

Abstract
Targeted delivery of therapeutic agent is an important way to improve therapeutic index and reduce side effects. To design nanoparticles for targeted delivery, both enhanced tumor tissue accumulation/retention and enhanced cellular internalization should be considered simultaneously. Unfortunately, so far, there have been very few nanoparticles with immutable structures can achieve this goal efficiently. Hierarchical targeting, a novel targeting strategy based on stimuli responsiveness, shows good potential to enhance both tumor tissue accumulation/retention and cellular internalization. In this review, we will introduce recent design and development of hierarchical targeting nanoplatforms, which are based on changeable particle sizes, switchable surface charges and activatable surface ligands. In general, the targeting moieties in these nanoplatforms are not activated during blood circulation for efficient tumor tissue accumulation and then re-activated by certain internal or external stimuli in tumor microenvironment for enhanced cellular internalization.
Keywords: Hierarchical targeting, Stimuli-responsive nanoplatform, Changeable particle size, Switchable surface charge, Activatable surface ligand
Graphical Abstract
Hierarchical targeting, which combines the advantages of both enhanced tumor tissue accumulation/retention and enhanced cellular internalization, is a promising strategy to improve tumor targeting efficiency of nanoparticles. This review summarized the different strategies include exploitation of changeable particle size, switchable surface charge and activatable surface ligand to develop hierarchical targeting nanoplatforms.
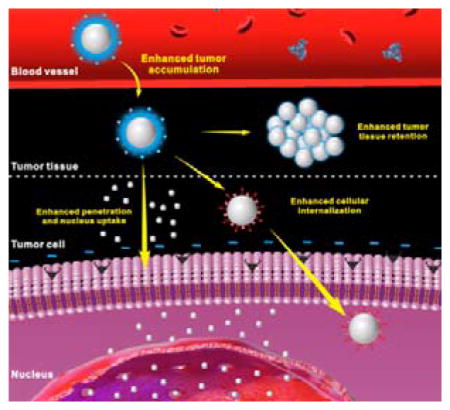
1. Introduction
Many effective tumor therapies, such as chemotherapy, photothermal therapy (PTT) and photodynamic therapy (PDT), are still limited by nonspecific tissue biodistribution and rapid renal clearance of therapeutic agents.[1] These limitations would lead to relatively low bioavailability of therapeutic agent and cause severe adverse effects to patients.[2] Nanoparticles (NPs) with tumor cell targeting ability have been actively developed to overcome these limitations for therapeutic agent delivery.[3] Tumor tissue accumulation/retention is usually based on the enhanced permeability and retention (EPR) effect, which exploits the leaky tumor vasculature and poor lymphatic drainage of tumors that allow NPs to accumulate in the tumor much more than that in normal tissues.[4] For many chemotherapeutic drugs, cellular internalization and even subsequent nucleus uptake is of critical importance because these drugs only act on tumor intracellular targets.[5] Cellular internalization is generally enhanced with the help of targeting ligands or positive charge onto the surface of NPs.[6] Thus the particles can bind to tumor cell membranes by specific ligand-receptor interactions or strong electrostatic interactions, resulting in improved tumor retention and subsequent cellular internalization.
Therefore, to develop an ideal targeted nanoparticular delivery system, both tumor tissue accumulation/retention and cellular internalization should be taken into consideration. To achieve this goal, the properties of NPs, such as size, surface charge and surface modification, should be properly designed. To date, however, most of the NP platforms are suboptimal. For example, positively charged or ligand modified NPs often display recognition and subsequent accelerated clearance by the reticuloendothelial system (RES), leading to relatively low tumor tissue accumulation (Figure 1A).[7] Neutral or negatively charged NPs modified with hydrophilic ‘stealth’ polymers, such as polyethylene glycol (PEG) and zwitterionic groups, may accumulate effectively into tumor tissue by EPR effect due to their high stability and long circulation time;[8] while the tumor retention and subsequent tumor cellular uptake of these NPs are unsatisfactory because the negative charge or ‘stealth’ polymer coating may hinder NP-cell interactions (Figure 1B).[9]
Figure 1.

Schematic illustration of the drug delivery procedure of positively charged or ligand modified nanoparticles (A), neutral or negatively charged nanoparticles modified with hydrophilic ‘stealth’ polymers (B) and hierarchical targeting nanoparticles (C) which can achieve enhanced tumor tissue retention (C1), enhanced cellular internalization (C2), enhanced tumor penetration and nucleus uptake (C3).
To address these problems, the designed NPs are expected to have the ability to change their properties in different environments. In recent years, various stimuli-responsive NPs[10] that respond to either tumor internal (e.g. pH,[11] redox,[12] enzyme[13]) or external (e.g. light,[14] temperature,[15] ultrasound,[16] magnetic field[17]) stimuli have been developed. These nanomaterials usually contain sensitive chemical groups, bonds, sequences or molecules that can realize stimuli-triggered responses, such as protonation, bond cleavage, conformation change, state transition, denaturation, degradation, and so on, to achieve the desired properties (Table 1). Hierarchical targeting, a novel targeting strategy that consists of two targeting stages including tumor tissue targeting based on EPR effect and tumor cell targeting based on targeting ligands or positive charge, shows great potential to enhance tumor targeted delivery of therapeutic agent. The hierarchical targeting nanoplatforms are generally based on stimuli-responsive nanomaterials and expected to show high stability during blood circulation and then transform into another form under special stimuli in the tumor tissue to achieve enhanced tumor retention, cellular internalization and even nucleus uptake (Figure 1C).
Table 1.
Examples of Stimuli, Chemicals, Responses, and Achieved Properties
| Stimuli | Chemicals | Responses | Achieved properties | Ref. | |
|---|---|---|---|---|---|
| Internal | pH | Amines | Protonation | Hierarchical targeting | [30, 73–74] |
| Protonation | Triggered drug release | [79] | |||
| Hydrazone bonds | Bond cleavage | Hierarchical targeting | [63, 66] | ||
| Bond cleavage | Triggered drug release | [80] | |||
| Benzoic-imine bond | Bond cleavage | Hierarchical targeting | [65] | ||
| Peptide sequence | Conformation change | Hierarchical targeting | [59–60] | ||
| Carbonate and phosphate | Degradation | Triggered drug or gene release | [81] | ||
|
| |||||
| Redox | Disulfide bonds | Bond cleavage | Triggered drug or gene release | [12, 82] | |
| Thioketal linkages | Bond cleavage | Triggered drug release | [83] | ||
|
| |||||
| Enzyme | Hyaluronic acid | Degradation | Hierarchical targeting | [71] | |
| Peptide sequence | Bond cleavage | Hierarchical targeting | [55–57] | ||
|
| |||||
| External | Light | o-nitrobenzyl (ONB) group | Bond cleavage | Hierarchical targeting | [48–49, 76] |
| DNA | Denaturation | Triggered drug release | [84] | ||
|
| |||||
| Temperature | Peptide sequence | Conformation change | Triggered drug release | [58] | |
| Carbonate | Degradation | Triggered drug release | [15] | ||
|
| |||||
| Ultrasound | Perfluorocarbon | State transition | Triggered drug release | [85] | |
|
| |||||
| Magnetic field | DNA | Denaturation | Triggered drug release | [17] | |
|
| |||||
| Electric field | Host-guest chemical | Disassociation | Triggered drug release | [86] | |
In this review, we will introduce recent advances in the design and development of hierarchical targeting nanoplatforms, which can have changeable particle size, switchable surface charge and/or activatable surface ligand (Figure 2). The perspectives and challenges of these nanoplatforms will also be discussed.
Figure 2.

Hierarchical targeting nanoplatforms based on changeable particle size (A), switchable surface charge (B) and activatable surface ligand (C).
2. Changeable particles size
Particle size is a key factor in the process of tumor tissue accumulation/retention, tumor penetration and cellular/nucleus internalization. For tumor tissue accumulation, particles with a diameter less than 400 nm can extravasate from leaky vasculature into tumor interstitium.[1] In addition, to reduce liver capture and renal filtration, the generally accepted size of the NPs is in the range of 10–100 nm.[18] Although this size range is also suitable for cellular internalization, smaller size is required for efficient nucleus uptake of particles. Small NPs show higher tumor penetration efficiency than large particles,[19] but they cannot be retained in tumor tissue effectively because they are able to re-enter the bloodstream;[20] large microparticles or fibers can be trapped in tumor tissue and hence achieve enhanced retention. However, microparticles or fibers are easily captured by the RES and have no chance to permeate in tumor tissue. Therefore, the changeable particles size maybe an efficient strategy to achieve enhanced tumor retention, penetration, cellular internalization and nucleus uptake.
2.1 Aggregation
The stimuli-triggered NP aggregation is an effective strategy to achieve enhanced tumor tissue retention. During blood circulation, the particles are small in size and can extravasate into tumor tissue. Once the small NPs arrive at tumor tissue, under certain stimuli, they would aggregate into larger particles and thus be trapped in tumor site. For example, Ji et al. reported a pH-triggered size-controlled NP for enhanced tumor retention.[21] Compared to normal healthy tissues (pH is approximately 7.4), the extracellular environment of many solid tumors displays weak acidity (pH is lower than 7.0).[22] It is contributed to the Warburg effect: fast-growing tumors cause increased glucose uptake and high rate of glycolysis followed by lactic acid fermentation in the tumor interstitium.[23] As shown in Figure 3A, the small sized gold NPs (~16 nm) were surface modified with zwitterionic mixed-charge monolayers, which enabled the NPs to be stable at physiological pH with stealth function. However, when the pH of surrounding environment decreased to 6.0–7.0, the NPs quickly aggregate to form large particles (Figure 3B). The aggregation transition pH values of this system can be tuned from 7.0 to 5.5 by adjusting the feed ratios of carboxylic group (C10-C) to quaternary ammonium group (C10-N4). The enhanced tumor retention effect of aggregatable NPs (16-AuNP-C10-CN4-5:5) was further evaluated in KB tumor-bearing nude mice. As shown in Figure 3C and 3D, at 24 h postinjection, the tumor tissue accumulation of aggregatable NPs was increased over 2-fold compared to that of PEG modified gold NPs (16 nm, 16-AuNP-PEG2000). Furthermore, the retention of aggregatable NPs is much better. At 72 h postinjection, the tumor accumulation of aggregatable NPs was ~80% of that at 24 h postinjection. However, the tumor accumulation of 16-AuNP-PEG2000 was only ~30% of that at 24 h postinjection. It may be attributed to the migration of these small NPs into surrounding tissues. Considering the excellent tumor retention, the aggregatable NP systems are promising nanoplatforms for delivering therapeutic agents.[24] In addition, the aggregation strategy also can be used to develop stimuli-responsive nanosystems for triggered drug release. For example, Gu et al. developed a liquid metal-based nanoplatform for antitumor drug delivery.[25] The liquid-metal NPs are able to fuse together into a larger NP under tumor acidic environment, thus promoting the drug release.
Figure 3.

