay using His-tagged C4BPα1-2 (Fig. 4a and Supplementary Fig. S12). Of the single-site substitutions, only F75A, which binds in the C4BPα1 nook, showed substantially decreased binding. Molecular dynamics (MD) simulations of this mutation highlighted the importance of M2 F75, with contacts between the R39 ‘hydrophobic nook’ and M2 diminishing and waters infiltrating into this site upon alanine-substitution of F75 (Supplementary Fig. S13 and Supplementary Videos S1 and S2). Strikingly, all other residues could be mutated to Ala without substantial losses in binding (Supplementary Fig. S12), and indeed in two cases, increased binding was observed (see below). Providing verification for the structural observations, two double substitutions resulted in substantial loss of binding: D62A/E68A, which removed two of the polar contacts to the C4BPα2 quadrilateral, and E76A/D79A, which removed the two salt bridges to C4BPα1 R39.
Figure 4. M2-C4BP interaction.
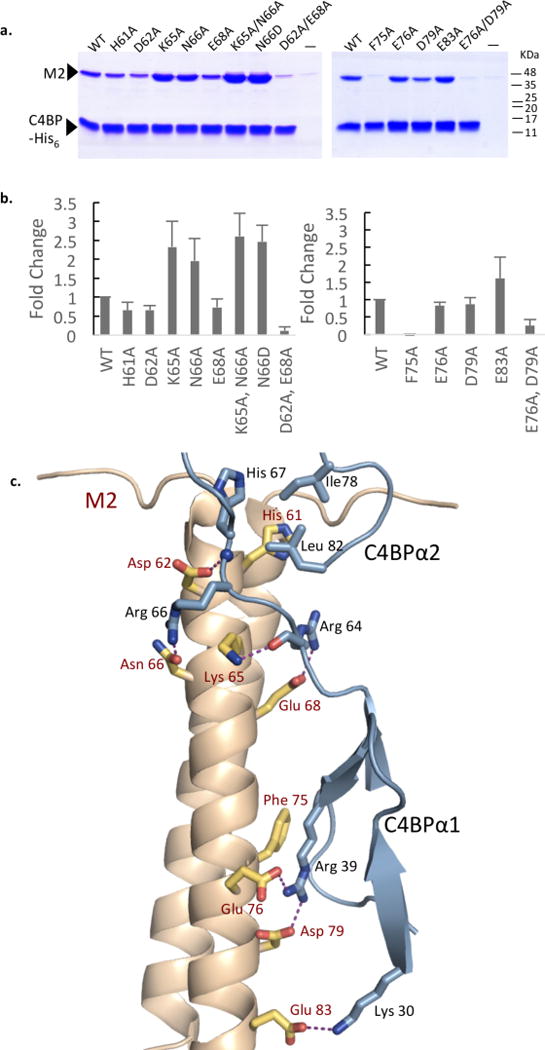
a. Association of His-tagged C4BPα1-2 with wild-type and mutant M2 HVR at 37 °C, as assessed by a Ni2+-NTA agarose coprecipitation assay and visualized by non-reducing, Coomassie-stained SDS-PAGE. Only bound fractions are shown here. Input samples shown in Fig. S12. This gel is representative of four experimental replicates. Molecular mass markers were not run on these particular gels; their positions are based on measurements from equivalent gels.
b. Quantification of the interaction between C4BPα1-2 and wild-type M2 or M2 mutants proteins. The values shown are averages of four experimental replicates, corrected for the level of background binding (i.e., no C4BPα1-2) and normalized to wild-type M2. Standard deviations are depicted.
c. Structure of M2 (gray ribbon representation with key side chains in bonds representation, in which carbons are yellow, oxygens red, and nitrogens blue) bound to C4BPα1-2 (cyan ribbon representation, with key side chains in bonds representation, in which carbons are cyan and nitrogens blue). Hydrogen bonds and salt bridges depicted by dashed magenta lines.
