

Abstract
Pharmaceutical intervention often requires therapeutics and/or their carriers to enter cells via endocytosis. Therefore, endocytic aberrancies resulting from disease represent a key, yet often overlooked, parameter in designing therapeutic strategies. In the case of lysosomal storage diseases (LSDs), characterized by lysosomal accumulation of undegraded substances, common clinical interventions rely on endocytosis of recombinant enzymes. However, the lysosomal defect in these diseases can affect endocytosis, as we recently demonstrated for clathrin-mediated uptake in patient fibroblasts with type A Niemann-Pick disease (NPD), a disorder characterized by acid sphingomylinase (ASM) deficiency and subsequent sphingomyelin storage. Using similar cells, we have examined if this is also the case for clathrin-independent pathways, including caveolae-mediated endocytosis and macropinocytosis. We observed impaired caveolin-1 enrichment at ligand-binding sites in NPD vs. wild type fibroblasts, corresponding with altered uptake of ligands and fluid-phase markers by both pathways. Similarly, aberrant lysosomal storage of sphingomyelin induced by pharmacological means also diminished uptake. Partial degradation of the lysosomal storage by untargeted recombinant ASM lead to partial uptake enhancement, while both parameters were restored to wild type levels by ASM delivery using model polymer nanocarriers specifically targeted to intercellular adhesion molecule-1 (anti-ICAM NCs). Carriers also restored caveolin-1 enrichment at ligand-binding sites and uptake through the caveolar and macropinocytic routes. These results demonstrate a link between lysosomal storage in NPD and alterations in clathrin-independent endocytosis, which could apply to other LSDs. Hence, this study shall guide the design of therapeutic approaches using viable endocytic pathways.
Keywords: Lysosomal storage disorders, type A Niemann-Pick disease, caveolae-mediated endocytosis, macropinocytosis, ICAM-1-targeted nanocarriers, enzyme delivery
Graphical Abstract
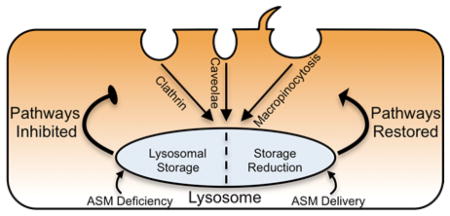
Introduction
Cells continuously internalize extracellular material and plasmalemma molecules via endocytosis, supporting a plethora of biological functions and providing a gateway for intracellular drug delivery 1–3. Budding of endocytic vesicles from the plasma membrane can occur constitutively, or can be induced by specific binding of ligands to their receptors on the cell surface, where both extracellular fluid and said molecules can be internalized by pinocytosis 3, 4. As such, therapeutics and/or their carriers may undergo uptake from the extracellular milieu as a part of the fluid-phase, or may target and induce uptake by association with plasmalemma components, most commonly endocytic receptors 2, 5.
Parameters such as the vesicle size, intracellular trafficking, and molecular players that regulate the fate of endocytosed substances vary among each of the three classical pinocytic mechanisms 2, 3: clathrin-mediated endocytosis, caveolin-mediated endocytosis, and macropinocytosis 3, 4. Each has been explored to varying degrees for uptake of therapeutics 1, 5. Clathrin-mediated endocytosis has been broadly observed in mammalian cells in association with a number of receptors (e.g., transferrin, low density lipoprotein, mannose-6-phosphate, etc.). This route has been widely targeted for delivery of therapeutics, drug conjugates, and carriers bearing clathrin-associated ligands 4, 6, 7. Alternative routes have also drawn considerable attention. For example, caveolae-mediated endocytosis occurs in most cell types 4, 5, 8 and is characterized by caveolin-1-rich invaginations of ~50–70 nm that form in lipid raft regions of the plasmalemma 8, 9. Uptake via this route has been achieved by exploiting caveolar ligands such as cholera toxin, simian virus 40, albumin, aminopeptidase P, etc., or antibodies to caveolar markers 2, 5, 7, 10–12. Alternatively, macropinocytosis is characterized by constitutive uptake of fluids in large vesicles (>500 nm in diameter) 13 and, although it is especially active in macrophages and dendritic cells, it can be transiently induced in non-hematopoietic cells by epidermal growth factor (EGF), therefore also playing a key role in biology and drug uptake 5, 13.
It has become increasingly apparent however, that normal endocytic trafficking behavior can be altered by disease, reducing the efficiency of therapies requiring endocytic uptake. Lysosomal storage disorders (LSDs) illustrate this paradigm. LSDs often arise from a genetic deficiency of a lysosomal hydrolase, causing abnormal lysosomal accumulation of metabolic substrates 14. Therapy by gene transfer via viral vectors (in clinical trial for Sanfilippo and Pompe disease) or replacement therapy with recombinant enzymes (clinically approved for Gaucher, Fabry, Pompe, and mucopolysaccharidosis types I, II, and VI) 15–17 both require endocytic uptake within cells 18. However, secondary cellular defects that arise from substrate storage (e.g., altered cytoskeletal rearrangement, diminished secretory trafficking, subcellular traffic jams, etc.) may impair uptake of such therapies 19–23. In particular, lysosomal accumulation of lipids in certain LSDs has been linked with aberrant trafficking of plasma membrane components and ligands associated with clathrin and caveolar pathways 21–25. This is the case for fibroblasts from patients with type A Niemann-Pick disease (NPD). NPD cells are characterized by acid sphingomyelinase (ASM) deficiency leading to sphingomyelin and, secondarily, cholesterol storage 26. In these cells, we have recently described impairment of clathrin-mediated endocytosis 27. This defect correlated with suboptimal uptake of therapeutic ASM 23, 27, an enzyme that naturally targets the clathrin route via the mannose-6-phosphate receptor 23, 26. Instead, enzyme delivery was more efficient and led to greater degradation of the lysosomal storage by bypassing this defunct pathway 27. This was achieved by delivering the enzyme via polymer nanocarriers targeted to a clathrin-, caveolae-, and macropinocytic-independent route called cell adhesion molecule (CAM)-mediated endocytosis, which was induced by targeting intercellular adhesion molecule -1 (ICAM-1) 28–31. In addition, the reduction of lysosomal sphingomyelin storage restored the activity of the clathrin pathway 27, demonstrating a link between lysosomal storage and altered clathrin-mediated endocytosis.
In this study, we have explored and found that a similar relationship exists between aberrant lysosomal storage in type A Niemann-Pick disease and clathrin-independent endocytosis via caveolar and macropinocytic routes.
Methods
Antibodies and Reagents
Alexa Fluor-594 cholera toxin B subunit (CTB), 10,000 MW Texas Red dextran, BODIPY-FL-C12-sphingomyelin, and fluorescent secondary antibodies were from Molecular Probes (Eugene, OR). Anti-CTB and anti-caveolin-1 were from Calbiochem (La Jolla, CA). Mouse monoclonal anti-human ICAM-1 (clone R6.5) was from the American Type Culture Collection (Manassas, VA). Non-fluorescent or green Fluoresbrite 100 nm-diameter polystyrene beads were from Polysciences (Warrington, PA). Recombinant human ASM 32 was kindly provided by Dr. Edward Schuchman (Dept. of Genetics and Genomics Sciences, Mount Sinai School of Medicine, New York, NY). Unless otherwise noted, all other reagents were from Sigma Aldrich (St. Louis, MO).
Cell Cultures
Wild type and type A NPD patient skin fibroblasts (homozygous for the R496L mutation) were kindly provided by Dr. Edward Schuchman. Cells were seeded on glass coverslips and incubated at 37°C, 5% CO2, and 95% relative humidity in Dulbecco’s Modified Eagle Medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Where indicated, ICAM-1 expression was stimulated as in inflammatory conditions pertaining to NPD, by incubating cells with 10 ng/mL tumor necrosis factor α (TNFα) overnight prior to assays 29.
