

Abstract
Background
Eosinophilic esophagitis is an allergic disease of increasing worldwide incidence. Complications are due to tissue remodeling and involve transforming growth factor-β1 (TGFβ1) mediated fibrosis. Plasminogen Activator Inhibitor-1 (PAI-1/serpinE1) can be induced by TGFβ1 but its role in EoE is not known.
Objective
To understand the expression and role of PAI-1 in EoE.
Methods
We used esophageal biopsy specimens and plasma samples from control and EoE subjects, primary human esophageal epithelial cells, and EoE fibroblasts in immunohistochemistry, quantitative PCR, and immunoassay experiments to understand the induction of PAI-1 by TGFβ1, the relationship between PAI-1 and esophageal fibrosis, and the role of PAI-1 in fibrotic gene expression.
Results
PAI-1 expression was significantly elevated in epithelial cells of active EoE biopsies as compared with inactive EoE or control biopsies (p<0.001). Treatment of primary esophageal epithelial cells with recombinant TGFβ1 increased PAI-1 transcription, intracellular protein expression, and secretion. Esophageal PAI-1 expression correlated with basal zone hyperplasia, a fibrosis, and markers of esophageal remodeling including vimentin, TGFβ1, collagen I, fibronectin, and matrix metalloproteases and plasma PAI-1 levels correlated with plasma TGFβ1. PAI-1 inhibition significantly decreased baseline and TGFβ1-induced fibrotic gene expression.
Conclusions
PAI-1 is significantly elevated in the EoE epithelium, reflects fibrosis, and its inhibition decreases TGFβ1-induced gene expression. Epithelial PAI-1 may serve as a marker of EoE severity and form part of a TGFβ1-induced pro-fibrotic network.
Keywords: Eosinophil, Esophagitis, SerpinE1, Remodeling, Fibrosis, Transforming growth factor beta-1
Introduction
Eosinophilic esophagitis (EoE) is a chronic antigen driven allergic disease of increasing worldwide incidence and prevalence (1). One mechanism for disease complications is fibrosis which occurs largely in the subepithelium, begins early in life, and is progressive without intervention (2–8). EoE esophageal remodeling includes epithelial changes of basal zone hyperplasia (BZH) and dilated intercellular spaces and subepithelial fibrosis and angiogenesis in the lamina propria (LP). Fibrosis is variably responsive to anti-inflammatory interventions (8–14). Since the subepithelial tissue is not always present for histologic evaluation, the presence and response of remodeling changes to intervention can be difficult to gauge. Without treatment EoE patients suffer from vomiting, failure to thrive, dysphagia, food impactions and consistently progress to esophageal strictures (1, 3, 4).
Studies from our laboratory and others have demonstrated that TGFβ1 can be a major regulator of childhood EoE remodeling (5, 15–20). A number of genes that are involved in changes in cellular function and trafficking are induced by TGFβ1 including periostin which increases eosinophil trafficking (18) and phospholamban which induces TGFβ1-mediated esophageal smooth muscle cell contraction in culture (17). Among the genes that are induced by TGFβ1 is the serine protease inhibitor, plasminogen activator inhibitor-1 (PAI-1/serpinE1) which promotes tissue fibrosis in asthma (21–24). However, the role of PAI-1, its induction in EoE, or its role in esophageal fibrosis has not been previously studied.
PAI-1 is elevated in human asthma and promotes airway remodeling in murine asthma models (21, 23–28). PAI-1 overexpression, triggered by viruses and allergens, is associated with airway fibrosis while PAI-1 deficiency protects mice from fibrotic airway remodeling (21, 23–26, 29). Mast cell derived TGFβ1 increases PAI-1 expression from immortalized airway epithelial cells in culture (22). In addition, a single nucleotide polymorphism 4G in the PAI-1 gene associates with human asthma risk (27, 28). These results define a role for PAI-1 expression in airway remodeling but the induction of PAI-1 in EoE and its role in esophageal fibrosis is not known.
Based on these findings and our prior work in EoE that shows that TGFβ1 promotes EoE associated remodeling, we explored the role of PAI-1 in EoE. Since EoE is associated with substantial tissue fibrosis that can lead to esophageal rigidity, food impactions, and strictures, determining the mechanisms and new potential targets for controlling remodeling is an issue of clinical interest. Herein we demonstrate that PAI-1 levels are increased in the active EoE esophagus as compared with inactive EoE, athat TGFβ1 significantly induces PAI-1 in esophageal epithelial cells, and that pharmacologic inhibition of PAI-1 decreases TGFβ1-induced gene expression. Our discovery that PAI-1 is part of a fibrotic network induced by TGFβ1 indicates that PAI-1 could be a new target in the therapy of esophageal remodeling.
Methods
EoE subjects/biopsies
We utilized our UCSD/RCHSD EoE database to select a cohort of EoE subjects who had active and inactive EoE defined as ≥15 eosinophils and <15 eosinophils per hpf, respectively. Inactive EoE was a consequence of standard of care EoE directed therapy of topical fluticasone or budesonide. Control subjects had no eosinophils in the esophagus and no endoscopic abnormalities. Archived biopsies were used for immunohistochemistry (IHC) and immunofluorescence (IF) and frozen biopsies were used for qPCR and were collected in RNAlater and kept at −80°C until use. Plasma samples were obtained at the time of EGD with biopsy. Written informed consent/assent was obtained for all samples under UCSD/RCHSD IRB approved protocol 091485. Procurement and use of epithelial cells from deceased organ donors was not considered human subjects research and approved under UCSD IRB protocol 130835. Clinical features of subjects whose biopsies were used in immunohistology experiments are shown Table E1.