Surprisingly, two of the single-site mutations, M2 K65A and N66A, increased binding, as did the K65A/N66A double mutant (Fig. 4a and Supplementary Fig. S12). The structure of M2 (K65A/N66A) in complex with C4BPα1-2 was determined to 2.29 Å resolution limit, and no re-ordering of the binding site was evident (RMSD 0.15 Å) (Supplementary Fig. S14). This result suggested that M2 K65 and N66 were tolerated in the binding site but not optimal. While M2 K65 formed a hydrogen bond to the main chain oxygen of C4BP R64, it was sandwiched between two positively charged side chains (i.e., C4BP R64 and R66), providing an explanation for why Ala substitution of K65 led to enhanced binding. MD simulations reinforced this interpretation, as Ala substitution of K65 led to better contacts between M2 and C4BP, especially evident in the increased frequency of hydrogen bonding between C4BP R66 and M2 N66 (Supplementary Table S2). The simulations suggested that the hydrogen bond between these two residues was otherwise infrequent (Supplementary Videos S3 and S4, Supplementary Table S2), and indeed these two residues had the highest B-factors in the binding site (Supplementary Fig. S15). In other M proteins belonging to the M2/M49 pattern, the equivalent of N66 is almost always Asp or Glu (Supplementary Fig. S9). Consistent with this trend, substitution of M2 N66 with Asp resulted in increased binding (Fig. 4a), and MD simulations provided evidence of the favorable interactions between C4BP R66 and negatively charged M protein amino acids (Supplementary Video S5 and Supplementary Table S3). Puzzlingly, substitution of M2 N66 with Ala (and thus loss of hydrogen bonding to C4BP R66) also resulted in better binding. It is worth noting that C4BP R66 had an even higher relative B-factor when contacting M2 (K65A/N66A) as compared to wild-type M2 (Supplementary Fig. S15). Thus, it appears that C4BP R66 prefers a salt bridge (e.g., N66D) or no interaction (e.g., N66A) to a hydrogen bond, because the salt bridge provides sufficient binding energy to relieve the entropic cost of ordering the Arg, whereas the hydrogen bond does not. In short, the mutagenesis experiments reinforced the notion that the ‘reading head’ in C4BP is highly tolerant to variation in the M protein.
Discussion
We have shown that broad recognition between M proteins and C4BP is not due to contacts to the main chain, as it is for MHC-peptide complexes. Instead, the breadth of recognition in M-C4BP complexes is explained by three unique attributes. First, the C4BP binding site is tolerant, due notably to the prevalence of arginines. The combination of a charged head and a long alkyl body enables arginine to engage in both electrostatic and hydrophobic interactions. As a result, only loose restrictions apply to M protein side chains that interact with C4BP arginines. For example, whereas negatively charged M protein side chains were preferred for the C4BPα2 quadrilateral arginines, a set of hydrophobic residues were accommodated in M49; and for C4BPα1 R39, hydrophobic M protein side chains were the commonality. Second, there appears to be no ‘hot spot’ for interaction and instead the binding energy appears to be dispersed broadly over the interaction site. No single amino acid substitution in M2, except for one, reduced binding to C4BP substantially. A similar observation has been made for M2212. Alanine substitution of M22 E65 (E24 in Persson et al.), which we found interacts with C4BP R64, did not change binding to C4BP but did alter recognition by antibodies12. Third, the M protein coiled coil can align with C4BP in multiple ways. This enables M protein side chains that interact with C4BP to reside at different positions of the heptad repeat. Two different arrangements were seen here, but there are likely more to be discovered.
C4BP is recruited by a large number of pathogens (including viruses and fungi), in order to prevent phagocytic uptake, formation of the membrane attack complex, and generation of immunostimulatory anaphylatoxins (e.g., C3a and C5a)13. The importance of C4BP recruitment to GAS infection was demonstrated in an M22 strain. Specific loss of C4BP binding in this strain was effected through a seven residue deletion in M22, which our results indicate eliminated interaction with C4BP R64 and R66. This C4BP binding-deficient M22 strain was ~3- to 13-fold more susceptible to elimination by human blood as compared to the wild-type M22 strain16,27. Further evidence for the importance of C4BP recruitment was recently garnered using transgenic mice expressing human C4BP17 (murine C4BP does not bind M protein21). In particular, human C4BP transgenic mice showed a much earlier time to death as compared to nontransgenic mice when infected by a C4BP-binding GAS strain. This and other effects, including bacterial burden and levels of proinflammatory cytokines, were exacerbated when these mice also expressed human factor H, another soluble negative regulator of the complement system. Interestingly, factor H, which is composed of CCP domains like C4BP, also binds M protein HVRs28. While M protein HVRs generally bind either C4BP or factor H28, the GAS strain used in this study produced Protein H17, which binds both29. Our results provide, to our knowledge, the first atomic-level understanding of the interaction between a negative regulator of the complement system and a microbial virulence factor, and open up possibilities for rational disruption of the M-C4BP interaction for therapeutic ends.