Ligand Uptake by Caveoli
Wild type and NPD fibroblasts were incubated with medium containing 2.9 μM red Alexa Fluor-594 CTB for varying periods of time (from fifteen minutes to five hours) at 37°C in order to measure the kinetics of uptake by caveolar endocytosis. Cells were then washed, fixed with 2% paraformaldehyde, and incubated with goat anti-CTB followed by green fluorescein isothiocyanate (FITC)-labeled rat anti-goat IgG. This protocol renders CTB bound to the cell surface double stained in red and green (yellow) vs. internalized CTB, which appears only red, as previously described 28. To verify that CTB uptake was mainly contributed by caveolar- vs. clathrin-mediated endocytosis, similar experiments were conducted in the presence of 1 μg/mL filipin (inhibits caveolar uptake 33), 50 μM monodansylcadaverine (MDC; inhibits clathrin pathways 34), or a mixture of both inhibitors. Cells had been exposed to said inhibitors also for thirty minutes prior to incubation with CTB.
All samples were washed, mounted on slides, and analyzed by fluorescence microscopy. This was done using an Eclipse TE2000-U microscope with a 60x PlanApo objective and filters optimized for Texas Red and FITC fluorescence (Nikon, Melville, NY). Images were taken with an Orca-ICCD camera (Hamamatsu, Bridgewater, NJ) using ImagePro 3.0 software (Media Cybernetics, Silver Spring, MD). To facilitate analysis using a customized algorithm that detects both fluorescent pixels and endosomal-sized vesicles, both surpassing a background threshold fluorescence 37, CTB was pseudocolored in green and the additional stains were pseudocolored in red. Therefore, surface CTB appear yellow vs. internalized counterparts that appear green, which were quantified as described previously 35. The percentage of internalized green fluorescent pixels over the total cell-associated fluorescence and the number of green fluorescent vesicles were quantified. Where indicated, these parameters were expressed per cell area (shown in mm2), which was obtained from observation of the cell borders by phase-contrast microscopy.
Caveolin-1 Distribution
To measure the colocalization of CTB with caveolin-1, cells were incubated with red Alexa Fluor-594 CTB as described above, washed, fixed, and permeabilized with 0.2% Triton X-100. Samples were then immunostained with mouse anti-human caveolin-1 and green FITC goat anti-mouse IgG. Cells not exposed to CTB were used as controls to discern the baseline distribution of caveolin-1. After image analysis using the equipment and software described above, CTB co-localizing or not with caveolin-1 appears yellow vs. green, respectively, which was quantified as described previously 35. In addition, the percentage of caveolin-1 fluorescence that localized within ~3 μm distance from the cell nucleus compared to the whole cell-associated fraction was calculated, where both the nucleus and cell borders were imaged by phase-contrast microscopy.
Fluid-Phase Uptake
To quantify bulk fluid-phase pinocytosis as well as the contribution of individual pathways to such uptake, wild type and NPD fibroblasts were pretreated for thirty minutes at 37°C with control media or media supplemented with either one or a combination of the following pharmacological inhibitors: 50 μM MDC (to inhibit clathrin endocytosis), 1 μg/mL filipin (to inhibit caveolar endocytosis), and 3 mM amiloride (to inhibit macropinocytosis). We have validated the specificity of these treatments elsewhere 28. Cells were then incubated for varying periods of time (from thirty minutes to eight hours) with 1 mg/mL Texas Red dextran, a fluid phase marker for endocytosis, in control media or for one hour in inhibitor-supplemented media (see a description of the inhibitors above). Alternatively, macropinocytosis was evaluated by incubating cells for varying periods of time (from fifteen minutes to eight hours) at 37°C with 1 mg/mL Texas Red dextran in media containing 100 ng/mL EGF, which stimulates the formation of membrane ruffles and macropinocytosis 13. Cell samples were washed, fixed, and the number of fluorescent dextran-filled compartments was quantified by fluorescence microscopy as described 27. Where indicated, this parameter was expressed per cell area (shown in mm2), which was obtained from observation of the cell borders by phase-contrast microscopy.
Role of Induced Lipid Storage
To mimic lysosomal storage of sphingomyelin as seen in type A NPD, wild type fibroblasts were incubated at 37°C for forty eight hours with 50 μM imipramine, which cleaves endogenous ASM, rendering it inactive 36. To track sphingomyelin levels in control vs. imipramine-treated wild type fibroblasts, or NPD fibroblasts, cells were incubated overnight with BODIPY-FL-C12-sphingomyelin, which fluoresces red (620 nm) at high concentrations and green (530 nm) at physiological concentrations 29. Cells were then analyzed by fluorescence microscopy to quantify sphingomyelin accumulation.
In parallel experiments, fluid-phase uptake of 1 mg/mL Texas Red dextran was examined in imipramine-treated wild type fibroblasts and compared to that of control wild type or NPD fibroblasts, as described above.
Preparation of ASM-Loaded Nanocarriers Targeted to ICAM-1
Model polymer nanocarriers were prepared by adsorbing either anti-ICAM, control non-specific IgG, or a mix containing 50:50 mass ratio of anti-ICAM:ASM (anti-ICAM NCs, IgG NCs and anti-ICAM/ASM NCs, respectively) on the surface of 100 nm-diameter polystyrene beads (non-fluorescent or green Fluoresbrite, as indicated for respective experiments), as described 27, 29. This antibody:enzyme ratio has been shown to produce carriers with efficient targeting properties and active ASM capable of degrading accumulated lysosomal substrates 27, 29. Unbound molecules were removed by centrifugation at 13,000 g for three minutes, carriers were re-suspended in phosphate buffered saline supplemented with 1% bovine serum albumin, and the solution was sonicated at low power to disengage carrier aggregates. To characterize the carrier coat, either anti-ICAM, IgG, or the enzyme cargo were conjugated to 125I, and the amount of radiolabeled antibody or enzyme per carrier was determined with a gamma counter. The diameter of all coated particles ranged between 185 and 225 nm, with polydispersity ranging from 0.16 to 0.19, as measured by dynamic light scattering. Carriers coated with anti-ICAM or IgG alone contained 262 ± 9 and 244 ± 10 antibody molecules per particle, respectively, while those carrying ASM and anti-ICAM contained 230 ± 24 enzyme molecules and 135 ± 17 antibody molecules per particle. These formulations have been shown to exhibit minimal antibody or enzyme release by mechanical stress (~10% release), storage (<5% release after three days in saline at 4°C), or physiological-like conditions (~10% release after five hours in serum at 37°C) 37, 38. Although not intended for clinical use, these prototype carriers have been demonstrated to render coating efficacy, in vivo targeting, and intracellular transport comparable to anti-ICAM NCs made of biodegradable poly(lactic-co-glycolic acid) 39, 40, and are therefore a valid model.
Targeting and Uptake of anti-ICAM Carriers
Wild type fibroblasts were pretreated overnight with TNFα to mimic inflammatory activation typical of NPD and other LSDs 17, 26. Cells were exposed for one hour at 37°C to green Fluoresbrite anti-ICAM NCs or control non-specific IgG NCs, then washed and fixed. Samples were then incubated with a secondary antibody labeled with Texas Red, which is only accessible to anti-ICAM on the coat of cell-surface located carriers (not internalized ones), as demonstrated before 35. This allows differential staining of cell-surface bound (green + red = yellow) vs. internalized (green alone) carriers. Total cell-associated carriers and the percentage of carriers internalized were quantified by fluorescence microscopy, as described 35.
Effects of Delivered ASM
Following overnight treatment with TNFα to mimic inflammatory activation observed in many LSDs 17, 26, NPD fibroblasts were incubated for one hour at 37°C with recombinant ASM, applied either as a free counterpart (32 nM vs. 320 nM, respectively) or coupled onto anti-ICAM NCs (32 nM ASM). We previously demonstrated partial vs. complete normalization of the sphingomyelin levels, respectively, by these treatments 27. The cells were washed to remove treatments and incubated in basal media for three hours to permit degradation of intracellular sphingomyelin stores. Bulk fluid-phase endocytosis, uptake of CTB, caveolin-1 distribution and co-localization with CTB, and EGF-stimulated macropinocytosis were then assessed as described above.
Statistics
Experiments involved two or three assays and microscopy analysis involved random selection of 8–15 regions located throughout the sample for image acquisition. Individual analysis of each cell contained in such images was pursued, where all vesicles contained per cell were included in the analysis (this represents a total between a thousand and three thousand vesicles per condition). Data were calculated as mean ± standard error of the mean (SEM), where statistical significance was determined as p<0.05 by Student’s t-test, since comparisons were aimed between two groups.