(A) Schematic of the pH-responsive AuNP-C10-CN4-X:Y. (B) Schematic and TEM images of the AuNPs at different pH. (C) Accumulation of the AuNPs in KB tumor-bearing mice at different time postinjection. (D) Normalized tumor accumulation of the AuNPs. Reproduced with permission.[21] Copyright 2013 American Chemical Society.
2.2 Supramolecular coassembly
In vivo stimuli-triggered supermolecular coassembly is another promising strategy to prolong the tumor retention. Chen et al. reported an in situ tumor-specific coassembly strategy of indocyanine green (ICG, 1) and alkaline phosphatase (ALP)-responsive peptide (NapFFKYp, 2) for photoacoustic (PA) imaging and PTT.[13] The mixture of 1 and 2 forms micelles (Figure 4A), which can efficiently accumulate into tumor tissue by EPR effect. Once the micelles (1 + 2) reach tumor tissue, they will be efficiently self-assembled into nanofibers (5) in the presence of overexpressed endogenous phosphatase (Figure 4B), then the resulting nanofibers (Figure 4C) will be retained in tumor tissue. As shown in the near-infrared (NIR) fluorescence images (Figure 4D), 1 was quickly excreted within 4 h and only a small fraction of 1 was retained in tumor at 24 h postinjection. In contrast, for the mice treated with 1 + 2, a strong fluorescence signal was observed in the tumor site and lasted over 48 h. Ex vivo fluorescence imaging at 24 h postinjection also showed effective tumor retention of 5. Due to the significantly enhanced tumor retention, 5 is a unique system for PA imaging and PTT. As shown in Figure 4E, the tumor PA signal of mouse injected with 5 was much stronger than that of mouse injected with 1 at 24 h, which can be attributed to the enhanced tumor retention of nanofibers. To investigate the in vivo PTT effect of 1 and 5, HeLa tumor-bearing mice were intravenously injected 1 or 1 + 2 and then subjected to PTT treatment at 24 h or 48 h postinjection. As shown in Figure 4F, the tumors treated with 5 were effectively ablated upon NIR laser irradiation.
Figure 4.

(A) TEM image of the micelles (scale bar: 1 μm). (B) Schematic of in situ supramolecular coassembly. (C) TEM image of the nanofibers (scale bar: 100 nm). (D) Fluorescence images of the ICG (1) and nanofibers (5) on HeLa tumor-bearing mice at different time postinjection. (E) 3D PA images of the 1 and 5 on HeLa tumor-bearing mice at 24 h postinjection. (F) Tumor growth curves and survival curves of different groups. Reproduced with permission.[13] Copyright 2015 American Chemical Society.
Although the strategy of stimuli-responsive aggregation or supramolecular coassembly can efficiently deliver therapeutic agents into tumor tissue with prolonged tumor retention, the tumor penetration and cellular uptake of the aggregated particles or assemblies may be impaired due to their huge diffusion hindrance of tumor interstitium. Therefore, we believe that these strategies are more suitable for the delivery of therapeutic agents without requiring tumor penetration (for example: photothermal agents, because of the transitivity of heat).
2.3 Size decrease
For the delivery of many chemtherapeutic drugs, the tumor penetration should also be considered to improve the therapeutic index. As mentioned above, the tumor penetration efficiency of large NPs is suboptimal due to the diffusion hindrance; in contrast, too small NPs suffer from rapid clearance by renal filtration, leading to low tumor accumulation. To address this issue, a size decrease strategy has been developed. The initial size of NPs is relatively large to reduce renal clearance and thus improving tumor accumulation. Once the NPs arrive at tumor sites, their diameter will be decreased into a smaller one with higher tumor penetration efficiency. In a recent study, Wang et al. reported a clustered NP (iCluster) which was prepared by self-assembly of polycaprolactone (PCL), PEG-b-PCL and platinum (Pt) prodrug-conjugated poly(amidoamine)-graft-PCL (PCL-CDM-PAMAM/Pt).[26] As shown in Figure 5A, the PAMAM/Pt (~ 5 nm) were conjugated to the large NP (~ 100 nm) by pH-sensitive bonds, the resultant iCluster/Pt has an initial size of ~100 nm and a PEG coating, which are benefit for tumor accumulation. However, in the tumor extracellular environment, the PAMAM/Pt would be released for deep tumor penetration. TEM images showed the pH-triggered PAMAM/Pt release in a pH 6.8 buffer solution (Figure 5B). To evaluate the tumor penetration of iCluster and nonresponsive Cluster, the large core NP was labeled with rhodamine B (RhB, red) and PAMAM was labeled with fluorescein (Flu, green). Then the intratumoral distribution of dual labeled RhBiClusterFlu and RhBClusterFlu were studied by using BxPC-3 xenograft tumor as a model. As shown in Figure 5C, for RhBClusterFlu treatment, both the red fluorescence and green fluorescence almost exclusively distributed in blood vessels and their nearby areas, which indicated that the large NP cannot penetrate into deep tumor space. However, in the RhBiClusterFlu treated group, the green fluorescence showed a uniform distribution in the tumor, suggested that the PAMAM were released from the iCluster and penetrated into tumor interstitium.
Figure 5.

(A) Schematic illustration showing the tumor stimuli-triggered structural change of iCluster/Pt. (B) TEM images iCluster/Pt with pH 6.8 solution treatment for 0, 4 and 24 h. (C) Confocal images of RhBiClusterFlu and RhBClusterFlu in BxPC-3 xenograft tumor at 4 h postinjection (scale bar: 50 μm). PAMAM: Flu (green); the core of the nanoparticles: RhB (red); blood vessels: platelet endothelial cell adhesion molecule 1 (PECAM-1) and CFL-647 secondary antibody (yellow). Reproduced with permission.[26] Copyright 2016 National Academy of Sciences.
The size decrease strategy can also be employed to overcome the multidrug resistant (MDR) issue. The overexpressed transporter proteins in the membranes of MDR tumor cells can expel drugs from the cells. Since many antitumor drugs, such as doxorubicin (DOX), act on the nucleus, NPs with the property of intranucleus drug delivery are expected to address the MDR issue of these drugs by bypassing the transporter proteins. In order to achieve intranucleus drug delivery, the NPs should be small enough (< 30 nm) to cross the nuclear envelope.[27] Meanwhile, too small NPs bear suboptimal tumor tissue accumulation/retention due to the quick renal filtration. To combine the advantages of both enhanced tumor tissue accumulation/retention of large NPs and enhanced nucleus uptake of small NPs, in a recent study, Tan and co-workers developed a size-photocontrollable NP by using DNA self-assembly.[28] As shown in Figure 6A, multiple DOX-loaded gold NPs with very small size were self-assembled onto the side edges of a relatively large gold-silver nanorod (NR) via DNA hybridization. The end edges of NR were modified with aptamers, which are artificial oligonucleotides that can enhance cellular internalization. Upon NIR light irradiation, the dehybridization of DNA linkers between NR and NPs, which is caused by photothermal effect of the NR, allows the release of NPs from the NP/NR nanoassembly. As shown in Figure 6B, the diameter of the NP/NR nanoassembly is about 60 nm, which is suitable for tumor accumulation by EPR effect. To investigate the cellular internalization and photocontrolled nucleus uptake of the NP/NR nanoassemblies, the NPs and NRs were labeled with TAMRA (TMR) and Cy5 fluorophores, respectively. Then the TMR-NP/Cy5-NR nanoassemblies were incubated with CEM cells with or without NIR light irradiation. As shown in the confocal images (Figure 6C), without NIR light irradiation, the signals of both TMR-NPs and Cy5-NRs were mainly observed in the cytoplasm, which indicated that the TMR-NP/Cy5-NR nanoassemblies cannot enter cell nucleus. However, when NIR light irradiation was applied, the signal of TMR-NPs was observed in the nucleus, while the signal of Cy5-NRs remained in the cytoplasm. This result indicated that the small sized NPs were released from the NP/NR nanoassemblies upon laser irradiation and then entered into the nucleus. The ability of the NP/NR nanoassemblies to overcome the MDR problem was investigated by using K562/D, a drug-resistant cell line. A specific aptamer, KK1B10, was modified to the end edges of NR as the targeting ligand. As shown in Figure 6D, compared with the cells incubated with DOX and 8.5-NP-DOX/NR-KK1B10, the cells incubated with 8.5-NP-DOX/NR-KK1B10 and laser irradiation showed significantly decreased viability. These results demonstrated that the size-changeable NP/NR nanoassemblies can effectively improve the therapeutic efficacy of DOX against MDR tumors through the enhanced nucleus uptake.
Figure 6.