Lastly, our work has implications for vaccine design. Broadly neutralizing antibodies (bNAbs) have been identified for several highly antigenically variable microbial pathogens, including HIV and influenza virus30,31,32,33. These antibodies target invariant structural regions that are often hidden due to various factors, including glycosylation, as in the case of HIV, or steric occlusion, as in the case of both influenza and HIV. The hypervariability of M proteins has hindered the development of a GAS vaccine. Our work shows that hidden within M protein hypervariability are sequence patterns that are conserved and utilized for interaction with C4BP. This finding suggests it may be possible, using appropriate antigens, to elicit bNAbs against GAS, which would mimic the broad specificity of binding to M protein HVRs observed in C4BP. In this regard, it is notable that the 30-valent M protein vaccine displays some measure of crossreactivity to M types not included in the vaccine10. Indeed, 14 of the 30 M proteins in the vaccine belong to pattern 1 or 2, and crossreactivity was seen to 16 M proteins not in the vaccine belonging to pattern 1 or 2. Further work will be necessary to determine if and which other conserved patterns exist in the set of M proteins that are not readily assignable to either pattern 1 or 2 but still bind C4BP. An alternative notion is transplantation of the C4BP loops that form the uniform and tolerant ‘reading head’ of C4BP to the antigen-combining site of an antibody. Such an antibody would be predicted to have broad specificity against M proteins, and would provide neutralization due to targeting of the M protein HVR. This passive form of immunity could be made active by screening for antigens that bind tightly to the antibody displaying the C4BP ‘reading head’.
A potential challenge in these approaches based on the mode of C4BP binding is that the antibodies obtained through such methods may also recognize C4b. However, differences in C4BP binding modes between M protein HVRs and C4b suggest that selectivity would be possible22. A second challenge may be escape from such antibodies through further M protein variation. However, M protein HVRs vary from strain to strain but are stable within the type34, suggesting that their overall sequence variation is limited by negative selection. Binding to C4BP appears to be a major evolutionary selective pressure for GAS17; thus, escape from such broadly neutralizing antibodies targeting M protein HVRs through further sequence variation may be limited by pressure to maintain C4BP interaction.
Materials and Methods
DNA manipulation
The coding sequences of mature M2 (amino acids 42–367), M22 (42–335), M28 (42–363), and M49 (42–359) proteins were cloned from GAS strains M2 (AP2), M22 (Sir22), M28 (strain 4039-05), and M49 (NZ131), respectively, into a modified version of the pET28a vector (Novagen) modified such that it encoded an N-terminal His6-tag followed by a PreScission™ protease (GE Healthcare) cleavage site. Constructs that encoded truncated versions of these proteins, which consisted of only the N-terminal 79, 86 or 100 amino acids, were generated through the insertion of an amber stop codon at an appropriate site by site-directed mutagenesis. Site-specific mutations were also introduced into the M2 coding sequence by site-directed mutagenesis. All site-directed mutagenesis was performed according to the Agilent QuikChange™ manual, except that 50 μL reactions were set up for polymerase chain reactions (PCR) instead of 12.5 μL reactions.
The coding sequence of the CCP1-2 domains of human C4BPα chain (C4BPα1-2)20 (a kind gift from G. Lindahl) was cloned into the modified pET28a vector described above, and also into a pET28b vector that encoded a non-cleavable C-terminal His6-tag. The cleavable N-terminal His6-tag version of C4BPα1-2 was used for crystallographic studies, and the non-cleavable C-terminal His6-tagged version for co-precipitation binding studies. To obtain selenomethionine (SeMet)-substituted protein to be used in phase determination, methionines were introduced in the coding sequence of C4BPα1-2 at amino acid positions 29, 46, and/or 71 by site-directed mutagenesis.
Protein Expression and Purification
M proteins were expressed in Escherichia coli BL21 (DE3) and purified as described5 with minor modifications to the procedure. Specifically, bacteria were lysed with a C-5 Emulsiflex (Avestin Inc., Ottawa, Canada) and ion exchange chromatography was omitted, and in the case of purification of M2 (wild-type and variants), imidazole was not included in the lysis and wash buffers.
C4BPα1-2 was expressed in E. coli Rosetta 2 (Novagen) cells. The protein was purified and refolded as described23, except for the use of a C-5 Emulsiflex for lysis. Where needed, the N-terminal His6-tags of M proteins and C4BPα1-2 were removed by PreScission™ protease cleavage according to manufacturer’s instructions, and the cleaved protein was purified by reverse Ni2+-NTA chromatography. M proteins and C4BPα1-2 were lastly purified by size-exclusion chromatography (Superdex 200) in a buffer composed of 150 mM NaCl, 50 mM Tris, pH 8.5. Proteins were then concentrated to ~20 mg/mL by ultrafiltration; protein concentrations were determined by absorbance at 280 nm using calculated molar extinction coefficients. Aliquots of concentrated protein were flash-frozen in liquid N2 and stored at −80 °C.
SeMet was incorporated into C4BPα1-2 (L29M/L46M), C4BPα1-2 (L29M/L71M), and C4BPα1-2 (L46M/L71M) using methionine pathway inhibition as described35. SeMet-labeled C4BPα1-2 was purified as described above.