Results
Altered Endocytosis and Distribution of Caveolar Ligands and Markers in NPD Fibroblasts
To build upon our recent study showing altered clathrin-mediated endocytosis in NPD fibroblasts 27, here we have focused on clathrin-independent routes. We first examined the uptake of fluorescent CTB, a molecule that associates with ganglioside GM1 on the plasmalemma and internalizes largely by caveolar-mediated endocytosis 41. Over the course of one hour, total CTB associated with wild type fibroblasts to a significantly greater extent than with NPD cells, and also localized to the perinuclear region vs. an endosomal-punctate like appearance observed in NPD (Figure 1a). Hence, CTB distribution in wild type cells was consistent with endoplasmic reticulum and Golgi trafficking, as expected for caveolar uptake of this toxin 8. Verifying this route (since clathrin-mediated endocytosis could contribute to CTB uptake 42), only treatment with filipin, a cholesterol-binding agent that inhibits caveolar endocytosis 33, but not monodansylcadaverine (MDC), an inhibitor of clathrin coated pits 34, significantly diminished CTB endocytosis in wild type fibroblasts: 47% vs. 81% of control, respectively (Supplementary Figure 1). Also, CTB uptake in the presence of both filipin and MDC did not differ from filipin alone (51% of control; Supplementary Figure 1). Importantly, total association of CTB with NPD fibroblasts was only 64% that of wild type cells (Figure 1a), indicating a defect in the caveolar route. In fact, filipin inhibition of CTB uptake, alone or in combination with MDC, was less acute in NPD cells (63% and 80% of control, respectively; Supplementary Figure 1) and no effect was found for NPD cells treated with MDC alone (111% of control), which is expected since NPD cells have impaired clathrin-mediated uptake 27.
Figure 1.

Binding and internalization of cholera toxin B (CTB) in NPD fibroblasts. A Fluorescence microscopy of wild type (Wt) and NPD fibroblasts incubated with CTB (green) for one hour at 37°C. Unbound CTB was washed off, cells were fixed, and surface CTB was immunostained (red+green=yellow; arrowheads) to distinguish it from internalized CTB (green; arrows). Dotted lines = cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. Total, internal, and surface CTB (fluorescent objects over background) were quantified. B Kinetics of uptake of CTB in Wt vs. NPD fibroblasts, examined as described in A, where the percentage of internalized fluorescence from the total cell-associated fluorescence is shown. Lines are regression curves, where R2 was 0.99 for Wt and 0.94 for NPD. A–B Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
The endocytic differences observed between NPD and wild type cells mostly applied to the internalized fraction of CTB (47% that of wild type fibroblasts), while the amount of CTB bound to the cell surface was similar in both cell types, suggesting that the defect may lie in the uptake process. Examination of the kinetics of CTB uptake suggested this event was slower in NPD vs. wild type cells: half time for maximal uptake was 26 minutes vs. 12 minutes, respectively, when comparing the percentage of CTB that was internalized from the total cell-associated fraction (Figure 1b). With time, this parameter reached a similar saturation (95% uptake for wild type and 94% uptake for NPD; Figure 1b). When quantifying the number of internalized vesicles containing CTB, the uptake kinetics was even more affected (half time of 20 minutes for wild type vs. 134 minutes for NPD; Supplementary Figure 2a), and when the number of internalized vesicles containing CTB was normalized to the cell-surface area (since different cell sizes would render different numbers of invaginating vesicles even if the rate of uptake was same) a similar pattern was observed, yet still more acute (Supplementary Figure 2b).
Since caveolin-1 is enriched in caveolar invaginations, caveolae-mediated uptake is characterized by co-localization of ligands internalizing via this route with caveolin-1 41. To evaluate if this was affected in NPD cells, we examined the intracellular distribution of caveolin-1. In fixed cells and under control conditions (absence of CTB), caveolin-1 localized to widespread peripheral regions in wild type cells, as expected, while it appeared to preferentially occupy a nuclear-like region in NPD cells (Figure 2a). As shown in Figures 2b and 2c, when wild type cells were exposed to CTB prior to fixation and then immunostained for caveolin-1, this marker was also found to be broadly distributed (only 28% localization within 3 μm from the cell nucleus) and clearly associated with CTB (76% colocalization), as expected for caveolar uptake of CTB. In contrast, caveolin-1 remained localized in the nuclear region in NPD cells after exposure to CTB (88% perinuclear), with only 8% of all CTB being colocalized with caveolin-1, which is consistent with the previous observation of diminished CTB uptake via caveoli.
Figure 2.

Distribution of caveolin-1 in NPD fibroblasts. A Fluorescence microscopy images of Wt and NPD fibroblasts showing the distribution of caveolin-1 in fixed cells that had been previously incubated in the absence of ligands (CTB) added to the cell media. B After incubation with CTB (pseudocolored green) for one hour at 37°C to induce caveolar uptake, cells were washed, fixed, permeabilized and caveolin-1 was immunostained and pseudocolored red. Black/white panels illustrate the distribution of caveolin-1, while color panels show the colocalization (yellow) of caveolin-1 (red) and CTB (green) in the same cells. A–B Ovals denote the nucleus and dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. C The percentage of CTB that colocalized with caveolin-1 throughout the entire cell and the percentage of caveolin-1 located within ~3 μm of the nucleus were quantified. Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
Reduced Fluid-Phase Uptake via Caveoli and Macropinocytosis in NPD Fibroblasts
Aberrant distribution of caveolin-1 and reduced CTB uptake support that the caveolae-mediated pathway is altered in NPD. This may affect not only endocytosis of ligands (illustrated by CTB) but also fluid-phase uptake via caveoli. Indeed, using fluorescent dextran as a fluid-phase marker, NPD cells exhibited reduced uptake: 60–70% compared to wild type cells (see one hour time point in Figure 3a, and solid and dashed horizontal lines in Figure 3b), similar to our previous report for these cells 27. As in the case of altered uptake of CTB, NPD cells showed slower dextran uptake kinetics as compared to wild type cells (half time for maximal internalization was 67 minutes vs. 29 minutes, respectively), yet maximal uptake per cell was not decreased (Figure 3a). However, when the number of dextran-containing vesicles was normalized to the cell size, maximal uptake at saturation seemed reduced for NPD (Supplementary Figure 3).
Figure 3.

Pinocytosis and its mediation by caveolae in NPD fibroblasts. A Microscopy images and quantification of the number of dextran-filled vesicles (fluorescent objects over background) associated with Wt or NPD fibroblasts incubated for varying periods of time at 37°C with media containing fluorescent dextran (microcopy images show one hour as an example). Dotted lines mark the cell borders, as determined from the corresponding phase-contrast images. Scale bar = 10 μm. B Dextran-filled vesicles were quantified after one hour incubation at 37°C in control untreated cells (either Wt Ctr or NPD Ctr) vs. cells treated with either 1 μg/mL filipin (Fil; to inhibit caveolar endocytosis) or with a cocktail of 50 μM MDC and 3 mM amiloride (MDC + Amil; to inhibit clathrin and macropinocytosis simultaneously). Uptake by control untreated Wt or NPD cells are shown as solid and dashed horizontal lines, respectively. A–B Data are the mean ± SEM. *Comparison between NPD and similarly treated Wt cells; #comparison with untreated (control without inhibitor) cells within each cell type (p<0.05 by Student’s t-test). In graph A, lines are regression curves, where R2 = 0.99 for WT and 0.97 for NPD.
Interestingly, treatment of wild type cells with filipin to inhibit caveoli resulted in a significant reduction in dextran pinocytosis: 50% reduction (compare wild type filipin black bar vs. its horizontal solid line control; Figure 3b), demonstrating the significant role of the caveolar pathway in basal fluid-phase uptake. In NPD cells however, filipin treatment only diminished dextran uptake by 17% (compare NPD filipin black bar vs. its horizontal dashed line control). This suggests that caveolar endocytosis contributed less to dextran uptake in NPD vs. wild type cells, in accordance with the defect observed above.
