

Abstract
MiR-34a, an important tumor-suppressing microRNA, is downregulated in several types of cancer; loss of its expression has been linked with unfavorable clinical outcomes in non-small-cell lung cancer (NSCLC), among others. MiR-34a represses several key oncogenic proteins, and a synthetic mimic of miR-34a is currently being tested in a cancer trial. However, little is known about the potential role of miR-34a in regulating DNA damage response and repair. Here, we demonstrate that miR-34a directly binds to the 3' untranslated region of RAD51 and regulates homologous recombination, inhibiting double-strand-break repair in NSCLC cells. We further demonstrate the therapeutic potential of miR-34a delivery in combination with radiotherapy in mouse models of lung cancer. Collectively, our results suggest that administration of miR-34a in combination with radiotherapy may represent a novel strategy for treating NSCLC.
Keywords: miR-34a, non-small-cell lung cancer, RAD51, radiation
Introduction
The cellular DNA damage response (DDR) maintains genomic integrity by modulating various signaling pathways, including those controlling cell cycling, replication bypass mechanisms, and DNA repair.1 Loss of the normal DDR is common in cancer, and defects in DNA repair are exploited by using DNA-damaging agents such as radiation for anticancer therapy. Radiation induces DNA double-strand breaks (DSBs) that can be efficiently repaired by homologous recombination (HR), which is regulated mainly by RAD51,2,3 among other mechanisms such as non-homologous end joining. HR commonly occurs between the S and G2 phases of the cell cycle.4 RAD51 is a key player in HR, catalyzing the transfer of the new DNA strand between a broken sequence and its homolog to resynthesize the damaged region.5 In response to ionizing radiation, RAD51 relocalizes within the nucleus to form distinct foci that depend on BRCA2 and the RAD51 paralogues.2,3 This highly orchestrated complex of proteins increases the DNA repair capacity via HR, which leads to resistance to radiotherapy.6,7,8,9 HR is also regulated by other proteins, including p5310 and MYC.11 p53 has been shown to directly interact with RAD51 to regulate the extent and timing of HR.12 Interestingly, MYC knockdown inhibits RAD51 expression, promoting cell death after treatment with radiation.11 Recently, several microRNAs (miR-155, miR-193b* and miR-148b* were shown to regulate HR via RAD51 downregulation.9,13 Another miRNA known to regulate DDR is the p53-regulated miRNA miR-34a.14,15 miR-34a is one of the most important tumor-suppressing miRNAs in cancer. miR-34a is downregulated in several types of tumors, and loss of its expression has been linked with unfavorable clinical outcomes in several types of cancers, including non-small-cell lung cancer (NSCLC).16,17 Based on this understanding, miR-34a replacement therapy via the liposomal formulation MRX34 (refs. 18,19) is currently being studied in a phase 1 clinical cancer trial. miR-34a has shown to regulate aging and sensitivity to radiotherapy in different models.15,20,21,22,23 However, the exact mechanism by which miR-34a regulates DNA repair is largely unknown. In this study, we showed that miR-34a regulates RAD51 by directly binding to its 3' untranslated region (UTR), controlling HR and promoting radiosensitivity in NSCLC cells. We further demonstrated that MRX34 plus radiotherapy effectively inhibits tumor growth in lung cancer mouse models, findings that highlight the potential of this approach as a viable clinical strategy to combat NSCLC.
Results
MiR-34a overexpression suppressed DNA repair after irradiation
Previous studies have shown that miR-34a modulates radiosensitivity in vitro.20,21,22,23,24 Here, we investigate the effect of miR-34a overexpression on DNA repair. We confirmed the effect of miR-34a on radioresistance in NSCLC cell lines in vitro as follows. Firstly, we assessed the endogenous expression of miR-34a in 20 NSCLC cell lines in order to select best suitable models for our in vitro studies (Supplementary Figure S1). We selected NSCLC cell lines with low expression of miR-34a (A549, H1299, H460, and Calu6) and one NSCLC with higher expression of miR-34a (H1944). We transiently transfected A549 and H1299 cells with miR-34a or scrambled control miRNA mimics and analyzed clonogenic survival. Cells were plated 48 hours after transfection and treated with 2, 4, or 6 Gy of radiation. A549 and H1299 cells transfected with miR-34a were significantly more sensitive to the cytotoxic effects of radiotherapy than were cells transfected with a scrambled control (Figure 1a,b). The sensitizing enhancement ratios (SER) for miR-34a were 1.6 in A549 cells and 1.5 in H1299 cells. Next, we investigated the effect of miR-34a on radiation-induced DNA DSBs and the kinetics of DNA repair. We detected a significantly higher number of γ-H2AX foci (indicative of DSBs) in A549 and H1299 cells overexpressing miR-34a 1 and 8 hours after exposure to 4 Gy of radiation than in cells with scrambled control (Figure 1c–f).