(A) Schematic of the size-photocontrollable NP/NR nanoassembly for enhanced nucleus uptake. (B) TEM image of the NP/NR nanoassembly. (C) Confocal images of CEM cells incubated with the TMR-NP/Cy5-NR nanoassembly without (1) or with (2) laser irradiation. (D) Viability of K562/D cells after different treatments. Reproduced with permission.[28] Copyright 2015 American Chemical Society.
3. Switchable surface charge
The surface charge of NPs plays a significant role in delivery and cellular internalization.[7a, 18a] Positively charged NPs often bear short blood circulation half-life due to high nonspecific adsorption rate and rapid renal filtration.[8b] Furthermore, some studies showed that strong positive charge may hinder the solid tumor penetration of NPs due to a ‘binding-site barrier’ phenomenon: the NPs bind to their first targets so strongly that their penetration ability is decreased.[29] To have prolonged blood circulation and efficient tumor uptake/penetration, NPs that are neutral or have negative surface charge are preferred. Once they reach tumor tissue, positive charges are necessary to have enhanced tumor retention and cellular internalization by strong electrostatic interactions with negatively charged cell membranes. Different surface charges of NPs are needed in the different delivery stages; therefore, the design of nanoplatforms with switchable surface charge is an important way to achieve hierarchical targeting.
3.1 Protonation
Many surface charge-switchable NPs have numerous surface groups (such as amines,[30] histidine[31] and morpholin[32]) that undergo protonation in response to the weak acidic environment of solid tumor. The surface of pH-sensitive NPs can switch from being neutral/negative at physiological pH to being positive at tumor tissue pH, induced by the protonation of surface groups. For example, glycol chitosan coated superparamagnetic iron oxide NPs (GC-SPIO) (Figure 7A).[30] are neutral at physiological pH (aggregate pKa ~6.5), which have minimal interaction with serum proteins and normal tissues. Under acidic condition (pH < 7.0), the surface of GC-SPIO becomes positively charged and then electrostatically interacts with the negatively charged cell membrane to achieve enhanced tumor retention of the GC-SPIO. As shown in Figure 7B, the zeta-potential of GC-SPIO continually increased from + 0.3 mV (pH = 7.4) to + 8.2 mV (pH = 6.15) with decreased pH. As shown in T2-weighted magnetic resonance (MR) images of cells (Figure 7C), the cells incubated with GC-SPIO at acidic pH values showed stronger T2 signal intensity than that incubated with GC-SPIO at physiological pH, which is attributed to the high retention of GC-SPIO in acidic conditions. However, in the control groups without pH sensitivity (Glycidol GC-SPIO and Dextran-SPIO), neither surface charge nor tumor cell retention efficiency was altered at different pH values.
Figure 7.

(A) Schematic of cellular delivery of the samples at different pH. (B) Zeta potential changes of the samples at different pH. (C) T2-weighted imaging of T6–17 cells after incubation with the samples at different pH. Reproduced with permission.[30] Copyright 2011 American Chemical Society.
In another study, N-acetyl histidine (NAcHis) conjugated D-α-tocopheryl PEG1K succinate (TPGS) (NAcHis-TPGS) chains were modified onto the surface of poly(lactic-co-glycolic acid) (PLGA) NPs. The resulting NAcHis-TPGS/PLGA NPs (NHTPNs) were capable of co-delivering DOX and indocyanine green (ICG, a photothermal agent).[33] As shown in Figure 8A, the NHTPNs can achieve tumor extracellular pH (pHe) triggered surface charge switch due to the enhanced protonation of NAcHis moieties at pHe. The cellular uptake results indicated that NHTPNs can be more effectively internalized by tumor cells under weak acidic conditions (pH 6.3). In contrast, the cellular uptake efficiency of TPNs (without NAcHis moieties) was not affected by pH (Figure 8B and 8C). It is also notable that the antitumor effect of ICG/DOX-loaded NHTPNs at pH 6.3 is appreciably better than that at pH 7.4 (Figure 8D).
Figure 8.

(A) Schematic of pHe-triggered surface charge switch of ICG/DOX-loaded NHTPNs. (B) Flow cytometric (FCM) analysis TRAMP-C1 cells incubated with ICG/DOX-loaded TPNs and NHTPNs at pH 7.4 or 6.3 for 2 h. (C) Fluorescence images of TRAMP-C1 cells incubated with ICG/DOX-loaded TPN and NHTPNs at pH 7.4 or 6.3 for 1.5 h. Cell cytoskeleton: F-actin marker (green); nuclei: Hoechst (blue). (D) Cell viability of TRAMP-C1 cells incubated with ICG-loaded NHTPNs and ICG/DOX-loaded NHTPNs at pH 7.4 or 6.3. Reproduced with permission.[33] Copyright 2016 Ivyspring International Publisher.
This strategy has also been employed to develop hierarchical targeted gene delivery systems.[34] For example, Shuai et al. developed a pH and reduction dual-sensitive polyplex for siRNA delivery.[35] By adjusting the N/P ratio, a charge-switchable NP is stable and shows slightly negative charge in blood circulation. Once the polyplex arrives at acidic tumor extracellular environment, their surface charge can be reversed from being negative to positive due to the protonation of amino groups in the polyplex, leading to effective tumor cellular internalization of these NPs. In vivo results indicate that this gene delivery system can achieve effective tumor specific gene silencing.
3.2 Positive charge exposure
To protect the unprotonated amino groups on the surface of NPs, negatively charged 2, 3-dimethylmaleic anhydride (DMMA) has been employed as the charge shield.[36] At physiological pH, the NPs are negatively charge because of the presence of abundant DMMA carboxyl groups. However, the amide bonds between DMMAs and amino groups would be cleaved in response to the tumor extracellular pH (~6.8) (Figure 9A). The 1H NMR spectra demonstrated that ~ 60% of the DMMA groups were detached within 60 min incubation (Figure 9B). The detachment of negatively charged carboxyl groups as well as the exposure of protonated amino groups led to the surface charge reversion. As shown in Figure 9C, the zeta-potential increased significantly within 10 min at pH 6.8, from −15 to +10 mV. The NPs remained negative after incubation at pH 7.4 for 2 h. The cellular uptake results further demonstrated that the charge-switchable NPs had higher tumor cellular internalization at the pHe than that at physiological pH. As shown in Figure 9D, at pH 6.8, the NPs were internalized and mainly distributed in the cytoplasm, which was not observed when incubated at pH 7.4. This charge-switch strategy has also been used to design the charge-reversal polypeptide micelles and fluorescence nanoprobe for tumor-specific imaging.[37]
Figure 9.

(A) Schematic of pH-triggered cellular internalization of PPC-Hyd-DOX-DA. (B) 1H NMR spectra of polymer incubated at pH 6.8 for different time. (C) Zeta potential changes of PPC-Hyd-DOX-DA at different pH values. (D) Confocal images of MDA-MB-231 cells incubated with DOX-loaded nanoparticle at different pH values for 1 h. Cell cytoskeleton: Alexa 488 (green); nuclei: DAPI (blue). Reproduced with permission.[36] Copyright 2011 American Chemical Society.
The negatively charged coating materials can also be added to the surface of positively charged NPs by electrostatic adsorption as charge shield. Under special conditions, the negatively charged layer will detach, followed by the exposure of positively charged layer.[38] For example, a pH-sensitive polymer (mPEG45-b-PAEP75-Cya-DMMA, PPC-DA), was introduced onto the surface of cationic poly(ethylenimine) (PEI)/siRNA complex via electrostatic interaction, so the PEI/siRNA complex was coated by PEG shell (Figure 10A).[39] At tumor extracellular pH, DMMA groups would be detached from PPC, and then the positively charged PPC would be shed because of electrostatic repulsion. Subsequently, the re-exposed PEI/siRNA complex would enter cells efficiently. Figure 10B shows the zeta-potential change of sheddable NPs (S-NP) and unsheddable NPs (unS-NP). At pH 6.8, the surface charge of S-NP was reversed from negative to positive within 60 min; however, the surface charge of S-NP at pH 7.4 and unS-NP at both pH values kept negative. As shown in Figure 10C and 10D, the S-NP incubated cells at pH 6.8 had higher cellular uptake than the other groups.
Figure 10.

(A) Schematic of the shedding process of PPC coating. At the pHe, the positively charged PPC would be shedded because of electrostatic repulsion and the PEI/siRNA would be re-exposed. (B) Zeta-potential changes of S-NP and unS-NP at different pH. (C) Confocal images of MDA-MB-231 cells incubated with S-NP and unS-NP at different pH. Nanoparticles carrying FAM-siRNA (green); cell cytoskeleton: Alexa Fluor 568 phalloidin (red); nuclei: DAPI (blue). (D, E) FCM analysis of MDA-MB-231 cells incubated with S-NP (D) and unS-NP (E) at pH 7.4 or 6.8. Reproduced with permission.[39] Copyright 2011 American Chemical Society.
3.3 Zwitterionic-to-cationic charge conversion
Another strategy to allow surface charge switch is using zwitterionic surfaces with pH-responsive zwitterionic-to-cationic charge conversion property. Various pH-responsive zwitterionic groups, such as carboxybetaine,[40] phosphorylcholine[41] and alkoxyphenyl acylsulfonamide,[42] have been employed for the surface modification of NPs. Among these groups, alkoxyphenyl acylsulfonamide is an attractive one because it can respond to weakly acidic pH of tumor environment with high sensitivity. A gold NP (AuNP) formula functionalized with alkoxyphenyl acylsulfonamide ligands was developed for enhanced tumor cellular uptake.[42] As shown in Figure 11A, two kinds of zwitterionic AuNPs were prepared; AuNP 1 with aryl acylsulfonamide groups and AuNP 2 with alkyl acylsulfonamide groups. Because of the presence of the surface zwitterionic structure, both 1 and 2 were neutral at physiological pH. However, at acidic pH, the surface charge of 1 became positive due to the protonation of negatively charged groups. In contrast, 2 retained a zwitterionic surface even when the pH was lowered to 5.5, which may be attributed to the insensitivity of alkyl acylsulfonamide groups to pH (Figure 11B). As positively charged AuNPs show higher cytotoxicity than zwitterionic AuNPs,[43] the cytotoxicities of 1 and 2 at different pH values were also evaluated to demonstrate the pH-induced charge-conversion (Figure 11C). At pH 7.4, both 1 and 2 were relatively non-toxic as a result of being zwitterionic. At pH 6.0, 1 showed increased cytotoxicity due to the enhanced cellular uptake of positively charged 1 induced by the surface charge conversion.
Figure 11.