Crystallization and Data Collection
For preparation of complexes, M2 (amino acids 42–141), M2 (K65A/N66A) (42–141), M22 (42–120), M28 (42–141), or M49 (42–127) protein was mixed with C4BPα1-2 (wild-type or SeMet-substituted mutant) at a 1:1 molar ratio (final concentration of complex ~5 mg/mL), and dialyzed overnight at 4 °C in 10 mM Tris, pH 8. The samples were then concentrated by ultrafiltration to ~20 mg/mL. Crystallization was performed by the hanging drop vapor-diffusion method.
The M2-C4BPα1-2, M2 (K65A/N66A)-C4BPα1-2, and M28-C4BPα1-2 complexes and the SeMet-labeled M2-C4BPα1-2 (L29M/L46M) and M2-C4BPα1-2 (L46M/L71M) complexes were co-crystallized at 20°C by mixing 1 μL of complex with 1 μL of the reservoir solution, which was 1.5 M (NH4)2SO4, 0.1 M Bis-Tris Propane, pH 7.0. These crystals were transferred to the reservoir solution supplemented with 20% ethylene glycol for cryopreservation, mounted in fiber loops, and flash-cooled in liquid N2. Crystals containing SeMet-labeled protein were treated similarly, except the reservoir solution was supplemented with freshly prepared 1 mM TCEP.
The M22-C4BPα1-2 complex was co-crystallized similarly, except the reservoir solution was 2 M (NH4)2SO4, 2% PEG 400, and HEPES pH 7.5. The SeMet-labeled M49-C4BPα1-2 L29M/L46M complex was also co-crystallized similarly, except the reservoir solution was 1.6 M Na/K PO4, pH 6.9. These two co-crystals were transferred to their respective reservoir solutions supplemented with 20% glycerol before being flash-cooled in liquid N2.
Diffraction data were collected from crystals under cryogenic conditions. Diffraction data for M2-C4BPα1-2 were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 9-2, for M22-C4BPα1-2 at the Advanced Photon Source (APS) beamline 24-ID-C, and for M2 (K65A/N66A)-C4BPα1-2 and M28-C4BPα1-2 at the Advanced Lightsource (ALS) beamline 8.2.1. Single-wavelength anomalous dispersion (SAD) data were collected from SeMet-labeled M2-C4BPα1-2 (L29M/L46M) and M2-C4BPα1-2 (L46M/L71M) at the APS beamline 19-ID, and from SeMet-labeled M49-C4BPα1-2 (L29M/L46M) at the APS beamline 24-ID-E.
Diffraction data from crystals of M22-C4BPα1-2 and M49-C4BPα1-2 (L29M/L46M) were indexed, integrated, and scaled using XDS36, while HKL200037 was used for data from all other crystals.
Structure Determination and Refinement: M2-C4BPα1-2
For structure determination of M2-C4BPα1-2, Se sites were located from SAD data of SeMet-labeled M2-C4BPα1-2 (L29M/L46M) and M2-C4BPα1-2 (L46M/L71M), and phases calculated for each data set using Autosol (within Phenix38). The two sets of phases were combined using the Reflection File Editor program (within Phenix). From the combined phase set, four Se sites, three at substituted methionines and one at the native Met 14, were identified per asymmetric unit, which contained one M2 α-helix and one C4BPα1-2 molecule.
Here and in all cases below, model building was carried out with Coot39 as guided by inspection of SAD-phased maps or σA-weighted 2mFo – DFc and mFo – DFc maps, and refinement was carried out with Refine (within Phenix) using default parameters. Between 15 and 75 iterative cycles of building and refinement, with each refinement step consisting of 1–10 rounds, were performed in each case. In later stages of refinement, TLS parameterization was used in Refine. Individual B-factors were refined isotropically. Water molecules were added in the final stages of refinement using Phenix with default parameters (3σ peak height in σA-weighted mFo – DFc maps).
In order to model M2-C4BPα1-2 (L29M/L46M/L71M), the NMR structure of C4BPα1-2 was manually fit into SAD-phased density, with the two domains of C4BPα1-2 being treated as individual rigid bodies. The M2 molecule was then built into density, with the register of the coiled coil being assigned from well defined density corresponding to large side chains (i.e., His 20, Phe 75, and His 85). The SeMet residues in the model were changed to leucines, and the model was then refined against the higher resolution (2.56 Å resolution limit) data collected from crystals of M2-C4BPα1-2. TLS parameterization involved the following groups: For M2, 53–57 and 58–86; for C4BPα1-2, 0–59 and 60–124. Continuous electron density was evident for the entire main chain of C4BPα1-2 and for residues 53–86 of the M2 protein. Here and in all cases below, electron density was visible for side chains of M protein residues, except for some solvent-exposed, flexible residues (i.e. Lys, Arg, or Glu) distant from the interface with C4BPα1-2. Electron density was also visible for side chains of C4BPα1-2, except for some residues in long loops that were also distant from the interface with M protein. An exception to this was C4BP R66, for which electron density for the side chain was broken. Long loops of C4BPα1-2 also contained some residues whose φ, ψ angles were in the outlier region of the Ramachandran plot. For the M2 protein, the only residue in the Ramachandran outlier region was A58, which is the N-terminal residue of the M2 model.