Figure 1.

MiR-34a overexpression suppressed DNA repair after irradiation. (a,b) A549 and H1299 transfected with miR-34a were more sensitive to the cytotoxic effects of radiation (2, 4, or 6 Gy) than were cells transfected with a scrambled control. (c–f) Histone H2AX phosphorylation on serine-139 (γ-H2AX) was upregulated in A549 and H1299 cells overexpressing miR-34a at 1 and 8 hours after exposure to 4 Gy. Representative images of γ-H2AX detection after irradiation of 4 Gy are shown. *P < 0.05 in a Student's t-test.
MiR-34a regulates several DNA repair and related proteins, including RAD51
We next investigated the mechanisms by which miR-34a modulates DNA damage and repair pathways in lung cancer cells. To do this, we transiently overexpressed miR-34a in A549 and H1299 cells and analyzed the expression of DNA damage and repair genes by using a custom 384-well panel quantitative polymerase chain reaction (qPCR) plate (58 genes). We found that miR-34a overexpression promoted the downregulation of RAD51, UBC, BARD1, UBA52, ATF1, CCNA2, USP1, FANCD2 USP1, ATF1, BARD1 mRNA and the upregulation of p53 (in A549 cells), RANBP2, MRE11A, RELA, RELB, NFKBIB, POLR2D, AKT1, NFKB1 and CCND1 mRNA (Figure 2a). In a next step, we investigated whether miR-34-induced gene expression could also be observed in 532 patients with NSCLC from The Cancer Genome Atlas data set. We confirmed that miR-34a expression was inversely correlated with RAD51, USP1, ATF1, BARD1 and CCNA2 genes (Figure 2b and Supplementary Figure S2). We next searched for binding sites in the 3' UTRs of the genes downregulated by miR-34a identified in Figure 2a. Our search revealed that the 3' UTRs of RAD51 contain binding sites specific for miR-34a. The miR-34-mediated regulation of RAD51 was further confirmed by western blot analysis, which showed downregulation of this gene in A549, H1299, H460, and Calu6 cells, after miR-34a enforced overexpression (Figure 2c,d), and upregulation of RAD51 in H1944 cells after depletion of miR-34a (Figure 2e,f).
Figure 2.

MiR-34a regulates several DNA repair and related proteins, including RAD51, in lung cancer cells. (a) Selected DNA Damage and Repair genes from a custom 384-well panel qPCR plate in A549 and H1299 cells transfected with miR-34a. (b) Correlation of miR-34a and RAD51 expression in samples from 532 patients with NSCLC from the Cancer Genome Atlas data set (LUAD). (c,d) miR-34a overexpression in A549, H1299, H460 and Calu6 downregulates RAD51 protein expression. NSCLC cell lines cell lysates were collected and subjected to SDS–polyacrylamide gel electrophoresis for western blotting. Actin was used as a loading control. (e,f) Inhibition of miR-34a with anti-miR-34a in H1944 cells upregulated the expression of RAD51 in those cells compared with scrambled control. (g) 3' untranslated region (UTR) of RAD51 with binding sites specific for miR-34a and deletion of 5 base pairs (red) in the mutant vector. (h) Luciferase activity was reduced in cells transfected with miR-34a and RAD51 3' UTR luciferase constructs compared with scrambled controls. Mutation of the predicted miR-34a binding site rescued the luciferase activity, thus confirming that miR-34a directly interacts with the RAD51 3′ UTRs. *P < 0.05 in a Student's t-test.
RAD51 is a direct target of miR-34a
To determine whether miR-34a interacts directly with the putative target gene RAD51, we cotransfected H1299 cells with miR-34a mimics and a reporter vector encoding the luciferase gene that was fused to the 3' UTR of RAD51. Luciferase activity was reduced in cells transfected with miR-34a and the RAD51 3' UTR luciferase construct compared with the scrambled control. Mutation of the miR-34a interaction site rescued the luciferase activity, thus confirming that miR-34a directly interacts with the RAD51 3́ UTR (Figure 2g,h).