(A) Chemical structure of the monolayer-protected AuNPs and schematic of pH-responsive delivery. (B) Zeta potential of 1 and 2 at different pH values. (C) Viability of HeLa cells after incubation with 1 and 2 for 72 h. Reproduced with permission.[42] Copyright 2015 Wiley.
Another strategy to achieve zwitterionic surface is to modify the NP with mixed charge species.[44] By adjusting the ratio of the positively and negatively charged groups on the surface of NPs, the pKa value can be tuned within a range. Based on this strategy, an amine/carboxyl-terminated PEG modified gold nanostar (GNS) formula was developed.[45] As shown in Figure 12A, both positively charged amines and negatively charged carboxylates were added to GNS through long-chain PEG linkers. By adjusting the amine/carboxyl ratios (3:1, 4:1, 5:1, 6:1, and 7:1), a series of mixed-charge GNSs, as well as complete amine or carboxyl modified GNSs, were prepared and termed as GNS-N/C 3, GNS-N/C 4, GNS-N/C 5, GNS-N/C 6, GNS-N/C 7, GNS-NH2 and GNS-COOH, respectively. The zeta potential measurements of GNSs under different pH conditions demonstrated that the pKa value of the mixed-charge GNSs could be regulated continuously (Figure 12B). Cellular uptake efficiency of GNS-N/C 4 and 5 showed obviously pH dependence (Figure 12C). To evaluate the therapeutic efficacy of the mixed-charge GNSs, HeLa cells were incubated with GNS-N/C 3-7, GNS-NH2 or GNS-COOH for 4 h and subsequently irradiated by NIR light for 3 min. As shown in Figure 12D, the cell killing effect of GNS-N/C 4 at pH 6.4 was significantly higher than that at pH 7.4, which may be attributed to the high cellular uptake efficiency of GNS-N/C 4 at pH 6.4.
Figure 12.

(A) Schematic of the preparation pH- controlled photothermal therapeutic efficiency of mixed-charge GNSs. (B) Zeta potentials of GNS-N/C 3-7, GNS-NH2 and GNS-COOH at different pH values. (C) Cellular uptake of GNSs after incubation with samples at different pH values for 4 h. (D) Viability of HeLa cells after incubation with different concentration of samples for 4 h at pH 7.4 (left) or 6.4 (right) and subsequent 808 nm laser irradiation (2 W cm−2, 3 min). Reproduced with permission.[45] Copyright 2015 Wiley.
4. Activatable surface ligand
Conventional active targeting strategy by connecting ligands onto the surface of NPs faces many challenges that limit their further in vivo applications. For instance, NPs with a ligand modified surface often show suboptimal tumor tissue accumulation due to unintended high uptake by normal tissues/cells expressing low levels of receptor.[46] An ideal active targeting strategy may include two steps: i) the surface targeting ligands are shielded during blood circulation to avoid opsonization and RES organ uptake, ii) the targeting function of NPs will be activated after being accumulated in tumor tissues, and followed by efficient internalization into tumor cells.
4.1 Uncage
Caging groups can deactivate the targeting function of ligands. Many targeting molecules, such as folate and cell-penetrating peptides (CPPs), have been safeguarded by various stimuli-responsive caging groups. Photolabile o-nitrobenzyl (ONB) group is a commonly used linker to connect ligands with caging groups for photo-caging, because the ONB group promises UV light-induced bond breaking.[47] Based on this approach, a targeting peptide (sequence: YIGSR) was caged with a 4,5-dimethoxy-2-nitrobenzyl (DMNB) group and then conjugated to the surface of polystyrene NPs (Figure 13A).[48] The targeting function of YIGSR was inactive until an UV light is applied, which led to the release of caging group (Figure 13B and 13C). Fluorescence images showed that a large number of cells with UV light irradiation were targeted by the particles (Figure 13D), while almost no targeting effect was observed in the cells without irradiation (Figure 13E). Based on a similar strategy, folate[49] and CPP[50] were also caged by UV light sensitive groups through covalent binding. In order to avoid UV damage to normal tissues as well as to increase the penetration of the applied light, NIR light was employed to replace the UV light with the help of upconversion NPs (UCNPs).[51] As shown in Figure 14A, 2-nitrobenzylamine (NBA) caged folate molecules conjugated with PEG on the surface of UCNP@SiO2 NPs was designed for DOX delivery. The folate was initially inactive so it could not recognize the folate receptor. Upon 980 nm NIR light illumination, the emitted UV light will cleave the linker between folate and the caging group, leading to the exposure of the folate. The photo-responsive targeting ability of UCNP@SiO2 NPs was further demonstrated at cellular level (Figure 14B). Without laser illumination, the fluorescence signal was detected in the cells incubated with folate-PEGylated UCNP@SiO2 NPs, but not with caged folate-PEGylated UCNP@SiO2 NPs treated cells. Upon NIR light illumination, the targeting function of the caged folate was re-activated, resulting in high cellular uptake of the uncaged folate-PEGylated UCNP@SiO2 NPs.
Figure 13.

(A) Schematic of a phototargeted NP. Upon UV light irradiation, the caged YIGSR will be activated, and the NP can bind to the cells. (B) FTIR spectra of caged NPs with or without UV light illumination. (C) Absorbance spectra of free DMNB. (D, E) Fluorescent images of cells incubated with caged NPs with (D) or without (E) UV light illumination. Reproduced with permission.[48] Copyright 2010 American Chemical Society.
Figure 14.

(A) Schematic of NIR light-induced cellular internalization of caged UCNP@SiO2. Upon NIR light irradiation, the caging molecules would be removed, leading to the activation of folate. (B) Confocal images of HeLa cells incubated with different samples: (a) cells alone, (b) uncaged DOX-loaded NPs, (c) caged DOX-loaded NPs, and (d) caged DOX-loaded NPs with an irradiation of NIR light. Cell cytoskeleton: Alexa 488 (green); nuclei: DAPI (blue). Reproduced with permission.[51b] Copyright 2013 American Chemical Society.
Aptamers are single-stranded oligonucleotides and have been widely explored as targeting ligands for cancer diagnosis and treatment.[52] In a recent study, Sgc8, an aptamer that can specifically bind to the cell-membrane protein tyrosine kinase-7 (PTK-7),[53] was conjugated onto the surface of gold nanorods (GNRs) and caged by a single-stranded DNA (ssDNA) with the complementary sequence (Figure 15A).[54] The targeting ability of Sgc8 was abrogated due to the formation of double-stranded DNA (dsDNA). Upon NIR light irradiation, the heat generated by GNRs raises the local temperature, which induces dehybridization of dsDNA. Then the exposure of Sgc8 enables the specific binding to PTK-7 on tumor cells. Dark-field scattering imaging was used to investigate the NIR light activated cell binding of GNRs-Apt/DNA. As shown in Figure 15B, while CCRF-CEM cells had prominent uptake of GNRs-Apt, these PTK-7 positive cells had almost no uptake of GNRs-Apt/DNA as the function of sgc8 was completely abrogated by ssDNA. When NIR light was applied to the cells incubated with GNRs-Apt/DNA, strong scattered light signal was observed, manifesting light-induced removal of caging ssDNA.
Figure 15.

(A) Schematic of the light-activable targeted nanorod conjugated with ssDNA caged aptamers. (B) Dark-field images of CCRF-CEM cells incubated with different samples: GNRs, GNRs-Apt, and GNRs-Apt/DNA with (+L) and without NIR light irradiation. Reproduced with permission.[54] Copyright 2015 Tsinghua University Press and Springer-Verlag Berlin Heidelberg.
In tumor tissues, many enzymes, such as legumain and matrix metalloproteinase (MMP), are overexpressed, which have also been exploited to design enzyme-activatable targeting.[55] For example, a recent study reported an inactivated trans-activator transcription (TAT) peptide prepared by conjugating a caging amino acid or dipeptide to its lysine units.[56] After the cleavage of caging groups with the specific enzymes (aminopeptidase N or dipeptidyl peptidase IV), the TAT peptide became activated, and thus enhanced the cellular internalization. Another method to cage polycationic CPP is by a neutralizing polyanion. A polycationic activatable CPP (ACPP) is coupled with a polyanion via a MMP-cleavable linker to form an ACPP-conjugated dendrimer (ACPPD).[57] The targeting effect of ACPP is invalidated by the polyanion until the linker is cleaved by MMP. Upon cleavage of the linker, the ACPP would be activated due to the release of caging polyanion, which can specifically bind to tumor cells. The in vivo fluorescence imaging demonstrated the better tumor targeting efficiency of cleavable ACPPD over non-cleavable ACPPD.
4.2 Conformation change
Stimuli-induced conformation change can also be exploited to achieve the specific functions. For example, a temperature-sensitive amphiphilic leucine zipper peptide was introduced to the lipid layers of liposome. When hyperthermia was applied, the conformation of leucine zipper peptide would unfold, and then a channel would be open in the liposome leading to drug release.[58] Another example is the so-called pH low insertion peptide (pHLIP), a pH-sensitive peptide that is water-soluble at neutral and physiological pH.[59] However, the hydrophobicity of pHLIP would be increased in an acidic environment because of the protonation of two Asp residues in the transmembrane region. Thus the pHLIP spontaneously changes to a rigid transmembrane α-helix conformation that inserts across lipid cell membrane. The pHLIP, together with europium luminescent complex (EuL) were conjugated to the surface of AuNP to give pHLIP•EuL•AuNP,[60] which could effectively enter cells within minutes. In another study, as shown in Figure 16A, pHLIP and chlorin e6 (Ce6) photosensitizer were conjugated onto the surface of hollow gold nanospheres (HAuNS) to form an antitumor therapeutic system.[61] The pHLIP signal of cells at pH 6.2 was higher than that at pH 7.4, confirming pH-responsive targeting ability of pHLIP (Figure 16B).
Figure 16.