We also evaluated uptake via the caveolar route in healthy vs. diseased fibroblasts when other pathways were inhibited. Wild type cells were treated simultaneously with MDC and amiloride to simultaneously inhibit clathrin- and macropinocytosis-mediated uptake, leaving only the caveolar route active. In these cells, dextran uptake was nearly equivalent to that of untreated, control cells (99% of control; Fig 3b), suggesting that the caveolar pathway may compensate for the loss of clathrin endocytosis and micropinocytosis function in wild type cells. This is in agreement with previous studies demonstrating compensatory roles between different endocytic pathways 43. In contrast, NPD fibroblasts treated with the MDC+amiloride cocktail did not exhibit increased uptake (84% of its control), suggesting that the caveolar route may not be able to compensate for decreased dextran uptake under these conditions.
In addition, since macropinocytosis is known to contribute to non-specific uptake of extracellular fluid, we also evaluated this route. We examined dextran uptake in wild type vs. NPD fibroblasts under induction with EGF, a treatment that transiently stimulates macropinocytosis 13. In this scenario, NPD cells internalized only 72% as much dextran as wild type cells in one hour (Figures 4a and 4b), demonstrating deficient macropinocytic activity as well. Both the kinetics associated to this endocytic route and maximal uptake level were affected: the half time for maximal internalization was 26 minutes in NPD cells vs. 17 minutes in wild type cells, with NPD cells reaching only 86% of the maximal uptake observed in wild type samples (Figure 4b). A yet more altered result was found when the number of internalized vesicles was normalized to the cell size: maximal uptake per mm2 in NPD was 67% of wild type (Supplementary Figure 4).
Figure 4.
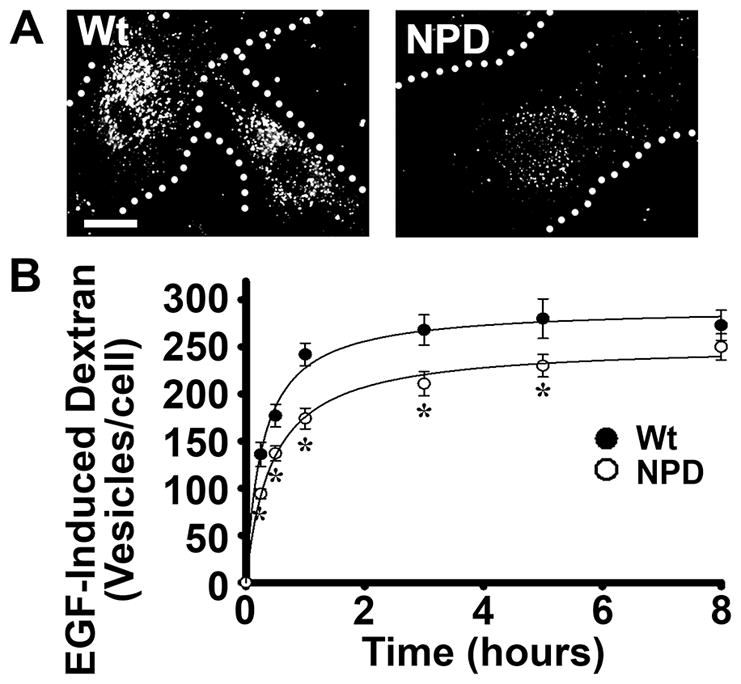
Macropinocytosis in NPD fibroblasts. A Microscopy images of Wt and NPD fibroblasts incubated for one hour at 37°C with media containing 100 ng/mL EGF (to stimulate macropinocytosis) and fluorescent dextran. Dotted lines mark the cell borders, as determined from the corresponding phase-contrast images. Scale bar = 10 μm. The number of dextran-filled vesicles was quantified as in Fig. 3. B Kinetics of EGF-induced uptake of dextran in Wt vs. NPD cells was assessed at the indicated times, as described in panel A. A–B Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test). In graph B, lines are regression curves, where R2 = 0.99 for both WT and NPD.
Uptake is Inversely Related to Lysosomal Storage and Can Be Rescued by Reducing Said Storage
NPD patient fibroblasts show alterations in endocytic uptake. Hence, we next examined if this defect is associated with aberrant lysosomal storage. We compared the sphingomyelin level and bulk uptake of dextran (reflecting the additive uptake through all endocytic pathways) in wild type fibroblasts treated with imipramine, and compared these parameters to that of untreated wild type and NPD cells. Imipramine is known to inhibit ASM activity, hence inducing in cells an NPD-like phenotype that mimics genetic ASM deficiency 36. As expected, imipramine-treated wild type cells accumulated sphingomyelin in perinuclear compartments, resulting in a 4-fold increase in sphingomyelin levels over control wild type cells (Figure 5a). The level and perinuclear distribution of the storage material were similar to that of NPD cells, which showed 5-fold enhanced sphingomyelin accumulation compared to control wild type cells (Figure 5a). Importantly, treatment of wild type cells with imipramine also resulted in a reduction of dextran uptake, which was diminished by 55% compared to control wild type cells, similar to that observed for NPD cells (50% uptake reduction; Figure 5b). These results correlate reduced pinocytosis with sphingomyelin storage.
Figure 5.

Induction of sphingomyelin storage in wild type fibroblasts decreases bulk uptake. A Wild type (Wt), NPD, and imipramine-treated Wt fibroblasts were incubated overnight with fluorescent BODIPY-FL-C12-sphingomyelin to allow incorporation and visualization of storage material. Sphingomyelin fluorescence was quantified and normalized to the Wt level. B Wt, NPD, and imipramine-treated Wt fibroblasts were incubated for one hour at 37°C with media containing fluorescent dextran. Fluorescent vesicles were quantified and the data were normalized to the Wt level. A–B Dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. Data are the mean ± SEM. *Comparison with Wt cells. Comparison between NPD and imipramine-Wt cells resulted in no significant findings. (p<0.05 by Student’s t-test).
Given the relationship between lysosomal storage and diminished uptake, it is expected that a reduction in storage would revive this function. We have reported that incubation of NPD fibroblasts with recombinant ASM added to the cell media resulted in only partial reduction of sphingomeylin storage, which correlated with reduced ASM uptake due to altered clathrin-mediated endocytosis in these cells 27. In contrast, delivery of recombinant ASM was improved by nanocarriers targeted to ICAM-1, which rely on the CAM-mediated pathway, resulting in normalization of sphingomyelin storage to wild type levels 27. The specificity of anti-ICAM NCs to enhance binding, uptake, lysosomal enzyme delivery and activity, storage reduction in cell culture, and biodistribution in mouse models, vs. naked ASM, control IgG NCs, or in the presence of ICAM-1 blockers, has been previously documented 28, 30–32, 37–40, 44. In accordance with previous work, targeting and endocytosis of anti-ICAM NCs vs. control IgG NCs was specific in these fibroblasts: 354 vs. 2 NCs associated per cell and 49% vs. 3% internalization, respectively, after one hour incubation in these cells (Supplementary Figure 5).
We then examined bulk uptake of dextran in cells treated with these different strategies, i.e., naked ASM vs. ASM coupled to anti-ICAM NCs. As shown in Figure 6, dextran uptake directly correlated with the therapies’ ability to attenuate storage: uptake was normalized (98% of wild type; 2-fold uptake over untreated NPD cells) when NPD fibroblasts were incubated with ASM coupled to ICAM-1-targeted nanocarriers (ASM NCs; low dose of 32 nM ASM). Treatment with similar dose of untargeted ASM did not exert any effect (48% of wild type uptake; no difference vs. untreated NPD cells). Only increasing the dose of untargeted ASM by 10-fold (320 nM) enhanced dextran uptake, yet still partially (68% of wild type; 1.4-fold over untreated NPD cells).
Figure 6.

Recovery of bulk uptake by enzyme replacement in NPD fibroblasts. NPD fibroblasts were treated for one hour at 37°C with media containing untargeted ASM (supplied at a low dose of 32 nM or a high dose of 320 nM) or ASM coated on the surface of anti-ICAM NCs (32 nM of ASM). Cells were then incubated for one hour with Texas Red dextran to examine endocytosis as in Fig. 3. A Fluorescence microcopy of dextran uptake in enzyme-treated cells. Dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. B Dextran vesicles were quantified for each treatment group and normalized to that of Wt cells. Relative uptake by untreated Wt and NPD cells are shown as horizontal solid and dashed lines, respectively. Data are the mean ± SEM. *Comparison with Wt cells. #Comparison with untreated NPD cells (p<0.05 by Student’s t-test).