MiR-34a is a negative regulator of HR and RAD51 foci formation
Next, we tested the effect of miR-34a overexpression on HR in HeLa-DR-13–9 cells, a model for studying HR in vitro.25 HeLa-DR-13–9 cells were transfected twice on different days with scrambled and miR-34a mimics. On day 6 after transfection, cells were collected and analyzed by flow cytometry. We found that miR-34a overexpression significantly reduced the HR process by 59% (Figure 3a). Accordingly, a fluorescent immunoassay revealed a reduction in radiation-induced RAD51 foci formation in A549 cells transfected with miR-34a compared with controls (Figure 3b). As previously shown in Figure 1, overexpression of miR-34a sensitized the cells to radiation, as measured by clonogenic survival assays. We confirmed that reintroduction of the RAD51 ORF could rescue this phenotype (Figure 3c).
Figure 3.

miR-34a inhibits HR and RAD51 foci formation. (a) Homologous recombination assay in HeLa-DR-13–9 cells overexpressing miR-34a. HeLa-DR-13–9 cells were transfected twice on different days with scrambled sequences and miR-34a mimics. On day 6 after transfection, cells were collected and analyzed by flow cytometry. (b) Inhibition of RAD51 foci in A540 cells overexpressing miR-34a 8 hours after irradiation of 4 Gy are shown. (c) Reintroduction of the RAD51 ORF rescued the radiosensitization caused by miR-34a overexpression as measured by clonogenic survival assays. Cells were transiently transfected with miR-34a mimic or scrambled (±ORF). Cell viability was assessed using the clonogenic survival assay in the presence of radiation (2, 4, or 6 Gy). *P < 0.05 in a Student's t-test.
MiR-34a therapeutic delivery sensitizes lung tumors to radiotherapy
Our findings thus far suggest that miR-34 augments γ-H2AX foci and inhibits HR and RAD51 foci formation and can enhance the effects of radiation. To explore whether this phenotype can be exploited therapeutically, we examined the effect of miR-34a on radiosensitivity in two mouse models of human lung cancer. To do so, we administered MRX34, a liposomal nanoparticle loaded with miR-34a mimics,18 in combination with fractionated irradiation to xenograft tumors in mice. Intramuscular tumors were created by inoculating 1 × 106 H460 or H1299 cells into the right leg of each mouse. When the tumors reached 8 mm in diameter, the mice were randomly assigned to one of four groups: control; MRX34 only; radiation; and MRX34 plus radiation. MRX34 was given as subcutaneous peritumoral injections at a dose of 1 mg/kg (H460) or 5 mg/kg (H1299), and local irradiation was given as five 4-Gy fractions over 5 days (total dose 20 Gy) starting when tumors were 8 mm. For the combination-therapy group, MRX34 was given 1 hour before the radiation. In agreement with our previous in vitro observations, in vivo tumor growth delay assays revealed that miR-34a significantly sensitized lung tumors to radiation (Figure 4a,b; Tables 1 and 2). Tumors collected 24 hours after the last MRX34 dose exhibited approximately 20–30 times higher miR-34a levels than did controls (Figure 4c,d). Interestingly, mRNA and protein levels of RAD51 were markedly reduced in H460 and H1299 tumors treated with MRX34 compared with controls (Figure 4h). In summary, these results indicate that the therapeutic delivery of miR-34a led to repression of RAD51 in lung tumors and consequently enhanced the effects of radiation on tumor growth inhibition.
Figure 4.

Therapeutic delivery of miR-34a sensitizes lung tumors to radiotherapy in mice. (a,b) Effect of MRX34 on radioresponse of implanted H460 (a) and H1299 (b) cells measured by tumor growth delay shows that miR-34a significantly sensitized lung tumor to radiation. (c,d) Accumulation of miR-34a mimics in tumor after subcutaneous injection. At 24 hours after the third dose of MRX34, two mice per group were killed, and tissues were harvested for total RNA extraction and subsequent evaluation of miR-34a levels. Increased miR-34a levels were detected in tumors from the mice given MRX34 or MRX34 plus radiation. (e,f) RAD51 mRNA expression levels in mice treated with MRX34 compared with control. (g) Western blots of RAD51 protein levels in H460 xenografts from two mice treated with MRX34. (h) Immunohistochemical stains of RAD51 in H1299 xenografts from mice treated with MRX34.
Table 1. Effect of miR-34a on radioresponse of H1299 cells measured by tumor growth delay.