(A) Schematic of HAuNS-pHLIP-Ce6 for pH induced translocation. (B) Fluorescent images of cells incubated with HAuNS-pHLIP-Ce6 at different pH for 4 h. Reproduced with permission.[61] Copyright 2015 American Chemical Society.
4.3 Ligand exposure
As mentioned above, modifying ligands or cationic groups onto the surface of NPs directly may lead to low tumor accumulation due to high normal tissues/cells uptake. Furthermore, some exposed targeting molecules are susceptible to degradation by enzymes in the blood.[62] To address this issue, Long ‘stealth’ polymer chains connected to the surface of NPs via sensitive bonds will form a sheddable surface coating to prevent the NPs from interacting with normal tissues/cells and blood enzymes. In this strategy, PEG is a commonly used coating material because it is able to improve the stability and prolong the circulation time of NPs. To date, various smart nanoplatforms with sheddable PEG chains and shielded targeting molecules such as folate,[63] TAT peptide,[64] RGD peptide[65] or positively charged quaternary ammonium salt[66] and lysine[67] have been developed. As shown in Figure 17A, PEG2K were connected onto the surface of NPs with pH-sensitive hydrazone bonds to form a thick outer corona, while the short folate chains were hidden in the inner layer.[63] So the targeting function of folate is turned off in the blood circulation and normal tissues at physiological pH of 7.4. Once the NPs reach the acidic tumor site by EPR effect, PEG chains are detached by the hydrolysis of hydrazone bonds to expose the folate receptor-mediated internalization. The cellular uptake studies showed that PBS buffer pretreated folate-PEG-coated polymeric lipid vesicles (FPPLVs) were difficult to enter cells. In contrast, the FPPLVs with pH 5.0 solution pretreatment had markedly improved cellular uptake efficiency, presumably due to the detachment of protective PEG layer and the exposure of folate ligands (Figure 17B).
Figure 17.

(A) Schematic of the shedding of PEG coating as well as the exposure of folate at acidic microenvironment. (B) Fluorescence images and FCM analysis of HeLa cells incubated with FITC-labeled FPPLVs. Nuclei: DAPI (blue). Reproduced with permission.[63] Copyright 2014 Royal Society of Chemistry.
The PEG chains also can be connected to the ligands directly by stimuli-responsive chemical bonds, and the ligands are attached to the NP surface by another functional group, so that the ligands are in the intercalation between PEG coating and the NP. Wang et al. developed a tumor pHe-responsive polymeric vector for siRNA delivery.[68] As shown in Figure 18A, a PEG-CPP-PCL block copolymer was synthesized, in which a pHe-labile linker was inserted between the PEG and positively charged CPP. The copolymers then formed a micelle structure and were used as siRNA delivery system (Dm-NPsiRNA). As shown in Figure 18B, the Dm-NP showed relatively high stability at pH 7.4 with less than 20% PEG release after 24 h incubation. In contrast, nearly 60% of PEG chains were released from the Dm-NP at pH 6.5. This PEG release was attributed to the cleavage of the pHe-labile linkers. As a result of PEG release and CPP exposure at pH 6.5, the surface charge of the Dm-NP was significantly changed (zeta potential increased from ~ 20 mV to ~ 35 mV). Conversely, the change of zeta potential of Dm-NP incubated at pH 7.4 was negligible (Figure 18C). The pH-triggered enhanced cellular uptake of the Dm-NPFAM-siRNA was then determined by flow cytometry. As shown in Figure 18D, the cells incubated with Dm-NP at pH 7.4 showed very weak FAM signals, indicating low cellular uptake efficiency. However, much stronger FAM signal was obtained when the cells were incubated with Dm-NP at pH 6.5, suggesting that CPP was exposed to the surface of the Dm-NP. With enhanced cellular internalization, pH-triggered down-regulation of mRNA was also observed (Figure 18E).
Figure 18.

(A) Schematic of the polymeric vector and its tumor pHe-reponsive change. (B) PEG release from the Dm-NP incubated at pH 7.4 and 6.5. (C) Zeta potential changes of NP and Dm-NP incubated at pH 7.4 and 6.5. (D) FCM analysis of A549 cells incubated with FAM-siRNA-loaded NP and Dm-NP at pH 7.4 and 6.5 for 2 h. (E) The relative mRNA expression of mutant A549 cells treated with different samples at pH 7.4 and 6.5. Reproduced with permission.[68] Copyright 2015 American Chemical Society.
Many macromolecules or polypeptides, such as hyaluronic acid (HA), can rapidly degraded by corresponding enzymes that expressed in tumor extracellular environment.[69] Based on this, hyaluronic acid (HA), a natural macromolecule with negative charge, was employed to develop hierarchical targeting nanoplatforms because it can be degraded rapidly by hyaluronidase (HAase).[70] HA coated positively charged CPP modified-liposomes (HA-CPP-Ls) were thus developed for drug delivery.[71]. After tumor accumulation of HA-CPP-Ls, the HA coating can be degraded by HAase, leading to the exposure of CPPs and the high cellular uptake of CPP-Ls.
In a recent study, Chan et al. reported a new strategy which exploits long DNA strands as coating materials to achieve dynamic control of targeting function.[72] As shown in Figure 19A, the nanostructure was formed by a large AuNP (orange), a medium AuNP (red) and several small AuNPs (black). Long DNA strands with small AuNPs were bound to short oligonucleotides on the surface of large AuNP, whereas medium AuNP only bear short oligonucleotides. In this morphology (Morphol. 1), the small AuNPs were distributed around the large AuNP as satellites (Figure 19B). If A1, a linker nucleotide, was added, it would anchor small AuNPs to the medium AuNP by DNA hybridization. Then L1comp, a complementary strand to the linker between large AuNP and small AuNPs, was added to detach the small AuNPs from the large AuNP core. As a result, small AuNPs were detached from the large AuNP and bound to the medium AuNP, converting the assemblies into a new morphology (Morphol. 2) in which large AuNP bear short oligonucleotides. These DNA-controlled assemblies can be further used to design controllable targeting nanoplatforms by adding ligands such as folic acid (FA) to the surface of large AuNP core. In Morphol. 1, the FA molecules were covered by the long DNA strands, thus the targeting function was in the “OFF” state. However, in the presence of A1 and L1comp, the morphology of the assemblies was converted into Morphol. 2. The FA molecules were exposed to achieve subsequent enhanced cellular internalization (Figure 19C). As shown in Figure 19D, the cellular uptake efficiency of FA-conjugated assemblies of Morphol. 2 was significantly higher than that of Morphol. 1, indicating DNA-triggered ligand exposure.
Figure 19.

(A) Schematic of the DNA-controlled morphology change of assemblies. (B) TEM images of the assemblies with different morphologies. (C) Schematic of the DNA-controlled cellular internalization of the assemblies. (D) Cellular uptake efficiencies of the assemblies without or with FA modification in different morphologies. Reproduced with permission.[72] Copyright 2016 American Association for the Advancement of Science.
Besides the sheddable coating strategy, the targeting molecules also can be hidden in the inner layer of NPs by stimuli-responsive hydrophobic anchors. The targeting function is re-activated when the hidden targeting molecules are moved out to the surface of particles under certain stimulus. The unbinding of targeting molecules can be realized by two methods: i) using sensitive anchors to achieve stimuli-induced change of water solubility; ii) using sensitive bonds between targeting molecules and anchors to achieve stimuli-induced exposure of targeting molecules. Based on the first method, a pH-responsive polymeric micelle has been developed by self-assembly of a synthesized amphiphilic polymer, in which a short poly(L-histidine) (polyHis) chain was used as a linker between hydrophilic PEG chain and biotin.[73] The polyHis chains are hydrophobic at physiological pH, so the biotin molecules are bound in the hydrophobic core of the polymeric micelles and are shielded by the PEG coating. In the tumor extracellular acidic environment, the polyHis chains become hydrophilic through the protonation of imidazole groups. Thus the completely hydrophilic PEG-polyHis-biotin chains would push the biotin molecules out to the surface of polymeric micelles (Figure 20A). The cellular uptake results (Figure 20B) showed almost no fluorescence in the cells incubated with DOX-loaded micelles at pH 7.4, indicating minimal internalization of the micelles; however, as the pH value was decreased, significantly increased fluorescence intensity was observed. Similar results were also observed when the biotin was replaced by TAT peptide.[74]
Figure 20.

(A) Schematic of the pH-triggered unbunding of biotin. (B) Confocal images of MCF-7 cells incubated with FITC-labeled DOX-loaded micelles at different pH. Reproduced with permission.[73] Copyright 2005 American Chemical Society.
The unbinding of targeting molecules can also be realized by the cleavage of linkers.[75] For example, an UV light responsive ONB group was inserted between hydrophilic PEG and hydrophobic octadecyl ester as a linker to synthesize an ABA-typed amphiphilic polymer C18-PEG-biotin-photo site-C18 (CPB-p-C), which can form a flower-like structure for the surface functionalization of Pluronic P 123 micelles (P123). Biotin is bound by octadecyl ester and hidden by folded PEG chains.[76] Upon UV irradiation triggered cleavage of the ONB groups, the flower-like structure of the functional accessory will be destroyed and thus the biotin molecules will be popped up to the surface of micelles, resulting in receptor-mediated specific targeted delivery (Figure 21A). Two types of mixed micelles, 10p (CPB-p-C/P123 = 1/10, w/w) and 20p (CPB-p-C/P123 = 2/10, w/w), were investigated for the photo-triggered targeting. As shown in Figure 21B, both P123 and 10p showed low cellular uptake efficiency without UV irradiation, suggesting that most biotin molecules were bound in the inner layer. While the 20p exhibited slightly higher cellular uptake even without UV irradiation, likely due to the relatively larger amount of unprotected biotins. Upon UV irradiation, almost no change in cellular uptake efficiency was found in the cells incubated with P123, while enhanced cellular internalization was observed in the cells treated with 10p or 20p (Figure 21C and 21D), which is attributed to the fact that unbound biotins were exposed for active targeting.
Figure 21.