Recovery of Caveolae- and Macropinocytic-Mediated Uptake by ICAM-1-Mediated ASM Delivery
Bulk uptake (examined using dextran) was normalized after incubation of NPD cells with recombinant ASM delivered by anti-ICAM NCs (Figure 6). This may be in part due to improvement in clathrin-mediated endocytosis by this treatment, which we previously reported 27. Hence, we examined if ICAM-1-mediated enzyme delivery also improves caveolae- and macropinocytic-mediated uptake, which we observed here to also be altered in NPD cells.
As per the caveolar pathway, the distribution of caveolin-1 in the presence of CTB was more widespread after treating cells with anti-ICAM/ASM NCs, with only 23% caveolin-1 remaining within 3 μm of the nucleus, comparable to wild type cells (Figure 7a and Supplementary Figure 6). Also, treatment with nanocarriers increased the colocalization of CTB with caveolin-1 to levels comparable to that of wild type cells (63% colocalization; 83% of wild type; Figure 7a and Supplementary Figure 6). In agreement with this recovery, total accumulation of CTB in nanocarrier treated cells, as well as CTB internalization and surface association were all restored and even enhanced as compared to wild type cells (1.6-, 1.5-, and 2.2-fold of wild type, respectively; Figure 7b). As per macropinocytosis, EGF-induced uptake of dextran in NPD cells was assessed after treatment with anti-ICAM/ASM NCs (Figure 7c and Supplementary Figure 7). Treatment resulted in a significant enhancement in this pathway as well, which was 4-fold greater than that of untreated NPD cells and 1.7-fold over that of the wild type.
Figure 7.

Recovery of clathrin-independent endocytosis in NPD fibroblasts treated with anti-ICAM/ASM NCs. A NPD fibroblasts were incubated for one hour at 37°C with fluorescent CTB (pseudocolored green) three hours after treatment with 32 nM ASM coated on anti-ICAM NCs and compared to untreated NPD cells similarly incubated with CTB. Caveolin-1 was immunostained and its distribution and colocalization with CTB were imaged as in Figure 2. The percent colocalization of CTB with caveolin-1 and the percent of caveolin-1 located within ~3 μm of the nucleus were quantified as in Figure 2. B Binding and uptake of fluorescent CTB were quantified as in Figure 1, and data are normalized to wild type levels (horizontal line). C NPD fibroblasts were incubated for one hour at 37°C with fluorescent dextran and EGF three hours after treatment with 32 nM ASM coated on anti-ICAM NCs, and compared to untreated NPD cells (as in Figure 4). Dextran-filled vesicles were quantified and normalized to wild type levels (horizontal line). A–C Data are the mean ± SEM. A*Comparison with untreated NPD cells (p<0.05 by Student’s t-test). B–C *Comparison with Wt cells (p<0.05 by Student’s t-test).
Discussion
Non-specific fluid-phase and receptor-mediated uptake are rather ubiquitous in mammalian cells, where they regulate a plethora of physiological functions and represent viable routes for intracellular drug delivery 2–4. Therefore, altered endocytic transport may affect cell and tissue homeostasis, as well as intracellular delivery of therapeutics aimed to treat such conditions. Yet, information on the relative activity of individual endocytic pathways in diseased cells is still scarce. Illustrating this, we recently described aberrant clathrin-mediated endocytosis in type A NPD fibroblasts, which limited the uptake of therapeutic ASM 27, an enzyme known to bind to the mannose-6-phospahte receptor for internalization via clathrin-mediated endocytosis 23, 26, 45. Targeting ASM to an independent receptor/pathway, CAM endocytosis mediated by ICAM-1, enhanced intracellular ASM delivery and reduction of lysosomal (sphingomyelin) storage, also restoring clathrin-mediated endocytosis to wild type levels 27. Here, we examine caveolae- and macropinocytic-mediated uptake in these cells, and the relationship between endocytic activity and lysosomal storage.
As for the clathrin route 27, uptake of a model ligand (CTB) or a model fluid-phase marker (dextran) via caveolae was also impaired in NPD fibroblasts. Since CTB levels at the cell surface were comparable with wild type cells, and since fluid-phase caveolar uptake does not associate with specific receptors, the observed defect seems due to an aberrant capability for internalization rather than abnormal availability of caveolar receptors, as described for receptors of the clathrin route 23, 27. Nevertheless, we have not compared the GM1 levels (to which CTB binds) on these cells, and it is possible that altered biosynthesis, trafficking to the cell surface, and/or recycling of plasma membrane components could also be contribute to these effects.
The cellular distribution of the machinery regulating caveolar vesicle formation (e.g., caveolin-1) also appeared disrupted in NPD, which may have contributed to decreased uptake via this pathway. Caveolin-1, essential for caveolar endocytosis 13, appeared located in the proximity of the nucleus in NPD cells and it barely colocalized with CTB when this ligand was present (Figure 2c), suggesting aberrant endogenous trafficking and/or recruitment to caveolar-binding ligands. Aberrant caveolin localization to sites of lipid accumulation has been observed in murine fibroblasts and macrophages loaded with cholesterol 46. It is possible that aberrant sphingomyelin storage in NPD, which is associated with increased cholesterol levels 26, results in a similar outcome. Notably, we still observed CTB uptake by NPD cells at a level 47% of the wild type, which seemed high considering the alteration in caveolin-1 distribution. However, our understanding of the relationship between caveolin-1 and the endocytosis of lipid raft-associated ligands is still evolving 13. This difference may be due to uptake contributed by compensatory pathways; for example, small fractions of GM1 could be taken up in clathrin-coated vesicles 41, 42. However, this route is altered in NPD fibroblasts 27 and would contribute only minimally to CTB uptake, as shown using single vs. combination inhibitors for the clathrin and caveolar routes (Supplementary Figure 1). An alternative pathway involves dynamin-independent vesicles forming in lipid rafts and trafficking to the endoplasmic reticulum, as observed for CTB and simian virus 40 47. Whether this pathway compensates for CTB uptake in NPD cells remains to be determined.
Our finding showing aberrant caveolar uptake activity in LSD cells is supported by previous studies suggesting aberrant intracellular trafficking via this route. For example, patient fibroblasts and Drosophila models of LSDs have altered trafficking of a lactosylceramide analog, a plasmalemma lipid internalized by a “caveolar-like” route, 25, 48. CTB is re-routed to endosomes rather than the Golgi apparatus in fibroblasts from four LSDs 25. In agreement with this, we found that internalized CTB distributed differently in wild type vs. NPD cells: in perinuclear vs. more punctate and widespread compartments, consistent with the Golgi vs. endosomes, respectively (Figure 1). In addition to the patho-physiological impact of this observation, since CTB and other strategies have been explored as a means to target drugs for caveolar endocytosis 2, 7, the practicality of exploiting caveolar uptake and/or trafficking for future lysosomal storage therapies must be considered. Diminished caveolar uptake may pose an obstacle to adequate delivery, while altered trafficking toward an endo-lysosomal route may be desirable for therapeutic intervention in the lysosome.
Apart from clathrin- and caveolar-mediated pathways, EGF-induced macropinocytosis was also decreased in NPD cells. Although there is less information on this direction, several reports have suggested alteration in related functions. For instance, several pathogens, such as ebola virus and some intracellular bacteria, exploit macropinocytosis-like routes to invade cells 49–54, where ASM activity (affected in NPD) modulates such pathogen-host cell interactions 49, 52–54. Also, ASM activity products, such as ceramide or its derived metabolites, regulate dynamic reorganization of the actin cytoskeleton 55, which is a key contributor to micropinocytosis-related formation of ruffling and invagination structures involved in this route 56. Macropinocytosis also relates to formation of foamy macrophages in lipid metabolic alterations 57, both of which are observed to occur in NPD 26. Hence, it is plausible that ASM deficiency resulting in lipid storage in NPD results in altered macropinocytic uptake, as observed here.