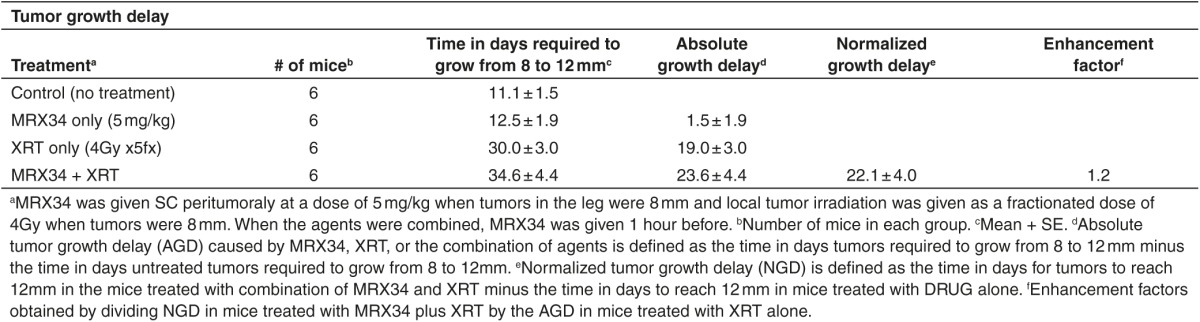
Table 2. Effect of miR-34a on radioresponse of H460 cells measured by tumor growth delay.

Discussion
In this study, we assessed the role of miR-34a in DNA damage and repair pathways and the potential application of therapeutic delivery of miR-34a-loaded liposomes (MRX34), currently being tested in a phase 1 clinical trial, in combination with radiotherapy in mouse lung cancer models.
Previous studies have shown that miR-34a overexpression sensitized NSCLC cells to radiation.21,23 We confirmed that miR-34a modulates radiosensitivity in NSCLC cell lines by using clonogenic assays. Although one study demonstrated that ectopic expression of miR-34a enhances radiosensitivity and promote apoptosis by suppressing the LyGDI pathway in NSCLC cells,20 and others showed that miR-34a regulates DDR pathways, little is known regarding its role in DNA repair signaling. Therefore, we aimed to investigate the effect of miR-34a overexpression on DNA repair in NSCLC cells. To do this, we tested if miR-34a modulates radiation-induced DSBs. In agreement with findings from our clonogenic assays, we found that the number of γ-H2AX foci in NSCLC cells overexpressing miR-34a was significantly higher than that in cells with scrambled control. These results indicate that miR-34a overexpression suppressed DNA repair after irradiation. We then investigated the potential mechanisms by which miR-34a regulates DNA repair. We profiled 59 genes from the DDR and repair pathways after enforced overexpression of miR-34a in NSCLC cell lines. We were able to confirm previously identified miR-34a targets and to discover new genes that may be regulated by miR-34a. We found that miR-34a overexpression downregulated several DNA damage and repair genes, including RAD51. We validated these results by analyzing patients with NSCLC from the The Cancer Genome Atlas data set. Accordingly, we found that miR-34a expression was inversely correlated with RAD51 and that miR-34a downregulated RAD51 protein expression in NSCLC cells via western blotting. We next identified RAD51 as a direct target of miR-34a. Our findings suggest that miR-34a modulates radiosensitivity via direct regulation of RAD51, among other mechanisms suggested by previous studies.15,20,21,22,23 In support of our observations, others have shown that overexpression of RAD51, which has a key role in HR, promotes resistance to radiation.26,27
We further examined the effect of miR-34a delivery, in combination with radiotherapy, on xenografted H460 and H1299 cell tumors in mouse models of lung cancer. We tested the liposomal nanoparticle MRX34,18 which contains a synthetic double-stranded mimic of the tumor suppressor miRNA miR-34a and is being tested in a phase 1 clinical trial. In both models, miR-34a delivery via MRX34 treatment promoted significant tumor growth delay compared with control. These findings suggest that delivery of miR-34a, in combination with radiotherapy, has potential as a therapeutic approach. In accordance with our previous in vitro findings, RAD51 mRNA and protein levels were downregulated in the mice treated with MRX34 compared with control. These findings suggest that miR-34a can overcome resistance to radiation by regulating RAD51 in lung cancer models. Because cancer cells with deficiencies in HR are sensitive to radiation, inhibition of HR via RAD51 represents a potential target for combined therapies.
In conclusion, we identified several DDR and repair proteins regulated by miR-34a, including RAD51. We demonstrated that miR-34a directly binds to RAD51 3' UTR and regulates HR, inhibiting DBSs repair in NSCLC cells. We further demonstrated the therapeutic potential of miR-34a delivery via MRX34 treatment in combination with RT in lung cancer in vivo models. Collectively, our results suggest that miR-34a, in combination with radiotherapy, may represent a novel approach for the treatment of NSCLC.
Materials and methods
Cell lines and irradiation. The established lung cancer cell lines A549, H460, H1299, Calu6, and H1944 were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum at 37 °C in a humidified 5% CO2 incubator. Cells were irradiated at room temperature with a Mark I 137Cs irradiator (JL Shepherd & Associates, San Fernando, CA) at a dose rate of 3.5 Gy/minute.