(A) Schematic of the UV light-activated, tumor-targeting drug delivery. (B) Confocal images of HeLa cells treated with P123, 10p and 20p with (UV+) or without (UV−) UV light irradiation. (C) FCM analysis of 10p (left) and 20p (right) with (gray filled) or without (black line) UV light irradiation. (D) Comparison of UV light-activated cell uptake of different samples (*p < 0.05). Reproduced with permission.[76] Copyright 2014 Wiley.
5. Conclusion and Perspective
To improve the therapeutic index and reduce the off-target toxicity, an ideal NP should efficiently accumulate and be retained in the tumor tissue and then specifically bind to tumor cells with enhanced cellular internalization. However, the positively charged or ligand modified NPs often display high adsorption rate with plasma proteins or normal tissues/cells, leading to reduced tumor accumulation efficiency. Neutral or negatively charged NPs modified with hydrophilic ‘stealth’ polymers may efficiently accumulate into tumor tissues by EPR effect, while the subsequent tumor cellular uptake of these NPs remains suboptimal. In order to improve tumor targeted delivery of therapeutic agent, hierarchical targeting, as a novel targeting strategy that consists of two targeting stages including tumor tissue targeting based on EPR effect and tumor cell targeting based on targeting ligands or positive charge, has been extensively studied.
This review briefly summarizes the strategies that are exploited to develop hierarchical targeting nanoplatforms based on stimuli-responsive nanomaterials, including changeable particle size, switchable surface charge and activatable surface ligand. Despite some exciting results obtained at the cellular level, little was done in live animals. Research on hierarchical targeting nanoplatforms is still in its infancy and faces many challenges. For example, to design the surface charge-switchable nanoplatforms, how to design the surface groups to achieve an appropriate pKa value which promises high sensitivity to respond to the different pH values between normal and tumor tissues, is a grand challenge. In general, the surface of NP is expected to be neutral or negatively charged during circulation (physiological pH) and becomes positively charged after arriving at the tumor site (pH around 6.5 at tumor extracellular environment), thus the pKa value in the range of 6.5–7.4 is helpful for tumor targeting. The strategy of using caged targeting molecules represents a promising hierarchical targeting strategy because of the off-on activity between the caged and uncaged ligand forms. However, this caging method usually requires two or more reaction points in one targeting molecule to link both the NP and the caging group. For the strategy that exploits surface coatings, the ratio of targeting molecules and coating materials should be optimized to achieve hierarchical target effect.
For the future research to improve the clinical applicability of the hierarchical targeting nanoplatforms, the following directions should be considered: 1) Design and fabrication of stimuli-responsive nanoplatforms with high sensitivity, which can realize rapid and significant structure changes in response to endogenous or exogenous stimuli. Most of the recent proof-of-principle studies were done at extreme conditions that are not relevant to pathophysiology, and the responses generally require long time (several hours). 2) Development of new strategies or amelioration of existing strategies. For instance, in the studies that involve ligand exposure, most of the stimuli-induced structure changes were only confirmed indirectly. Recently, by labeling with small NPs, the structural change of nanoassemblies in response to stimuli can be directly observed by TEM.[72, 77] This method can be expanded to design more hierarchical targeting nanoplatforms with direct evidences of structure changes. 3) Combination of hierarchical targeting and stimuli-triggered drug release to attain smart drug delivery. The release behaviors of many therapeutic agents should be considered in the design of drug delivery systems because these therapeutic agents can work only at the released state. In the past several years, stimuli-responsive nanomaterials have been employed to prepare drug delivery systems with triggered drug release behaviors.[78] However, NPs that can satisfy both improved targeting and controlled drug release are not common. 4) Assessment of toxicity, biocompability, immunogenicity, pharmacokinetics, and biodistribution of stimuli-responsive nanoplatforms. It should be noted that practical use of the reported hierarchical targeting systems is still farfetching. More systematic evaluations are still needed to justify the clinical translation of hierarchical targeting nanosystems.
Acknowledgments
This work was supported by the National Science Foundation of China (81401465, 51573096), and the Intramural Research Program (IRP) of the NIBIB, NIH.
Biographies

Sheng Wang received his PhD in Materials Science and Engineering from Tianjin University in 2015. He then worked with Prof. Peng Huang and Prof. Xiaoyuan (Shawn) Chen at Shenzhen University and National Institutes of Health (NIH) as a postdoctoral fellow in 2015. Currently, his research is focused on the design and development of stimuli-responsive materials for biomedical applications.

Peng Huang received his PhD in Biomedical Engineering from the Shanghai Jiao Tong University in 2012. Then he joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN) at the National Institutes of Health (NIH) as a postdoctoral fellow under the supervision of Prof. Xiaoyuan (Shawn) Chen. In 2015, he moved to Shenzhen University as a Distinguished Professor. His research is focused on the design, synthesis, and biomedical applications of functional nanomaterials.