The structure of M2 (K65A/N66A)-C4BPα1-2 was determined by difference Fourier synthesis using the refined structure of M2-C4BPα1-2. The set of reflections used for Rfree calculations for the refinement of M2-C4BPα1-2 were maintained. TLS parameterization was equivalent to that for M2-C4BPα1-2.
Structure Determination and Refinement: M28-C4BPα1-2
The structure of M28-C4BPα1-2 was determined by molecular replacement using the program Phaser (within Phenix38). The C4BPα1-2 molecule from the structure of the M2-C4BPα1-2 complex served as the search model. The molecular replacement solution had a log-likelihood gain score of 379. The asymmetric unit contained one C4BPα1-2 molecule and one M28 α-helix, whose register was determined by well defined density corresponding to large side chains (i.e. Tyr 62, Tyr 76, Tyr 77). The model was first subjected to cycles of rigid body refinement, followed by the refinement protocol described above. TLS parameterization involved the following groups: For M28, 55–83; for C4BPα1-2, 0–59, 60–86, and 87–124. Continuous electron density was evident for the entire main chain of C4BPα1-2, except for breaks in some of its longer loops, and for amino acids 53–83 of M28.
Structure Determination and Refinement: M22-C4BPα1-2
The structure of the M22-C4BPα1-2 complex was determined by molecular replacement using the program Phaser. The search model consisted of an M28 α-helical, dimeric coiled-coil in complex with a single C4BPα1-2 molecule. The solution, which had a log-likelihood gain score of 166, resulted in two copies of the search model in the asymmetric unit, while the solvent content suggested that the asymmetric unit was composed of two M22 α-helical, dimeric coiled-coils and four C4BPα1-2 molecules; this latter composition was found to be accurate. After refinement of the initial molecular replacement model, two additional C4BPα1-2 molecules became evident in electron density maps, and were placed stepwise into density, with the two domains of C4BPα1-2 being treated as individual rigid bodies, between rounds of iterative refinement. Both these additional copies had similar conformations to one another, and had a tilted orientation of the C4BPα1 and C4BPα2 domains relative to these domains in unbound C4BPα1-2. This tilted orientation differs from the 180° rotation observed in the two other copies of C4BPα1-2 bound to M22, as well as in copies of C4BPα1-2 bound to M2, M28, and M49. Side chains for M22 were subsequently built into density, with the register being assigned based on well defined density corresponding to large side chains (i.e., Tyr 66 and Tyr 67). The model was then subjected to cycles of rigid body refinement followed by the refinement procedures described above. TLS parameterization involved the following groups: For M22 chain A, 52–80; for M22 chain C, 52–79; for M22 chain E, 52–79; for M22 chain G, 52–80; for C4BPα1-2 chain B, 1–13, 14–27, 28–59, 60–73, 74–86, 87–102, 103–109, 110–115, and 116–124; for C4BPα1-2 chain D, 0–59 and 60–124; for C4BPα1-2 chain F, 1–59 and 60–124; for C4BPα1-2 chain H, 0–13, 14–33, 34–47, 48–59, 60–74, 75–86, 87–109, and 110–124. Continuous electron density was evident for the entire main chain of C4BPα1-2, except for breaks in some of the longer loops, and for residues 52–79 (or 80, depending on the chain) of M22.