PCR array analysis of genes involved in DNA damage and repair-induced responses. Total RNA was isolated from A549 and H1299 cells transfected with miR-34a or scrambled control with Triazol (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol. RNA quality and quantity were assessed with a Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA). Next, mRNA was reverse-transcribed by using a Superscript III kit (Life Technologies), diluted 1:5 with nuclease-free water, and loaded into a custom 384-well PCR plate with a panel of DNA Damage Induced Responses genes (Supplementary Table S2) (Biorad, Hercules, CA) according to the manufacturer's protocol. Reactions were run in a CFX96 Touch Real-Time PCR Detection System with SYBR Green (Life Technologies). Gene expression was analyzed with CFX Manager software (Biorad). The List of the 58 DNA damage and repair genes analyzed using a custom 384-well PCR array is described in Supplementary Table S1.
Clonogenic survival assay. NSCLC cells transfected with miR-34a or their respective controls for 48 hours were seeded in triplicate in 60-mm dishes and incubated overnight at 37 °C in a humidified chamber containing 5% CO2. The next day, the cells were irradiated using 0, 2, 4, or 6 Gray units (Gy), counted, seeded in 60-mm dishes, and incubated for 12 days to allow macroscopic colony formation. Cells were fixed and stained for 5 minutes with 0.5% crystal violet (Sigma-Aldrich, St. Louis, MO) in methanol. Colonies containing more than 50 viable cells formed in each treatment group were counted. Survival was calculated relative to that of unirradiated cells by using the formula survival = (plating efficiency of treated cells)/(plating efficiency of control cells) where plating efficiency = (number of colonies formed by treated cells)/(number of colonies formed by untreated cells).
Detection of γ-H2AX foci. For this assay, 200,000 cells were plated on coverslips, placed in 35-mm dishes, and allowed to attach overnight. Cells were then irradiated (4 Gy) 48 hours after transfection with miR-34a, incubated for 4–24 hours, and then fixed with 1% paraformaldehyde for 10 minutes, followed by a 10-minute wash in 70% ethanol at room temperature. Cells were then treated with 0.1% NP40 in phosphate-buffered saline (PBS) for 20 minutes, washed in PBS four times, and then blocked with 5% bovine serum albumin in PBS for 30 minutes. The cells were then incubated with anti-γ-H2AX and RAD51 antibody (Millipore, Billerica, MA) in 5% bovine serum albumin in PBS overnight. The next day, the cells were incubated with fluorescein-isothiocyanate–labeled secondary antibody at a dilution of 1:300 in 5% bovine serum albumin in PBS for 30 minutes. Then, cells were incubated in the dark with 4 4′,6-diamidino-2-phenylindole dihydrochloride (1 mg/ml) in PBS for 5 minutes, and coverslips were mounted on a slide with an antifade solution (Molecular Probes; Invitrogen, Waltham, MA). Slides were examined with a fluorescence microscope (Leica, Buffalo Grove, IL), and images were captured by a charge-coupled device camera and imported into the Advanced Spot Image analysis software package. For each treatment condition, the numbers of γ-H2AX foci were determined in at least 50 cells.
Identifying potential microRNA targets. Potential miR-34a targets were identified by using the target prediction databases miRNA body map (http://www.mirnabodymap.org/; Ghent University, Belgium)28 and miRwalk.29 These databases compare predicted targets from mirBase release 14, TargetScan 5.1, miRDB 3.0, MicroCosm v5, DIANA 3.0, TarBase v.5c, PITA catalog v6, RNA22 (August 2007), and miRecords v2.
Transfection. Pre-microRNAs miR-34a and anti-miR-34a and their respective negative controls (scrambled oligos) (Life Technologies) were reverse-transfected into A549, H460, H1299, Calu6, and H1944 with Lipofectamine 2000 (Life Technologies) at a final concentration of 100 nmol/l.
Homologous recombination assay. On day 1, HeLa-DR cells (4.0 × 104 in a 1.5-cm well) were transfected with 50 pmol oligos (control precursor/pre-miR-34a) in the presence of 0.5 μl of Lipofectamine 2000 (Life Technologies). On day 2, the transfected cells were transferred to 3.5-cm well dishes. On day 3, the cells were transfected with 50 pmol oligos (control precursor/pre-miR-34a) plus 3 μg of I-SceI expression plasmid (pCBASceI) in the presence of 2.5 μl of Lipofectamine 2000. On day 6, cells were trypsinized and 10,000 cells from each well were counted by flow cytometry using a Becton Dickinson FACSCalibur instrument (Franklin Lakes, NJ). The percentage values of cells with recombined loci encoding green fluorescence protein are the averages of three independent experiments. Values are shown as mean ± SD.