Beyond the alterations found in specific uptake routes, there appears to be a generalized defect in fluid-phase pinocytic activity, illustrated by lower uptake of dextran in NPD vs. wild type cells. This suggests that the endocytic defect may stem from a common cause, e.g., the aberrant lysosomal storage and/or the particular lipid accumulation. Indeed, we also observed diminished bulk uptake of dextran in wild type fibroblasts with imipramine-induced sphingomyelin storage (Figure 5). In turn, uptake was revived in NPD cells in proportion to the attenuation of storage by recombinant ASM (Figure 6). Similarly, colocalization of caveolin-1 and clathrin heavy chain with their ligands (CTB and transferrin, respectively), as well as ligand uptake via these routes were enhanced proportionally to lysosomal storage reduction (Figure 7 and 27). Recent work using neuronal ceroid lipofuscinosis models have also demonstrated endocytic dysfunctions, despite genetic and lysosomal storage profiles different from NPD 58, 59. Numerous factors may play a role, such as cytoskeletal abnormalities, impaired cytosolic diffusion of molecules, and/or altered lipid metabolism 19, 20, 22, 25, 48, 60. In particular, NPD-associated changes of the lipid composition (sphingomyelin and cholesterol 26) in membranes along the biosynthetic and endocytic routes could disrupt the biophysical properties of lipid raft domains and other regions, altering the association of integral and peripheral proteins that act as receptors, adaptors, signaling molecules, and others. This may cause disturbance in elements associated with vesicular fusion/fission, sorting, trafficking, etc. 21, 58, 61. In fact, endocytosis of recombinant ASM mediated by the mannose-6-phosphate receptor was decreased in cells from ASM knockout mice 23 and also lysosomal exocytosis was disrupted in LSDs, which has been linked to aberrant regulation of lysosomal ion channels by sphingomyelin 61.
Approaches to bypass altered routes or enhance endocytic signaling may help improve therapeutic efficacy. ICAM-1 is an attractive target due to its widespread expression and activation during inflammation (a hallmark of NPD and other LSDs 26, 62), because it undergoes uptake via a clathrin- and caveolae-independent pathway 28, 29, and because ICAM-1 has no endocytic ligands endogenous to the body, hence, there are no competitors for the induction and progress of this pathway. Multivalent binding of carriers displaying multiple copies of anti-ICAM to the cell surface may be more efficient in receptor clustering and inducing endocytic signals than classical non-multivalent binding strategies 63. Nevertheless, it is important to note that we observed endocytosis of ICAM-1-targeted carriers to also be decreased in ASM-deficient cells, since secreted ASM activity is required for ceramide enrichment and derived signaling events upon carrier binding on the cell surface 44. As said above, ASM activity is required for intracellular invasion by certain pathogens, including major class human rhinovirus which enter cells via ICAM-1 64. However, the mechanism by which the endocytic defect arises in the CAM pathway vs. clathrin or caveolar pathways likely differ. For instance, ASM activity has not been linked to clathrin pits; in fact, ceramide is displaced from these invaginations during uptake via the mannose-6-phosphate receptor 65. When ASM is coupled to carriers binding to ICAM-1 its activity is exogenously provided, ceramide enrichment at binding sites is rescued and so is CAM endocytosis 44. Since this mechanism does not apply to uptake of the mannose-6-phosphate receptor, this would explain why exogenous ASM cannot restore the clathrin or caveolar routes as efficiently as the CAM pathway.
Delivery of ASM via the CAM pathway vs. the classical route used by naked ASM resulted in enhanced degradation of lysosomal storage material 27 and recovery of the clathrin 27, caveolar, and macropinocytic routes (Figure 7). In fact, a rebound effect was observed, where uptake after treatment resulted in uptake values greater than wild type cells. Induction of endocytosis by multivalent anti-ICAM NCs leads to uptake of over three hundred NCs per cell (Supplemental Figure 5), with large cytoskeletal remodeling 44, 66, which may promote an endocytic phenotype. In fact, significant dextran uptake was observed in the presence of anti-ICAM NCs 67. This cytoskeletal remodeling activity returns to its basal state a few hours after uptake 66. Hence, this effect may be temporary, yet this requires further investigation.
Along with other works 23–25, 58–60, these findings indicate that endocytic dysfunction occurs rather widely in LSDs, although this will likely depend on the particular pathology, tissue, etc. Relevant implications derive from this premise with regard to LSD therapies. This applies to enzyme replacement, including administration of recombinant enzymes and viral vectors23, 27,18,68 that enter cells by receptor-mediated uptake, or other agents whose uptake may rely on pinocytosis. As an example, endocytosis of recombinant ASM via the mannose-6-receptor pathway, which is mediated via clathrin-coated pits, was decreased in NPD models 23. Combining such therapies with strategies to decrease lysosomal storage or restore endocytic trafficking by other means may be beneficial. It is possible that this lower uptake via altered routes may still render significant therapeutic outcomes in virtue of cumulative effects acquired as a result of such chronic treatments. Yet, alternative or combination strategies improving endocytic uptake may allow therapies to act faster and/or upon administration of lower doses, reducing cost. Also noteworthy, it has been postulated that lysosomal accumulation of undegraded materials in LSDs may offer protection against the potential effects of storing said materials elsewhere in the cell. If this is the case, improved endocytosis and lysosomal enzyme delivery, resulting in degradation of such materials, may provide a means to restore lysosomal trafficking from other cellular sites (e.g., the plasmalemma, the biosynthetic route, the autophagocytic pathway, etc.69), attenuating the storage in these places too. Hence, characterizing the activity of uptake pathways in healthy and diseased models shall inform the selection of appropriate therapeutic approaches for these diseases.
Supplementary Material
Supplementary Figure 1. Mechanism of endocytosis of cholera toxin B (CTB) by fibroblasts. Uptake of fluorescent CTB upon incubation for one hour at 37°C with wild type (Wt) or NPD fibroblasts was assessed as described in Figure 1. Incubations were conducted in the presence of 1 μg/mL filipin (to inhibit caveolar endocytosis), 50 μM MDC (to inhibit clathrin-mediated uptake), or a mixture of both inhibitors. The number of internalized vesicles containing CTB was quantified as in Figure 1 and expressed as a percentage of the uptake found in their respective controls (absence of inhibitors). Data are the mean ± SEM. *Comparison of inhibitor-treated cells vs. control condition; #comparison of MDC or filipin+MDC mix vs. filipin alone (p<0.05 by Student’s t-test).
Supplementary Figure 2. Kinetics of uptake of cholera toxin B (CTB) in NPD fibroblasts. A Kinetics of uptake of CTB in wild type (Wt) vs. NPD fibroblasts, examined as described in Figure 1, where the total number of CTB-positive vesicles internalized per cell is shown. Lines depict regression curves, where R2 was 0.99 for wild type and 0.97 for NPD. B CTB uptake kinetics as in A, where the number of internalized vesicles was normalized per cell area is shown, to account for the different size of cells. Lines depict regression curves, where R2 was 0.99 for wild type and 0.97 for NPD. A–B Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
Supplementary Figure 3. Kinetics of bulk pinocytosis in NPD fibroblasts. Kinetics of uptake of the fluid phase marker Texas Red dextran in wild type (Wt) vs. NPD fibroblasts, examined as described in Figure 3. The number of internalized dextran-positive vesicles per cell area is shown. Lines depict regression curves, where R2 was 0.97 for wild type and 0.95 for NPD. Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
Supplementary Figure 4. Kinetics of macropinocytosis in NPD fibroblasts. The kinetics of EGF-induced uptake of dextran in Wt vs. NPD cells was assessed at the indicated times, as described in Figure 4, yet data was expressed per cell area, to account for the different sizes of cells. Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test). Lines are regression curves, where R2 = 0.98 for WT and 0.99 for NPD.
Supplementary Figure 5. Specificity of targeting and uptake of anti-ICAM NCs. A Wild type fibroblasts were pre-treated with TNFα to mimic inflammation-like conditions typical in NPD, followed by incubation for one hour at 37°C with fluorescent anti-ICAM NCs vs. control IgG NCs. Surface bound vs. internalized NCs were distinguished using a immunolabeling technique (see Methods) that renders surface-bound NCs marked in yellow (red + green; arrowheads) vs. internalized NCs marked in green (arrows). Dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. B The total number of NCs associated per cell and the percent internalization was calculated from A. Data are the mean ± SEM. *Comparison with anti-ICAM NCs (p<0.05 by Student’s t-test).