Structure Determination and Refinement: M49-C4BPα1-2
For structure determination of M49-C4BPα1-2, Se sites were located from SAD data collected for SeMet-labeled M49-C4BPα1-2 (L29M/L46M), and phases calculated using the program Autosol. Six Se sites were identified per asymmetric unit, which was found to contain an M49 α-helical, coiled-coil dimer and two C4BPα1-2 molecules. This is consistent with the total of two SeMet substitutions introduced into C4BPα1-2. The crystal structure of C4BPα1-2 from the M2-C4BPα1-2 co-crystal structure was manually fit into SAD-phased density, with the two domains of C4BPα1-2 being treated as individual rigid bodies. A model of the M49 protein was then built into density, with the amino acid register for the coiled coil being assigned based on well defined density corresponding to large side chains (i.e., His 20, Phe 75, and His 85). TLS parameterization involved the following groups: For M49 chain A, 56–60 and 61–126; for M49 chain C, 56–126; for C4BPα1-2 chain B, 0–10, 11–62 and 63–124; for C4BPα1-2 chain D, 0–13, 14–27, 28–33, 34–44, 45–53, 54–62, 63–73, 74–86, 87–102, and 103–124. Continuous electron density was evident for most of the main chain of C4BPα1-2, except for some of the longer loops of the C4BPα1 domain, and for amino acids 56–124 (or 126, depending on the chain) of M49. The M49 residue A106 of chain A had φ, ψ angles that were in the outlier region of the Ramachandran plot; this residue was distant from the interface with C4BPα1-2.
Validation of Structures
Structural models were validated with MolProbity40 (Supplementary Table S1). Molecular figures were made with PyMol (http://pymol.sourceforge.net).
Co-Precipitation Assays
Forty μg of C4BPα1-2-His6 protein was mixed with 120 μg of intact M2 protein (wild-type or mutant) in 50 μL of phosphate buffered saline (PBS) at 37 °C for 30 min. Fifty μL of Ni2+-NTA agarose beads were equilibrated in PBS, then added to the protein mix in a 1:1 beads:PBS (100 μL) slurry and incubated for 30 min at 37 °C under agitation. The beads were washed three times with 0.5 mL of PBS supplemented with 15 mM imidazole, and eluted with 40 μL PBS supplemented with 500 mM imidazole. Proteins in the input and eluted fractions were resolved by non-reducing SDS-PAGE and visualized by Coomassie-staining. Gels were scanned and ImageJ41 was used to quantify band intensities. A total of four independent coprecipitation experiments were quantified, and band intensities were verified to be within the linear range of measurement. The intensity of the band from the lane containing no C4BPα1-2 was subtracted as background from other measurements. Values were normalized to the value of wild-type M2.
Molecular Dynamics: System Preparation
Heavy atom coordinates were taken from the co-crystal structures of M protein-C4BPα1-2 complexes. Structures of complexes containing M2 substitution mutants were created by computational point mutations at the desired amino acid(s). Due to the varying resolutions of crystal structures, crystallographic waters were removed prior to solvating the system. Each structure was prepared for simulation using the Amber14SB force field42,43,44. The ionization states of titratable residues at pH 7 were predicted using PROPKA 3.145,46 and visually inspected to ensure the accuracy of assigned states. Free cysteine residues were converted to disulfide-bonded pairs manually and built using tLeap, a system preparation program from the Amber Tools 2015 package44. The C-termini of proteins were capped to remove charges. The solvent was modeled explicitly using the TIP4P water model47 and a 0.15 M NaCl concentration was applied after neutralizing the overall charge of the protein complexes. The Particle Mesh Ewald electrostatic summation method48,49 was employed to evaluate electrostatics during simulation. In total, eight different M proteins in complex with C4BPα1-2 were simulated: 1) M2 (amino acids 53–86), 2) M2 K65A (53–86), 3) M2 N66D (53–86), 4) M2 N66A (53–86), 5) M2 K65A/N66A (53–86), 6) M2 F75A (53–86), M22 (52–79), 8) M28 (55–80), and 9) M49 (56–126). All systems contained residues 1 to 124 of C4BPα.
Molecular Dynamics: Minimization, Equilibration, and Production Molecular Dynamics
The NAMD simulation package50,51 was used to minimize, heat, equilibrate, and simulate each system using a 2 fs time-step. Every system underwent a series of separate minimization, heating, and equilibration stages in preparation for production runs. The minimization spanned five stages in 10 ps intervals using the NVT ensemble: 1) 5,000 steps of hydrogen-only minimization, 2) 5,000 steps of solvent minimization, 3) 5,000 steps of side-chain minimization, 4) 5,000 steps of protein-backbone minimization, and 5) 5,000 steps of full-system minimization. Following minimization, the Langevin thermostat52,53 was used to slowly heat the system to 310 K using the NVT ensemble over 250,000 steps (500 ps). The system was then subjected to three sequential equilibration stages using the NPT ensemble for 125,000 steps/stage (250 ps/stage). The pressure was set to 1 atm and maintained using the Beredensen barrostat54. In the first MD production run, atoms were assigned a random starting velocity, and sequential steps carried over the velocities from the previous step. Five replicates of each system were performed to enhance sampling of the conformational landscape55 and the total simulation time for each system was 25 ns/replicate (40 ns/replicate for M2 F75A). Therefore, the total aggregate simulation time for each system was 125 ns (200 ns for M2 F75A).