Xiaoyuan (Shawn) Chen received his PhD in Chemistry from the University of Idaho in 1999. He was a faculty at the University of Southern California and Stanford University before joining NIH as a Senior Investigator and Chief of the Laboratory of Molecular Imaging and Nanomedicine (LOMIN). His current research interests include development of molecular imaging toolbox for better understanding of biology, early diagnosis of disease, monitoring therapy response, and guiding drug discovery/development. His lab puts special emphasis on high-sensitivity nanosensors for biomarker detection and theranostic nanomedicine for imaging, gene and drug delivery, and monitoring of treatment.
Contributor Information
Dr. Sheng Wang, Guangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, Department of Biomedical Engineering, School of Medicine, Shenzhen University, Shenzhen 518060, China. Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland 20892, United States
Prof. Peng Huang, Email: peng.huang@szu.edu.cn, Guangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, Department of Biomedical Engineering, School of Medicine, Shenzhen University, Shenzhen 518060, China
Prof. Xiaoyuan Chen, Email: shawn.chen@nih.gov, Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland 20892, United States
References
- 1.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338:903. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petros RA, DeSimone JM. Nat Rev Drug Discovery. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387. [PubMed] [Google Scholar]
- 5.a) Sherlock SP, Tabakman SM, Xie L, Dai H. ACS Nano. 2011;5:1505. doi: 10.1021/nn103415x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Q, Liang C, Wang C, Liu Z. Adv Mater. 2015;27:903. doi: 10.1002/adma.201404308. [DOI] [PubMed] [Google Scholar]; c) Wang W, Cheng D, Gong F, Miao X, Shuai X. Adv Mater. 2012;24:115. doi: 10.1002/adma.201104066. [DOI] [PubMed] [Google Scholar]
- 6.a) Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB. Nat Mater. 2011;10:389. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang X, Grailer JJ, Rowland IJ, Javadi A, Hurley SA, Matson VZ, Steeber DA, Gong S. ACS Nano. 2010;4:6805. doi: 10.1021/nn101670k. [DOI] [PubMed] [Google Scholar]; c) Chow EKH, Ho D. Sci Transl Med. 2013;5:rv4. doi: 10.1126/scitranslmed.3005872. [DOI] [PubMed] [Google Scholar]
- 7.a) Ernsting MJ, Murakami M, Roy A, Li SD. J Control Release. 2013;172:782. doi: 10.1016/j.jconrel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McNeeley KM, Annapragada A, Bellamkonda RV. Nanotechnology. 2007;18:385101. [Google Scholar]
- 8.a) Lankveld DP, Rayavarapu RG, Krystek P, Oomen AG, Verharen HW, Van Leeuwen TG, De Jong WH, Manohar S. Nanomedicine. 2011;6:339. doi: 10.2217/nnm.10.122. [DOI] [PubMed] [Google Scholar]; b) Gullotti E, Yeo Y. Mol Pharm. 2009;6:1041. doi: 10.1021/mp900090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci U S A. 2008;105:11613. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Small. 2011;7:1322. doi: 10.1002/smll.201100001. [DOI] [PubMed] [Google Scholar]; c) Wang S, Zhang L, Dong C, Su L, Wang H, Chang J. Chem Commun. 2015;51:406. doi: 10.1039/c4cc08178a. [DOI] [PubMed] [Google Scholar]
- 10.a) Wang Y, Shim MS, Levinson NS, Sung HW, Xia Y. Adv Funct Mater. 2014;24:4206. doi: 10.1002/adfm.201400279. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blum AP, Kammeyer JK, Rush AM, Callmann CE, Hahn ME, Gianneschi NC. J Am Chem Soc. 2015;137:2140. doi: 10.1021/ja510147n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He C, Zhuang X, Tang Z, Tian H, Chen X. Adv Healthcare Mater. 2012;1:48. doi: 10.1002/adhm.201100008. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Yang W, Cui J, Li X, Dou Y, Su L, Chang J, Wang H, Zhang B. Biomater Sci. 2016;4:338. doi: 10.1039/c5bm00328h. [DOI] [PubMed] [Google Scholar]
- 12.Shim MS, Bhang SH, Yoon K, Choi K, Xia Y. Angew Chem Int Ed. 2012;124:12069. doi: 10.1002/anie.201206595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang P, Gao Y, Lin J, Hu H, Liao H-S, Yan X, Tang Y, Jin A, Song J, Niu G, Zhang G, Horkay F, Chen X. ACS Nano. 2015;9:9517. doi: 10.1021/acsnano.5b03874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan Q, Zhang Y, Chen T, Lu D, Zhao Z, Zhang X, Li Z, Yan C-H, Tan W. ACS Nano. 2012;6:6337. doi: 10.1021/nn3018365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Chen KJ, Chaung EY, Wey SP, Lin KJ, Cheng F, Lin CC, Liu HL, Tseng HW, Liu CP, Wei MC. ACS Nano. 2014;8:5105. doi: 10.1021/nn501162x. [DOI] [PubMed] [Google Scholar]; b) Chen KJ, Liang HF, Chen HL, Wang Y, Cheng PY, Liu HL, Xia Y, Sung HW. ACS Nano. 2013;7:438. doi: 10.1021/nn304474j. [DOI] [PubMed] [Google Scholar]
- 16.Paris JL, Cabañas MV, Manzano M, Vallet-Regí M. ACS Nano. 2015;9:11023. doi: 10.1021/acsnano.5b04378. [DOI] [PubMed] [Google Scholar]
- 17.a) Thomas CR, Ferris DP, Lee JH, Choi E, Cho MH, Kim ES, Stoddart JF, Shin JS, Cheon J, Zink JI. J Am Chem Soc. 2010;132:10623. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]; b) Ruiz-Hernandez E, Baeza A, Vallet-Regí Ma. ACS Nano. 2011;5:1259. doi: 10.1021/nn1029229. [DOI] [PubMed] [Google Scholar]
- 18.a) Wang J, Byrne JD, Napier ME, DeSimone JM. Small. 2011;7:1919. doi: 10.1002/smll.201100442. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Danhier F, Feron O, Preat V. J Control Release. 2010;148:135. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 19.a) Tong R, Chiang HH, Kohane DS. Proc Natl Acad Sci U S A. 2013;110:19048. doi: 10.1073/pnas.1315336110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tang L, Yang X, Yin Q, Cai K, Wang H, Chaudhury I, Yao C, Zhou Q, Kwon M, Hartman JA, Dobrucki IT, Dobrucki LW, Borst LB, Lezmi S, Helferich WG, Ferguson AL, Fan TM, Cheng J. Proc Natl Acad Sci U S A. 2014;111:15344. doi: 10.1073/pnas.1411499111. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huo S, Ma H, Huang K, Liu J, Wei T, Jin S, Zhang J, He S, Liang XJ. Cancer Res. 2013;73:319. doi: 10.1158/0008-5472.CAN-12-2071. [DOI] [PubMed] [Google Scholar]
- 20.Larsen EK, Nielsen T, Wittenborn T, Birkedal H, Vorup-Jensen T, Jakobsen MH, Ostergaard L, Horsman MR, Besenbacher F, Howard KA, Kjems J. ACS Nano. 2009;3:1947. doi: 10.1021/nn900330m. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Chen Y, Li H, Huang N, Jin Q, Ren K, Ji J. ACS Nano. 2013;7:6244. doi: 10.1021/nn402201w. [DOI] [PubMed] [Google Scholar]
- 22.Cheng H, Zhu J-Y, Xu X-D, Qiu W-X, Lei Q, Han K, Cheng Y-J, Zhang X-Z. ACS Appl Mater Interfaces. 2015;7:16061. doi: 10.1021/acsami.5b04517. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira LM. Exp Mol Pathol. 2010;89:372. doi: 10.1016/j.yexmp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 24.a) Wu W, Zhang Q, Wang J, Chen M, Li S, Lin Z, Li J. Polym Chem. 2014;5:5668. [Google Scholar]; b) Wang Y, Du J, Jin Q, Ji J. Chem Commun. 2015;51:2999. doi: 10.1039/c4cc09274k. [DOI] [PubMed] [Google Scholar]; c) Li H, Liu X, Huang N, Ren K, Jin Q, Ji J. ACS Appl Mater Interfaces. 2014;6:18930. doi: 10.1021/am504813f. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Hu Q, Lin Y, Pacardo DB, Wang C, Sun W, Ligler FS, Dickey MD, Gu Z. Nat Commun. 2015;6:10066. doi: 10.1038/ncomms10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li HJ, Du JZ, Du XJ, Xu CF, Sun CY, Wang HX, Cao ZT, Yang XZ, Zhu YH, Nie S, Wang J. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1522080113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Tkachenko AG, Xie H, Coleman D, Glomm W, Ryan J, Anderson MF, Franzen S, Feldheim DL. J Am Chem Soc. 2003;125:4700. doi: 10.1021/ja0296935. [DOI] [PubMed] [Google Scholar]; b) Pan L, He Q, Liu J, Chen Y, Ma M, Zhang L, Shi J. J Am Chem Soc. 2012;134:5722. doi: 10.1021/ja211035w. [DOI] [PubMed] [Google Scholar]
- 28.Qiu L, Chen T, Ocsoy I, Yasun E, Wu C, Zhu G, You M, Han D, Jiang J, Yu R, Tan W. Nano Lett. 2015;15:457. doi: 10.1021/nl503777s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Nomura T, Koreeda N, Yamashita F, Takakura Y, Hashida M. Pharm Res. 1998;15:128. doi: 10.1023/a:1011921324952. [DOI] [PubMed] [Google Scholar]; b) Lieleg O, Baumgärtel RM, Bausch AR. Biophys J. 2009;97:1569. doi: 10.1016/j.bpj.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crayton SH, Tsourkas A. ACS Nano. 2011;5:9592. doi: 10.1021/nn202863x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong Y, Yang J, Liu H, Wang T, Tang S, Zhang J, Zhang X. Theranostics. 2015;5:890. doi: 10.7150/thno.11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Wang C, Xu C, Yang C, Zhang Z, Yan H, Liu K. Chem Commun. 2013;49:7286. doi: 10.1039/c3cc43334j. [DOI] [PubMed] [Google Scholar]
- 33.Hung C-C, Huang W-C, Lin Y-W, Yu T-W, Chen H-H, Lin S-C, Chiang W-H, Chiu H-C. Theranostics. 2016;6:302. doi: 10.7150/thno.13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Xiao H, Fang J, Yu X, Su Z, Cheng D, Shuai X. Chem Commun. 2016;52:1194. doi: 10.1039/c5cc09181k. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Yu X, Wang Y, Yuan Y, Xiao H, Cheng D, Shuai X. Adv Mater. 2014;26:8217. doi: 10.1002/adma.201403877. [DOI] [PubMed] [Google Scholar]
- 36.a) Du JZ, Du XJ, Mao CQ, Wang J. J Am Chem Soc. 2011;133:17560. doi: 10.1021/ja207150n. [DOI] [PubMed] [Google Scholar]; b) Du JZ, Sun TM, Song WJ, Wu J, Wang J. Angew Chem Int Ed. 2010;122:3703. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 37.a) Han SS, Li ZY, Zhu JY, Han K, Zeng ZY, Hong W, Li WX, Jia HZ, Liu Y, Zhuo RX, Zhang XZ. Small. 2015;11:2543. doi: 10.1002/smll.201402865. [DOI] [PubMed] [Google Scholar]; b) Dong C, Liu Z, Zhang L, Guo W, Li X, Liu J, Wang H, Chang J. ACS Appl Mater Interfaces. 2015;7:7566. doi: 10.1021/am509011y. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Tian B, Li Y, Lei T, Meng J, Yang L, Zhang Y, Chen F, Zhang H, Xu H, Zhang Y, Tang X. ACS Appl Mater Interfaces. 2015;7:25147. doi: 10.1021/acsami.5b06488. [DOI] [PubMed] [Google Scholar]
- 39.Yang X-Z, Du J-Z, Dou S, Mao C-Q, Long H-Y, Wang J. ACS Nano. 2011;6:771. doi: 10.1021/nn204240b. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Xue H, Gao C, Carr L, Wang J, Chu B, Jiang S. Biomaterials. 2010;31:6582. doi: 10.1016/j.biomaterials.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 41.Jin Q, Xu J-P, Ji J, Shen J-C. Chem Commun. 2008:3058. doi: 10.1039/b801959b. [DOI] [PubMed] [Google Scholar]