Supplementary Figure 6. Normalization of caveolin-1 distribution in NPD fibroblasts treated with anti-ICAM/ASM NCs. A NPD fibroblasts were incubated for one hour at 37°C with fluorescent CTB (pseudocolored green) three hours after treatment with 32 nM ASM coated on anti-ICAM NCs and compared to untreated NPD cells similarly incubated with CTB. Caveolin-1 was immunostained and its distribution and colocalization with CTB were imaged as in Fig. 2. Black/white panels illustrate the distribution of caveolin-1, while color panels show the colocalization (yellow) of caveolin-1 (red) and CTB (green) in the same cells. Ovals denote the nucleus and dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm.
Supplementary Figure 7. Normalization of macropinocytosis in NPD fibroblasts treated with anti-ICAM/ASM NCs. A NPD fibroblasts were incubated for one hour at 37°C with media containing 100 ng/mL EGF (to stimulate macropinocytosis) and fluorescent dextran three hours after treatment with 32 nM ASM coated on anti-ICAM NCs. These were compared to untreated NPD cells similarly incubated with EGF and dextran. Dotted lines mark the cell borders, as determined from the corresponding phase-contrast images. Scale bar = 10 μm.
Acknowledgments
The authors thank Dr. Edward Schuchmann (Mount Sinai School of Medicine, New York, NY) for providing recombinant human ASM and NPD fibroblasts. This study was funded by NIH and NSF grants R01-HL098416 and CBET-1402756, respectively, to S. Muro, and by a Howard Hughes Medical Institute fellowship to J. Rappaport under the University of Maryland Undergraduate Science Education Program.
Abbreviations
- ASM
acid sphingomyelinase
- CTB
cholera toxin B subunit
- EGF
epidermal growth factor
- ICAM-1
intercellular adhesion molecule-1
- NCs
nanocarriers
- NPD
type A Niemann-Pick disease
Footnotes
Supporting Information Available
This information is available free of charge via the Internet at http://pubs.acs.org/
References
- 1.Duncan R, Richardson SC. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol Pharm. 2012;9(9):2380–402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 2.Muro S. Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release. 2012;164(2):125–37. doi: 10.1016/j.jconrel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 4.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 5.Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–96. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12(8):517–33. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 7.Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–58. doi: 10.1016/j.addr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stan RV. Structure and function of endothelial caveolae. Microsc Res Tech. 2002;57(5):350–64. doi: 10.1002/jemt.10089. [DOI] [PubMed] [Google Scholar]
- 9.Nichols B. Caveosomes and endocytosis of lipid rafts. J Cell Sci. 2003;116(Pt 23):4707–14. doi: 10.1242/jcs.00840. [DOI] [PubMed] [Google Scholar]
- 10.Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3(5):311–20. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 11.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3(5):473–83. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 12.McIntosh DP, Tan XY, Oh P, Schnitzer JE. Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery. Proc Natl Acad Sci U S A. 2002;99(4):1996–2001. doi: 10.1073/pnas.251662398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Q, Huntsman C, Ma D. Clathrin-independent internalization and recycling. J Cell Mol Med. 2008;12(1):126–44. doi: 10.1111/j.1582-4934.2007.00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5(7):554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 15.Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3(12):954–66. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 16.Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121(1):1–22. doi: 10.1007/s00439-006-0280-4. [DOI] [PubMed] [Google Scholar]
- 17.Muro S. Strategies for delivery of therapeutics into the central nervous system for treatment of lysosomal storage disorders. Drug Deliv and Trans Res. 2012;2:169–86. doi: 10.1007/s13346-012-0072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2(2):189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59(4):700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 20.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793(4):684–96. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, Ballabio A. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. Embo j. 2010;29(21):3607–20. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Simons K, Gruenberg J. Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol. 2000;10(11):459–62. doi: 10.1016/s0962-8924(00)01847-x. [DOI] [PubMed] [Google Scholar]
- 23.Dhami R, Schuchman EH. Mannose 6-phosphate receptor-mediated uptake is defective in acid sphingomyelinase-deficient macrophages: implications for Niemann-Pick disease enzyme replacement therapy. J Biol Chem. 2004;279(2):1526–32. doi: 10.1074/jbc.M309465200. [DOI] [PubMed] [Google Scholar]
- 24.Marks DL, Pagano RE. Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol. 2002;12(12):605–13. doi: 10.1016/s0962-8924(02)02399-1. [DOI] [PubMed] [Google Scholar]
- 25.Puri V, Watanabe R, Singh RD, Dominguez M, Brown JC, Wheatley CL, Marks DL, Pagano RE. Clathrin-dependent and -independent internalization of plasma membrane sphingolipids initiates two Golgi targeting pathways. J Cell Biol. 2001;154(3):535–47. doi: 10.1083/jcb.200102084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuchman E, Desnick R. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. McGraw-Hill; New York: 2000. pp. 3589–610. [Google Scholar]
- 27.Rappaport J, Garnacho C, Muro S. Clathrin-mediated endocytosis is impaired in type A–B Niemann-Pick disease model cells and can be restored by ICAM-1-mediated enzyme replacement. Mol Pharm. 2014;11(8):2887–95. doi: 10.1021/mp500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval M. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116(Pt 8):1599–609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 29.Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther. 2006;13(1):135–41. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 30.Calderon AJ, Muzykantov V, Muro S, Eckmann DM. Flow dynamics, binding and detachment of spherical carriers targeted to ICAM-1 on endothelial cells. Biorheology. 2009;46(4):323–41. doi: 10.3233/BIR-2009-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhowmick T, Berk E, Cui X, Muzykantov VR, Muro S. Effect of flow on endothelial endocytosis of nanocarriers targeted to ICAM-1. J Control Release. 2012;157(3):485–92. doi: 10.1016/j.jconrel.2011.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He X, Miranda SR, Xiong X, Dagan A, Gatt S, Schuchman EH. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim Biophys Acta. 1999;1432(2):251–64. doi: 10.1016/s0167-4838(99)00069-2. [DOI] [PubMed] [Google Scholar]
- 33.Orlandi PA, Fishman PH. Filipin-dependent Inhibition of Cholera Toxin: Evidence for Toxin Internalization and Activation through Caveolae-like Domains. J Cell Biol. 1998;141(4):905–15. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlegel R, Dickson RB, Willingham MC, Pastan IH. Amantadine and dansylcadaverine inhibit vesicular stomatitis virus uptake and receptor-mediated endocytosis of alpha 2-macroglobulin. Proc Natl Acad Sci U S A. 1982;79(7):2291–5. doi: 10.1073/pnas.79.7.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muro S, Muzykantov VR, Murciano JC. Characterization of endothelial internalization and targeting of antibody–enzyme conjugates in cell cultures and in laboratory animals. In: Niemeyer CM, editor. Bioconjugation protocols : strategies and methods. Humana Press; Totowa, N.J: 2004. [DOI] [PubMed] [Google Scholar]
- 36.Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. 1994;375(7):447–50. doi: 10.1515/bchm3.1994.375.7.447. [DOI] [PubMed] [Google Scholar]
- 37.Hsu J, Northrup L, Bhowmick T, Muro S. Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine. 2012;8(5):731–9. doi: 10.1016/j.nano.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, Garnacho C, Muro S. Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149(3):323–31. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, Muzykantov VR. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther. 2006;317(3):1161–9. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 40.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, Muzykantov V, Muro S. Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther. 2008;325(2):400–8. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 41.Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1745(3):273–86. doi: 10.1016/j.bbamcr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Torgersen ML, Skretting G, van Deurs B, Sandvig K. Internalization of cholera toxin by different endocytic mechanisms. J Cell Sci. 2001;114(Pt 20):3737–47. doi: 10.1242/jcs.114.20.3737. [DOI] [PubMed] [Google Scholar]