Quantitative polymerase chain reaction. Total RNA was isolated from cells with Triazol (Life Technologies) for miRNA analysis according to the manufacturer's protocol. To analyze expression of mature microRNA, total RNA was reverse-transcribed with miRNA-specific primers using a TaqMan MicroRNA Reverse Transcription kit (Life Technologies), followed by qPCR with Taqman MicroRNA assays according to the manufacturer's protocol. For studies of RAD51 expression, mRNA was reverse-transcribed using a Superscript III kit (Life Technologies) and analyzed by qPCR using SYBR Green (Life Technologies) with specific primers (Supplementary Table S2) according to the manufacturer's protocol. The comparative Ct method was used to calculate the relative abundance of miRNA and mRNAs compared with U6 and ACTB expression, respectively.
Protein extraction and western blot analysis. Total protein was extracted and resolved on denaturing gels as previously described.30 Membranes were probed with the following antibodies: primary antibodies, anti-RAD51, anti-GAPDH (Cell Signaling Technologies, Beverly, MA), anti-actin, anti-vinculin (Sigma, St. Louis, MO), and secondary antibody labeled by horseradish peroxidase (Amersham GE Healthcare, Cleveland, OH). The secondary antibody was visualized by using a chemiluminescent reagent Pierce ECL kit (Thermo Fisher Scientific, Waltham, MA).
Luciferase assay. H1299 cells were plated in 96-well dishes at 4 × 104 cells/well. Cells were transfected with miR-34a or scrambled constructs (100 nmol/l) with RAD51 3′ UTR constructs (wild-type or mutant) purchased from SwitchGear Genomics (Carlsbad, CA); at 48 hours after transfection, cells were incubated for 30 minutes with 100 µl/well of LightSwitch Luciferase Assay Reagent (SwitchGear Genomics, Carlsbad, CA). Firefly luciferase activity was measured sequentially by using luciferase assays (SwitchGear Genomics) with a Fluostar Optima plate reader (BMG Lab Technologies GmbH, Durham, NC). Three independent experiments were performed, and values are shown as means ± standard error of the mean.
In vivo tumor model and administration of MRX34. The mice used in this study were bred and maintained in our own institutional specific pathogen-free mouse colony. Male nude (nu/nu) mice were used for H1299 and H460 xenograft studies. Five animals per cage were housed in facilities approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current regulations and standards of the US Department of Agriculture and Department of Health and Human Services. Before tumor cell injection, tumor cell suspensions were prepared from cells grown in monolayers in vitro. H460 and H1299 tumors were generated by intramuscular injection of 1 × 106 cells in a volume of 20 µl into the right hind leg of 40 male nude Nu/Nu specific pathogen-free mice 3–4 months old. When tumors grew to 8 mm (range 7.8–8.3 mm) in diameter, mice were assigned to one the following groups (10 mice each): Control (no treatment), MRX34 (five doses of 1 or 5 mg/kg each given as subcutaneous peritumoral injections three times a week), radiation only (4 Gy once per day for 5 days, total 20 Gy), or MRX34 (five 5 mg/kg doses as described above) plus radiation. The miRNA was given 1–2 hours before radiation. Mice were irradiated with a 137Cesium device (dose rate 4 Gy/min) as previously described.30 For the growth-control assays, mice were euthanized when tumors reached 14–15 mm in diameter. Tumor growth delay was defined as the time for tumors to grow from 8 to 12 mm in diameter for treated mice as compared with tumor growth in untreated mice.
Biodistribution of MRX34. Twenty-four hours after the eighth injection of MRX34, two mice per group were euthanized and tumor was harvested for total RNA extraction and subsequent evaluation of tissue miR-34a levels. miRNAs were detected by isolating and analyzing total RNA from flash-frozen mouse tissues as previously described.30
Immunohistochemical analysis of RAD51. Formalin-fixed samples were routinely processed in an automatic tissue processor, embedded in paraffin (Peloris, Leica, Vista, CA), and sectioned at 4-μm thickness. Immunohistochemical staining was done with an automated staining system (Leica Bond Max, Leica Microsystems, Vista, CA). Briefly, slides were deparaffinized and hydrated, and antigen was retrieved by incubating in citrate buffer, pH 6.0, for 1 hour with RAD51 (Abcam, Cambridge, UK #ab213, dilution 1:25).