- 42.Mizuhara T, Saha K, Moyano DF, Kim CS, Yan B, Kim Y-K, Rotello VM. Angew Chem Int Ed. 2015;54:6567. doi: 10.1002/anie.201411615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arvizo RR, Miranda OR, Thompson MA, Pabelick CM, Bhattacharya R, Robertson JD, Rotello VM, Prakash Y, Mukherjee P. Nano Lett. 2010;10:2543. doi: 10.1021/nl101140t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pillai PP, Huda S, Kowalczyk B, Grzybowski BA. J Am Chem Soc. 2013;135:6392. doi: 10.1021/ja4001272. [DOI] [PubMed] [Google Scholar]
- 45.Wang S, Teng Z, Huang P, Liu D, Liu Y, Tian Y, Sun J, Li Y, Ju H, Chen X, Lu G. Small. 2015;11:1801. doi: 10.1002/smll.201403248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a) Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. Clin Cancer Res. 2003;9:6551. [PubMed] [Google Scholar]; b) Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Anal Biochem. 2005;338:284. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 47.a) Cui L, Zhang F, Wang Q, Lin H, Yang C, Zhang T, Tong R, An N, Qu F. J Mater Chem B. 2015;3:7046. doi: 10.1039/c5tb00709g. [DOI] [PubMed] [Google Scholar]; b) Alonso-Cristobal P, Oton-Fernandez O, Mendez-Gonzalez D, Fernando Diaz J, Lopez-Cabarcos E, Barasoain I, Rubio-Retama J. ACS Appl Mater Interfaces. 2015;7:14992. doi: 10.1021/acsami.5b03881. [DOI] [PubMed] [Google Scholar]; c) Li S, Ji S, Zhou Z, Chen G, Li Q. Macromol Chem Phys. 2015;216:1192. [Google Scholar]
- 48.Dvir T, Banghart MR, Timko BP, Langer R, Kohane DS. Nano Lett. 2010;10:250. doi: 10.1021/nl903411s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan N-C, Cheng F-Y, Ho J-aA, Yeh C-S. Angew Chem Int Ed. 2012;51:8806. doi: 10.1002/anie.201203339. [DOI] [PubMed] [Google Scholar]
- 50.Shamay Y, Adar L, Ashkenasy G, David A. Biomaterials. 2011;32:1377. doi: 10.1016/j.biomaterials.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 51.a) Li Z, Zhang Y, Jiang S. Adv Mater. 2008;20:4765. [Google Scholar]; b) Chien YH, Chou YL, Wang SW, Hung ST, Liau MC, Chao YJ, Su CH, Yeh CS. ACS Nano. 2013;7:8516. doi: 10.1021/nn402399m. [DOI] [PubMed] [Google Scholar]; c) Jayakumar MKG, Idris NM, Zhang Y. Proc Natl Acad Sci U S A. 2012;109:8483. doi: 10.1073/pnas.1114551109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.a) Zhu G, Niu G, Chen X. Bioconjugate Chem. 2015;26:2186. doi: 10.1021/acs.bioconjchem.5b00291. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang R, Zhu G, Mei L, Xie Y, Ma H, Ye M, Qing FL, Tan W. J Am Chem Soc. 2014;136:2731. doi: 10.1021/ja4117395. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Cancer Res. 2004;64:7668. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Zhu G, You M, Song E, Shukoor MI, Zhang K, Altman MB, Chen Y, Zhu Z, Huang CZ, Tan W. ACS Nano. 2012;6:5070. doi: 10.1021/nn300694v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y, Liu J, Sun X, Feng L, Zhu W, Liu Z, Chen M. Nano Research. 2016;9:139. [Google Scholar]
- 55.a) Liu Z, Xiong M, Gong J, Zhang Y, Bai N, Luo Y, Li L, Wei Y, Liu Y, Tan X, Xiang R. Nat Commun. 2014;5:4280. doi: 10.1038/ncomms5280. [DOI] [PubMed] [Google Scholar]; b) Gao H, Zhang S, Cao S, Yang Z, Pang Z, Jiang X. Mol Pharm. 2014;11:2755. doi: 10.1021/mp500113p. [DOI] [PubMed] [Google Scholar]; c) Xia H, Gu G, Hu Q, Liu Z, Jiang M, Kang T, Miao D, Song Q, Yao L, Tu Y, Chen H, Gao X, Chen J. Bioconjugate Chem. 2013;24:419. doi: 10.1021/bc300520t. [DOI] [PubMed] [Google Scholar]
- 56.Bode SA, Hansen MB, Oerlemans RAJF, van Hest JCM, Lowik DWPM. Bioconjugate Chem. 2015;26:850. doi: 10.1021/acs.bioconjchem.5b00066. [DOI] [PubMed] [Google Scholar]
- 57.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Proc Natl Acad Sci U S A. 2010;107:4311. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Ahmady ZS, Al-Jamal WT, Bossche JV, Bui TT, Drake AF, Mason AJ, Kostarelos K. ACS Nano. 2012;6:9335. doi: 10.1021/nn302148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.a) Reshetnyak YK, Andreev OA, Segala M, Markin VS, Engelman DM. Proc Natl Acad Sci U S A. 2008;105:15340. doi: 10.1073/pnas.0804746105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reshetnyak YK, Yao L, Zheng S, Kuznetsov S, Engelman DM, Andreev OA. Mol Imaging Biol. 2011;13:1146. doi: 10.1007/s11307-010-0457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Andreev OA, Karabadzhak AG, Weerakkody D, Andreev GO, Engelman DM, Reshetnyak YK. Proc Natl Acad Sci U S A. 2010;107:4081. doi: 10.1073/pnas.0914330107. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Onyango JO, Chung MS, Eng CH, Klees LM, Langenbacher R, Yao L, An M. Angew Chem Int Ed. 2015;54:3658. doi: 10.1002/anie.201409770. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Schach DK, Rock W, Franz J, Bonn M, Parekh SH, Weidner T. J Am Chem Soc. 2015;137:12199. doi: 10.1021/jacs.5b06720. [DOI] [PubMed] [Google Scholar]
- 60.Davies A, Lewis DJ, Watson SP, Thomas SG, Pikramenou Z. Proc Natl Acad Sci U S A. 2012;109:1862. doi: 10.1073/pnas.1112132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu M, Guo F, Wang J, Tan F, Li N. ACS Appl Mater Interfaces. 2015;7:17592. doi: 10.1021/acsami.5b05763. [DOI] [PubMed] [Google Scholar]
- 62.Koren E, Apte A, Sawant RR, Grunwald J, Torchilin VP. Drug Delivery. 2011;18:377. doi: 10.3109/10717544.2011.567310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang S, Wang H, Liu Z, Wang L, Wang X, Su L, Chang J. Nanoscale. 2014;6:7635. doi: 10.1039/c4nr00843j. [DOI] [PubMed] [Google Scholar]
- 64.a) Zhu L, Kate P, Torchilin VP. ACS Nano. 2012;6:3491. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koren E, Apte A, Jani A, Torchilin VP. J Control Release. 2012;160:264. doi: 10.1016/j.jconrel.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao D, Jia H-Z, Zhang J, Liu C-W, Zhuo R-X, Zhang X-Z. Small. 2014;10:591. doi: 10.1002/smll.201301926. [DOI] [PubMed] [Google Scholar]
- 66.Ding M, Li J, He X, Song N, Tan H, Zhang Y, Zhou L, Gu Q, Deng H, Fu Q. Adv Mater. 2012;24:3639. doi: 10.1002/adma.201200954. [DOI] [PubMed] [Google Scholar]
- 67.Wang S, Zhang S, Liu J, Liu Z, Su L, Wang H, Chang J. ACS Appl Mater Interfaces. 2014;6:10706. doi: 10.1021/am502579e. [DOI] [PubMed] [Google Scholar]
- 68.Sun CY, Shen S, Xu CF, Li HJ, Liu Y, Cao ZT, Yang XZ, Xia JX, Wang J. J Am Chem Soc. 2015;137:15217. doi: 10.1021/jacs.5b09602. [DOI] [PubMed] [Google Scholar]
- 69.a) Stern R, Jedrzejas MJ. Chem Rev. 2006;106:818. doi: 10.1021/cr050247k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mo R, Jiang T, DiSanto R, Tai W, Gu Z. Nat Commun. 2014;5:3364. doi: 10.1038/ncomms4364. [DOI] [PubMed] [Google Scholar]; c) Stern R. Semin Cancer Biol. 2008;18:275. doi: 10.1016/j.semcancer.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 70.Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Adv Funct Mater. 2014;24:2295. [Google Scholar]
- 71.Jiang T, Zhang Z, Zhang Y, Lv H, Zhou J, Li C, Hou L, Zhang Q. Biomaterials. 2012;33:9246. doi: 10.1016/j.biomaterials.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 72.Ohta S, Glancy D, Chan WC. Science. 2016;351:841. doi: 10.1126/science.aad4925. [DOI] [PubMed] [Google Scholar]
- 73.Lee ES, Na K, Bae YH. Nano Lett. 2005;5:325. doi: 10.1021/nl0479987. [DOI] [PubMed] [Google Scholar]
- 74.Lee ES, Gao Z, Kim D, Park K, Kwon IC, Bae YH. J Control Release. 2008;129:228. doi: 10.1016/j.jconrel.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.a) Yuan Z, Que Z, Cheng S, Zhuo R, Li F. Chem Commun. 2012;48:8129. doi: 10.1039/c2cc34225a. [DOI] [PubMed] [Google Scholar]; b) Wang W, Liu Q, Zhan C, Barhoumi A, Yang T, Wylie RG, Armstrong PA, Kohane DS. Nano Lett. 2015;15:6332. doi: 10.1021/acs.nanolett.5b01325. [DOI] [PubMed] [Google Scholar]
- 76.Yuan Z, Zhao D, Yi X, Zhuo R, Li F. Adv Funct Mater. 2014;24:1799. [Google Scholar]
- 77.Parak WJ. Science. 2016;351:814. doi: 10.1126/science.aaf0751. [DOI] [PubMed] [Google Scholar]
- 78.Mura S, Nicolas J, Couvreur P. Nat Mater. 2013;12:991. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 79.a) Gao Z, Lee D, Kim D, Bae Y. J Drug Targeting. 2005;13:391. doi: 10.1080/10611860500376741. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee ES, Na K, Bae YH. J Control Release. 2003;91:103. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 80.a) Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, Li Y. ACS Nano. 2013;7:5858. doi: 10.1021/nn4010796. [DOI] [PubMed] [Google Scholar]; b) Xiong XB, Lavasanifar A. ACS Nano. 2011;5:5202. doi: 10.1021/nn2013707. [DOI] [PubMed] [Google Scholar]; c) Cui J, Yan Y, Wang Y, Caruso F. Adv Funct Mater. 2012;22:4718. [Google Scholar]; d) Zhu L, Wang D, Wei X, Zhu X, Li J, Tu C, Su Y, Wu J, Zhu B, Yan D. J Control Release. 2013;169:228. doi: 10.1016/j.jconrel.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 81.a) Rim HP, Min KH, Lee HJ, Jeong SY, Lee SC. Angew Chem Int Ed. 2011;50:8853. doi: 10.1002/anie.201101536. [DOI] [PubMed] [Google Scholar]; b) Li J, Chen YC, Tseng YC, Mozumdar S, Huang L. J Control Release. 2010;142:416. doi: 10.1016/j.jconrel.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li J, Yang Y, Huang L. J Control Release. 2011 [Google Scholar]; d) Ke CJ, Su TY, Chen HL, Liu HL, Chiang WL, Chu PC, Xia Y, Sung HW. Angew Chem Int Ed. 2011;123:8236. doi: 10.1002/anie.201102852. [DOI] [PubMed] [Google Scholar]
- 82.a) Meng F, Hennink WE, Zhong Z. Biomaterials. 2009;30:2180. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]; b) Wu L, Zou Y, Deng C, Cheng R, Meng F, Zhong Z. Biomaterials. 2013;34:5262. doi: 10.1016/j.biomaterials.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 83.Shim MS, Xia Y. Angew Chem Int Ed. 2013;52:6926. doi: 10.1002/anie.201209633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao Z, Ji C, Shi J, Pridgen EM, Frieder J, Wu J, Farokhzad OC. Angew Chem Int Ed. 2012;51:11853. doi: 10.1002/anie.201204018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam K-H. J Control Release. 2009;138:268. doi: 10.1016/j.jconrel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan Q, Yuan J, Cai Z, Xin Y, Kang Y, Yin Y. J Am Chem Soc. 2010;132:9268. doi: 10.1021/ja1027502. [DOI] [PubMed] [Google Scholar]