- 43.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 44.Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. ICAM-1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler Thromb Vasc Biol. 2012;32(5):1178–85. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16(1):77–84. [PubMed] [Google Scholar]
- 46.Frank PG, Cheung MW, Pavlides S, Llaverias G, Park DS, Lisanti MP. Caveolin-1 and regulation of cellular cholesterol homeostasis. Am J Physiol Heart Circ Physiol. 2006;291(2):H677–86. doi: 10.1152/ajpheart.01092.2005. [DOI] [PubMed] [Google Scholar]
- 47.Damm EM, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol. 2005;168(3):477–88. doi: 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hortsch R, Lee E, Erathodiyil N, Hebbar S, Steinert S, Lee JY, Chua DS, Kraut R. Glycolipid trafficking in Drosophila undergoes pathway switching in response to aberrant cholesterol levels. Mol Biol Cell. 2010;21(5):778–90. doi: 10.1091/mbc.E09-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med. 2011;208(5):909–21. doi: 10.1084/jem.20102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moulder JW. Comparative biology of intracellular parasitism. Microbiol Rev. 1985;49(3):298–337. doi: 10.1128/mr.49.3.298-337.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falkow S, Isberg RR, Portnoy DA. The interaction of bacteria with mammalian cells. Annu Rev Cell Biol. 1992;8:333–63. doi: 10.1146/annurev.cb.08.110192.002001. [DOI] [PubMed] [Google Scholar]
- 52.Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. 2012;86(14):7473–83. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grassme H, Becker KA, Zhang Y, Gulbins E. Ceramide in bacterial infections and cystic fibrosis. Biol Chem. 2008;389(11):1371–9. doi: 10.1515/BC.2008.162. [DOI] [PubMed] [Google Scholar]
- 54.Grassme H, Riethmuller J, Gulbins E. Biological aspects of ceramide-enriched membrane domains. Prog Lipid Res. 2007;46(3–4):161–70. doi: 10.1016/j.plipres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 55.Zeidan YH, Jenkins RW, Hannun YA. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J Cell Biol. 2008;181(2):335–50. doi: 10.1083/jcb.200705060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mooren OL, Galletta BJ, Cooper JA. Roles for actin assembly in endocytosis. Annu Rev Biochem. 2012;81:661–86. doi: 10.1146/annurev-biochem-060910-094416. [DOI] [PubMed] [Google Scholar]
- 57.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280(3):2352–60. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 58.Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci. 2013;33(46):18065–79. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aby E, Gumps K, Roth A, Sigmon S, Jenkins SE, Kim JJ, Kramer NJ, Parfitt KD, Korey CA. Mutations in palmitoyl-protein thioesterase 1 alter exocytosis and endocytosis at synapses in Drosophila larvae. Fly (Austin) 2013;7(4):267–79. doi: 10.4161/fly.26630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liscum L, Faust JR. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J Biol Chem. 1987;262(35):17002–8. [PubMed] [Google Scholar]
- 61.Samie MA, Xu H. Lysosomal exocytosis and lipid storage disorders. J Lipid Res. 2014;55(6):995–1009. doi: 10.1194/jlr.R046896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muro S. Intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. In: Aird W, editor. Endothelial Biomedicine. Cambridge University Press; New York: 2007. pp. 1058–70. [Google Scholar]
- 63.Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem. 1992;61:307–30. doi: 10.1146/annurev.bi.61.070192.001515. [DOI] [PubMed] [Google Scholar]
- 64.Grassme H, Riehle A, Wilker B, Gulbins E. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J Biol Chem. 2005;280(28):26256–62. doi: 10.1074/jbc.M500835200. [DOI] [PubMed] [Google Scholar]
- 65.Shakor AB, Atia MM, Kwiatkowska K, Sobota A. Cell surface ceramide controls translocation of transferrin receptor to clathrin-coated pits. Cell Signal. 2012;24(3):677–84. doi: 10.1016/j.cellsig.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 66.Muro S, Mateescu M, Gajewski C, Robinson M, Muzykantov VR, Koval M. Control of intracellular trafficking of ICAM-1-targeted nanocarriers by endothelial Na+/H+ exchanger proteins. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L809–17. doi: 10.1152/ajplung.00311.2005. [DOI] [PubMed] [Google Scholar]
- 67.Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105(2):650–8. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 68.Kirkegaard T. Emerging therapies and therapeutic concepts for lysosomal storage diseases. Expert Opinion on Orphan Drugs. 2013;1(5):385–404. [Google Scholar]
- 69.Vidal-Donet JM, Cárcel-Trullols J, Casanova B, Aguado C, Knecht E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8(2):e55526. doi: 10.1371/journal.pone.0055526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Mechanism of endocytosis of cholera toxin B (CTB) by fibroblasts. Uptake of fluorescent CTB upon incubation for one hour at 37°C with wild type (Wt) or NPD fibroblasts was assessed as described in Figure 1. Incubations were conducted in the presence of 1 μg/mL filipin (to inhibit caveolar endocytosis), 50 μM MDC (to inhibit clathrin-mediated uptake), or a mixture of both inhibitors. The number of internalized vesicles containing CTB was quantified as in Figure 1 and expressed as a percentage of the uptake found in their respective controls (absence of inhibitors). Data are the mean ± SEM. *Comparison of inhibitor-treated cells vs. control condition; #comparison of MDC or filipin+MDC mix vs. filipin alone (p<0.05 by Student’s t-test).
Supplementary Figure 2. Kinetics of uptake of cholera toxin B (CTB) in NPD fibroblasts. A Kinetics of uptake of CTB in wild type (Wt) vs. NPD fibroblasts, examined as described in Figure 1, where the total number of CTB-positive vesicles internalized per cell is shown. Lines depict regression curves, where R2 was 0.99 for wild type and 0.97 for NPD. B CTB uptake kinetics as in A, where the number of internalized vesicles was normalized per cell area is shown, to account for the different size of cells. Lines depict regression curves, where R2 was 0.99 for wild type and 0.97 for NPD. A–B Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
Supplementary Figure 3. Kinetics of bulk pinocytosis in NPD fibroblasts. Kinetics of uptake of the fluid phase marker Texas Red dextran in wild type (Wt) vs. NPD fibroblasts, examined as described in Figure 3. The number of internalized dextran-positive vesicles per cell area is shown. Lines depict regression curves, where R2 was 0.97 for wild type and 0.95 for NPD. Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test).
Supplementary Figure 4. Kinetics of macropinocytosis in NPD fibroblasts. The kinetics of EGF-induced uptake of dextran in Wt vs. NPD cells was assessed at the indicated times, as described in Figure 4, yet data was expressed per cell area, to account for the different sizes of cells. Data are the mean ± SEM. *Comparison with Wt cells (p<0.05 by Student’s t-test). Lines are regression curves, where R2 = 0.98 for WT and 0.99 for NPD.
Supplementary Figure 5. Specificity of targeting and uptake of anti-ICAM NCs. A Wild type fibroblasts were pre-treated with TNFα to mimic inflammation-like conditions typical in NPD, followed by incubation for one hour at 37°C with fluorescent anti-ICAM NCs vs. control IgG NCs. Surface bound vs. internalized NCs were distinguished using a immunolabeling technique (see Methods) that renders surface-bound NCs marked in yellow (red + green; arrowheads) vs. internalized NCs marked in green (arrows). Dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm. B The total number of NCs associated per cell and the percent internalization was calculated from A. Data are the mean ± SEM. *Comparison with anti-ICAM NCs (p<0.05 by Student’s t-test).
Supplementary Figure 6. Normalization of caveolin-1 distribution in NPD fibroblasts treated with anti-ICAM/ASM NCs. A NPD fibroblasts were incubated for one hour at 37°C with fluorescent CTB (pseudocolored green) three hours after treatment with 32 nM ASM coated on anti-ICAM NCs and compared to untreated NPD cells similarly incubated with CTB. Caveolin-1 was immunostained and its distribution and colocalization with CTB were imaged as in Fig. 2. Black/white panels illustrate the distribution of caveolin-1, while color panels show the colocalization (yellow) of caveolin-1 (red) and CTB (green) in the same cells. Ovals denote the nucleus and dotted lines mark the cell borders, as observed by phase-contrast microscopy. Scale bar = 10 μm.
Supplementary Figure 7. Normalization of macropinocytosis in NPD fibroblasts treated with anti-ICAM/ASM NCs. A NPD fibroblasts were incubated for one hour at 37°C with media containing 100 ng/mL EGF (to stimulate macropinocytosis) and fluorescent dextran three hours after treatment with 32 nM ASM coated on anti-ICAM NCs. These were compared to untreated NPD cells similarly incubated with EGF and dextran. Dotted lines mark the cell borders, as determined from the corresponding phase-contrast images. Scale bar = 10 μm.