Molecular Dynamics: Percent Occupancy (Footprinting) Analysis
The five replicates comprising each system (125 ns total for all systems except 200 ns total for M2 F75A) were combined using cpptraj56, a simulation processing software in the AmberTools package44. Trajectories were aligned against the first frame and an average structure was calculated using all atoms in the appropriate protein complex. The average conformation was used to realign the trajectories with respect to Cα atoms. The average conformation was then used to calculate the root mean squared fluctuation (Å) (RMSF) of individual residues in the protein complex. A single concatenated 125 ns (200 ns for M2 F75A) trajectory consisting of the five replicates was written by cpptraj and used for the following analysis.
Using VMD57, the radial distribution function (RDF) of pairwise interactions for a number of protein-protein contacts was calculated over the duration of the concatenated trajectory58. Distances in the RDF analysis were explicitly calculated for the following heavy atoms of residues: backbone nitrogen of histidine; Cβ of alanine and valine; Cγ of aspartate, leucine, and isoleucine; Cδ of glutamate; and Cζ of arginine. A 5 Å cutoff was applied to all pairwise interactions to include salt bridges and hydrogen bonds between hydrogen atoms and heavy atoms that were not explicitly analyzed. This was done to capture interactions between equivalent atoms, e.g. Oδ and Oδ’ of aspartate interacting with Hω and Hω’ of arginine.
Supplementary Material
Acknowledgments
We thank Olivia Ghosh for help on the project. This works was supported by NIH grant T32 GM007240 (CZB), AHA Predoctoral Fellowship 14PRE18320032 (CZB), NIH R01 AI096837 (PG and VN), and NIH R01 AI077780 (VN). The work was also funded in part by the National Biomedical Computation Resource, NIH P41 GM103426, NIH Director’s New Innovator Award Program DP2-OD007237 and through the NSF XSEDE Supercomputer resources grant RAC CHE060073N to REA. SPH was supported by the Interfaces multi-scale analysis of Biological Structure and function training grant NIH T32 EB009380.
Footnotes
Author Contributions
CZB, VN, and PG conceived of the experiments; CZB, AJB-S, and TB carried out the structure determinations; CZB and AJB-S carried out the binding studies; SPH and REA carried out and analyzed the molecular dynamics simulations; and CZB and PG wrote the paper with input from all authors.
Accession codes
Protein Data Bank
5HYU
5HYP
5HYT
5HZP
5I0Q
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 3.Dale JB, et al. Group A streptococcal vaccines: paving a path for accelerated development. Vaccine. 2013;31(Suppl 2):B216–222. doi: 10.1016/j.vaccine.2012.09.045. [DOI] [PubMed] [Google Scholar]
- 4.Good MF, Pandey M, Batzloff MR, Tyrrell GJ. Strategic development of the conserved region of the M protein and other candidates as vaccines to prevent infection with group A streptococci. Expert Rev Vaccines. 2015;14:1459–1470. doi: 10.1586/14760584.2015.1081817. [DOI] [PubMed] [Google Scholar]
- 5.McNamara C, et al. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science. 2008;319:1405–1408. doi: 10.1126/science.1154470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh P. The nonideal coiled coil of M protein and its multifarious functions in pathogenesis. Adv Exp Med Biol. 2011;715:197–211. doi: 10.1007/978-94-007-0940-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandin C, Carlsson F, Lindahl G. Binding of human plasma proteins to Streptococcus pyogenes M protein determines the location of opsonic and non-opsonic epitopes. Mol Microbiol. 2006;59:20–30. doi: 10.1111/j.1365-2958.2005.04913.x. [DOI] [PubMed] [Google Scholar]
- 8.Penfound TA, Ofek I, Courtney HS, Hasty DL, Dale JB. The NH2-terminal region of Streptococcus pyogenes M5 protein confers protection against degradation by proteases and enhances mucosal colonization of mice. J Infect Dis. 2010;201:1580–1588. doi: 10.1086/652005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lannergard J, et al. The hypervariable region of Streptococcus pyogenes M protein escapes antibody attack by antigenic variation and weak immunogenicity. Cell Host Microbe. 2011;10:147–157. doi: 10.1016/j.chom.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Dale JB, Penfound TA, Chiang EY, Walton WJ. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine. 2011;29:8175–8178. doi: 10.1016/j.vaccine.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMillan DJ, et al. Updated model of group A Streptococcus M proteins based on a comprehensive worldwide study. Clin Microbiol Infect. 2013;19:E222–229. doi: 10.1111/1469-0691.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persson J, Beall B, Linse S, Lindahl G. Extreme sequence divergence but conserved ligand-binding specificity in Streptococcus pyogenes M protein. PLoS Pathog. 2006;2:e47. doi: 10.1371/journal.ppat.0020047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ermert D, Blom AM. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunol Lett. 2016;169:82–92. doi: 10.1016/j.imlet.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blom AM, Hallstrom T, Riesbeck K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol. 2009;46:2808–2817. doi: 10.1016/j.molimm.