Analysis of DNA damage and repair mRNAs and miR-34a levels in patients from The Cancer Genome Atlas (TCGA). mRNA and microRNA expression levels for 532 NSCLC patients included in the TCGA cohort can be obtained from the TCGA portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). RNA-Seq and miRNA data for these tumors were downloaded directly from the same source and used for subsequent analyses. For NSCLC samples included in the TCGA cohort, experimental procedures regarding RNA extraction from tumors, mRNA library preparation, sequencing (on the Illumina HiSeq platform), miRNA-Seq, quality control, and subsequent data processing for quantification of gene expression have been previously reported.31 Statistical analyses were conducted using the R system for statistical computing and Spearman correlation. All reported P values are two-tailed and for all analyses; P ≤ 0.05 is considered statistically significant.
Statistical analysis. Statistical comparisons were done with Student's t-tests. P values <0.05 were considered to represent statistically significant differences. All statistical analyses and graphing were done with Graph Pad (GraphPad Prism, La Jolla, CA) and Excel (Microsoft, Redmond, WA). Enhancement in radiosensitization was calculated for all clonogenic assays based on the Linear-Quadratic (L-Q) model for biological effect.32 The coefficients of determination (R2) calculated for each L-Q fitted curve were always better than 0.996. From the L-Q curves obtained for control or drug-treated samples, the classic radiosensitization enhancement factor was calculated as the ratio of radiation doses required to reduce the surviving populations to 10%. OriginLab version 8.1 (OriginLab, Northampton, MA) was used for mathematical calculations.
SUPPLEMENTARY MATERIAL Figure S1. Endogenous levels of miR-34a in non-small cell lung cancer lines (NSCLC). Figure S2. Correlation of miR-34a expression and DNA damage and repair genes in 532 patients with NSCLC from The Cancer Genome Atlas data set. Table S1. List of the 58 DNA damage and repair genes analyzed using a custom 384-well PCR array. Table S2. Primers designed for gene expression analysis by quantitative polymerase chain reaction.
Acknowledgments
The authors thank Christine F Wogan, MS, ELS, of MD Anderson's Division of Radiation Oncology, for editorial contributions; Jeffrey D. Parvin, Ohio State University, Columbus, OH, for sharing the HeLa-DR cells and the plasmid expressing the I-SceI endonuclease; and Kevin Kelnar for providing materials from Mirna to MD Anderson. This work was supported by Doctors Cancer Foundation Grant; The Lung Cancer Research Foundation; Cancer Center Support (Core) Grant CA016672 to The University of Texas MD Anderson Cancer Center; the Mabuchi Research fund; the family of M. Adnan Hamed and the Orr Family Foundation to MD Anderson Cancer Center's Thoracic Radiation Oncology program; an MD Anderson Knowledge Gap award; Department of Defense (BATTLE award W81XWH-06-1-0303, PROSPECT award W81XWH-07-1-03060); and the Wiegand Foundation. H.J.P. and A.G.B. were supported by a commercialization grant from the Cancer Prevention Research Institute of Texas (CPRIT). J.V.H. is supported by R01 CA168484-02 and the Lung Spore 5P50 CA070907. The authors declared no conflict of interest.
Supplementary Material
Endogenous levels of miR-34a in non-small cell lung cancer lines (NSCLC).
Correlation of miR-34a expression and DNA damage and repair genes in 532 patients with NSCLC from The Cancer Genome Atlas data set.
List of the 58 DNA damage and repair genes analyzed using a custom 384-well PCR array.
Primers designed for gene expression analysis by quantitative polymerase chain reaction.