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Carlsson F, Berggard K, Stalhammar-Carlemalm M, Lindahl G. Evasion of phagocytosis through cooperation between two ligand-binding regions in Streptococcus pyogenes M protein. J Exp Med. 2003;198:1057–1068. doi: 10.1084/jem.20030543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ermert D, et al. Virulence of group A Streptococci is enhanced by human complement inhibitors. PLoS Pathog. 2015;11:e1005043. doi: 10.1371/journal.ppat.1005043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 1992;257:919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 19.Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992;70:1035–1048. doi: 10.1016/0092-8674(92)90252-8. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins HT, et al. Human C4b-binding protein, structural basis for interaction with streptococcal M protein, a major bacterial virulence factor. J Biol Chem. 2006;281:3690–3697. doi: 10.1074/jbc.M511563200. [DOI] [PubMed] [Google Scholar]
- 21.Accardo P, Sanchez-Corral P, Criado O, Garcia E, Rodriguez de Cordoba S. Binding of human complement component C4b-binding protein (C4BP) to Streptococcus pyogenes involves the C4b-binding site. J Immunol. 1996;157:4935–4939. [PubMed] [Google Scholar]
- 22.Blom AM, et al. Human C4b-binding protein has overlapping, but not identical, binding sites for C4b and streptococcal M proteins. J Immunol. 2000;164:5328–5336. doi: 10.4049/jimmunol.164.10.5328. [DOI] [PubMed] [Google Scholar]
- 23.Andre I, et al. Streptococcal M protein: structural studies of the hypervariable region, free and bound to human C4BP. Biochemistry. 2006;45:4559–4568. doi: 10.1021/bi052455c. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 25.Sanderson-Smith M, et al. A systematic and functional classification of Streptococcus pyogenes that serves as a new tool for molecular typing and vaccine development. J Infect Dis. 2014;210:1325–1338. doi: 10.1093/infdis/jiu260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins SJ, Chung LP, Reid KB. Unusual ultrastructure of complement-component-C4b-binding protein of human complement by synchrotron X-ray scattering and hydrodynamic analysis. Biochem J. 1986;233:799–807. doi: 10.1042/bj2330799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berggard K, et al. Binding of human C4BP to the hypervariable region of M protein: a molecular mechanism of phagocytosis resistance in Streptococcus pyogenes. Mol Microbiol. 2001;42:539–551. doi: 10.1046/j.1365-2958.2001.02664.x. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson MC, et al. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog. 2013;9:e1003323. doi: 10.1371/journal.ppat.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ermert D, et al. Binding of complement inhibitor C4b-binding protein to a highly virulent Streptococcus pyogenes M1 strain is mediated by protein H and enhances adhesion to and invasion of endothelial cells. J Biol Chem. 2013;288:32172–32183. doi: 10.1074/jbc.M113.502955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou T, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 35.Doublie S. Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol Biol. 2007;363:91–108. doi: 10.1007/978-1-59745-209-0_5. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearlman DA, et al. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 43.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 44.Case DA, et al. AMBER 2015. University of California; San Fransisco: 2015. [Google Scholar]
- 45.Olsson MHM, Sondergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical pK(a) predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 46.Sondergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pK(a) Values. J Chem Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 47.Li PF, Roberts BP, Chakravorty DK, Merz KM. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J Chem Theory Comput. 2013;9:2733–2748. doi: 10.1021/ct400146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darden T, York D, Pedersen L. Particle mesh Ewald - an N.Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 49.Essmann U, et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 50.Nelson MT, et al. NAMD: A parallel, object oriented molecular dynamics program. Int J Supercomput Ap. 1996;10:251–268. [Google Scholar]
- 51.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD. Langevin stabilization of molecular dynamics. J Chem Phys. 2001;114:2090–2098. [Google Scholar]
- 53.Jiang W, et al. High-Performance scalable molecular dynamics simulations of a polarizable force field based on classical drude oscillators in NAMD. J Phys Chem Lett. 2011;2:87–92. doi: 10.1021/jz101461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 55.Caves LSD, Evanseck JD, Karplus M. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Science. 1998;7:649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 57.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 58.Levine BG, Stone JE, Kohlmeyer A. Fast analysis of molecular dynamics trajectories with graphics processing units-Radial distribution function histogramming. J Comput Phys. 2011;230:3556–3569. doi: 10.1016/j.jcp.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.