References
- Hoeijmakers, JH (2001). Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374. [DOI] [PubMed] [Google Scholar]
- Jasin, M (2002). Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 21: 8981–8993. [DOI] [PubMed] [Google Scholar]
- Jensen, RB, Ozes, A, Kim, T, Estep, A and Kowalczykowski, SC (2013). BRCA2 is epistatic to the RAD51 paralogs in response to DNA damage. DNA Repair (Amst) 12: 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata, M, Sasaki, MS, Sonoda, E, Morrison, C, Hashimoto, M, Utsumi, H et al. (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17: 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudeau, C, Lundin, C and Helleday, T (2001). DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307: 1235–1245. [DOI] [PubMed] [Google Scholar]
- Rinaldo, C, Bazzicalupo, P, Ederle, S, Hilliard, M and La Volpe, A (2002). Roles for Caenorhabditis elegans rad-51 in meiosis and in resistance to ionizing radiation during development. Genetics 160: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, LQ, Wang, Y, Wang, H, Cao, J, Liu, Q and Fan, FY (2011). Knockdown of Rad51 expression induces radiation- and chemo-sensitivity in osteosarcoma cells. Med Oncol 28: 1481–1487. [DOI] [PubMed] [Google Scholar]
- Mo, N, Lu, YK, Xie, WM, Liu, Y, Zhou, WX, Wang, HX et al. (2014). Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncol Rep 32: 1905–1912. [DOI] [PubMed] [Google Scholar]
- Gasparini, P, Lovat, F, Fassan, M, Casadei, L, Cascione, L, Jacob, NK et al. (2014). Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci USA 111: 4536–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Lopez, C, Lazaro-Trueba, I, Kerr, P, Lord, CJ, Dexter, T, Iravani, M et al. (2006). p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep 7: 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoto, KR, Meng, AX, Wasylishen, AR, Zhao, H, Coackley, CL, Penn, LZ et al. (2010). Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res 70: 8748–8759. [DOI] [PubMed] [Google Scholar]
- Stürzbecher, HW, Donzelmann, B, Henning, W, Knippschild, U and Buchhop, S (1996). p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J 15: 1992–2002. [PMC free article] [PubMed] [Google Scholar]
- Choi, YE, Pan, Y, Park, E, Konstantinopoulos, P, De, S, D'Andrea, A et al. (2014). MicroRNAs down-regulate homologous recombination in the G1 phase of cycling cells to maintain genomic stability. Elife 3: e02445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur, S, Pagliarini, R, Bunz, F, Rago, C, Diaz, LA Jr, Kinzler, KW et al. (2009). A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA 106: 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N, Fu, H, Tie, Y, Hu, Z, Kong, W, Wu, Y et al. (2009). miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett 275: 44–53. [DOI] [PubMed] [Google Scholar]
- Wiggins, JF, Ruffino, L, Kelnar, K, Omotola, M, Patrawala, L, Brown, D et al. (2010). Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res 70: 5923–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo, E, Navarro, A, Viñolas, N, Marrades, RM, Diaz, T, Gel, B et al. (2009). miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 30: 1903–1909. [DOI] [PubMed] [Google Scholar]
- Kelnar, K, Peltier, HJ, Leatherbury, N, Stoudemire, J and Bader, AG (2014). Quantification of therapeutic miRNA mimics in whole blood from nonhuman primates. Anal Chem 86: 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader, AG (2012). miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 3: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, W, Xu, Y, Dong, Y, Cao, L, Tong, J and Zhou, X (2013). Ectopic expression of miR-34a enhances radiosensitivity of non-small cell lung cancer cells, partly by suppressing the LyGDI signaling pathway. J Radiat Res 54: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghawanmeh, T, Thunberg, U, Castro, J, Murray, F and Laytragoon-Lewin, N (2011). miR-34a expression, cell cycle arrest and cell death of malignant mesothelioma cells upon treatment with radiation, docetaxel or combination treatment. Oncology 81: 330–335. [DOI] [PubMed] [Google Scholar]
- Liu, C, Zhou, C, Gao, F, Cai, S, Zhang, C, Zhao, L et al. (2011). MiR-34a in age and tissue related radio-sensitivity and serum miR-34a as a novel indicator of radiation injury. Int J Biol Sci 7: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, M, Paranjape, T, Müller, RU, Ullrich, R, Nallur, S, Gillespie, E et al. (2009). The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene 28: 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofman, AV, Kim, J, Park, SY, Dupart, E, Letson, C, Bao, Y et al. (2013). microRNA-34a promotes DNA damage and mitotic catastrophe. Cell Cycle 12: 3500–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransburgh, DJ, Chiba, N, Ishioka, C, Toland, AE and Parvin, JD (2010). Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 70: 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vispé, S, Cazaux, C, Lesca, C and Defais, M (1998). Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 26: 2859–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas, M, Davies, AA and West, SC (2004). RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci 359: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh, P, Lefever, S, Pattyn, F, Ridzon, D, Fredlund, E, Fieuw, A et al. (2011). The microRNA body map: dissecting microRNA function through integrative genomics. Nucleic Acids Res 39: e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweep, H, Sticht, C, Pandey, P and Gretz, N (2011). miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform 44: 839–847. [DOI] [PubMed] [Google Scholar]
- Cortez, MA, Valdecanas, D, Zhang, X, Zhan, Y, Bhardwaj, V, Calin, GA et al. (2014). Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol Ther 22: 1494–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz, HP, Coucke, PA and Mirimanoff, RO (1991). A linear-quadratic model of cell survival considering both sublethal and potentially lethal radiation damage. Radiother Oncol 21: 273–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Endogenous levels of miR-34a in non-small cell lung cancer lines (NSCLC).
Correlation of miR-34a expression and DNA damage and repair genes in 532 patients with NSCLC from The Cancer Genome Atlas data set.
List of the 58 DNA damage and repair genes analyzed using a custom 384-well PCR array.
Primers designed for gene expression analysis by quantitative polymerase chain reaction.