Immunostaining and histologic assessment
H&E stained, formalin fixed, paraffin embedded specimens were scored by a single pathologist blinded to the diagnosis and treatment (R.N.) using our previously published standardized histology scoring tool (30). The numbers of epithelial and lamina propria (LP) eosinophils, the severity of basal zone hyperplasia (BZH), and the LP fibrosis score were quantified (30). Fibrosis severity was scored 0–3 based on the collagen bundle thickness, collagen accumulation, and number of fibroblasts. BZH was graded on the percent of the epithelial height that was comprised of basal cells from 0–3 (30). Subjects whose biopsies were studied were chosen from the database using a random number generator and used if there was well oriented epithelium and the presence of adequate LP for analysis (active EoE subjects).
Tissue sections (5μM) were deparaffanized and hydrated prior to immunostaining as previously described (5). Following antigen retrieval, slides were incubated with anti-PAI-1 (1:200 AbCam ab66705, Cambridge, MA) alone or in combination with anti-Ki67 (1:100, DAKO, Carpinteria, CA) or anti-cytokeratin 14 (1:400, AbCam ab7800, Cambridge MA). Samples were processed for IF or IHC using the appropriate species-specific secondary antibodies as previously described (5). Isotype control antibodies were used to ensure staining specificity. Epithelial PAI-1 was quantified using color detection and image analysis with Image J software for the height of positively staining cells relative to the total epithelial height (the same index utilized for calculating BZH). All images were quantified and analyzed under identical light or fluorescence microscopic conditions, including magnification, gain, camera position, and background illumination.
Cell culture and treatment
Human organ transplant donor esophagi provided by the National Disease Research Interchange and the Arkansas Regional Organ Recovery Agency were processed for epithelial cells using collagenase D to digest isolated mucosa and cultured initially in DMEM/F12 with 10% fetal bovine serum and later in complete EpiCM2 media (ScienCell, Carlsbad CA). Cells were placed in growth factor reduced media overnight and stimulated with recombinant human TGFβ1 (10ng/ml, R&D, Minneapolis MN) in basal media for 48 hours prior to collection of RNA, protein, and supernatants. EoE fibroblasts were isolated using collagenase I, cultured in SMCM (ScienCell, Carlsbad CA) pre-treated with 75μM TM5275 for 2 hours followed by 24 hours of TGFβ1 (5ng/ml, R&D, Minneapolis MN).
Quantitative PCR
RNA was isolated from human esophageal epithelial cells or frozen EoE/control biopsy specimens stored in RNAlater, converted to cDNA using the manufacturer’s instructions, and subjected to quantitative real time (RT qPCR) using SYBR green and normalized to the housekeeping gene RPL13A. All dissociation curves were single peak. Primer sequences are listed in supplemental Table E21.
ELISAs and Immunoblots
ELISAs for PAI-1, TGFβ1, and PF4 were completed using the manufacturer’s instructions (DY1786, DY240, and DY795, R&D, Minneapolis MN) and cell supernatants or plasma samples. Plasma was collected in citrate plasma separator tubes and platelets were depleted using two centrifugation spins at 1800×g for 30 and 10 minutes. PAI-1 and TGFβ1 plasma levels were normalized for platelet activation using PF4 ELISA. For immunoblotting, cells were harvested on ice using RIPA lysis buffer (Teknova, Hollister, CA) with phosphatase and protease inhibitors (Roche Applied Science, Indianapolis, IN). Equivalent amounts of total protein was loaded into each well and electrophoresed on NuPAGE 4–12% Bis-Tris gels (Life Technologies, Grand Island, NY). Proteins were transferred to 0.2 um pore nitrocellulose membrane (Life Technologies, Grand Island, NY), blocked with 5% dry milk, incubated overnight with PAI-1 or Gap antibody and probed with the appropriate species specific HRP-linked secondary antibody. Immunoblots were developed with Immuno-Star WesternC Kit (BioRad, Hercules, CA), imaged, and analyzed using the GBOX XRQ and its accompanying software (Syngene, Frederick, MD).
Statistical analysis
All statistical analyses and graphing were done using GraphPad Prism (San Diego, CA). Comparisons between two groups with Gaussian distribution were done using a student’s t-test for unpaired variables; non-Gaussian data/non-continuous variables were compared using a Mann-Whitney test. Pre-post comparisons were done using a t-test for paired variables for Gaussian data/continuous variables or Wilcoxon’s test for non-parametric paired data. Spearman’s coefficient was calculated for correlations. A two tailed p value <0.05 was considered statistically significant.
Results
PAI-1 expression in the esophagus of EoE subjects
We utilized 14 paired archived biopsies to analyze PAI-1 expression in the active versus inactive state. Twenty-seven additional active EoE biopsies and 11 control biopsies were used for comparison of active to non-diseased state. In the total group, the subjects had a mean age of 7 years (range 1–17 years), 831% were male, and 40% of subjects failed PPI monotherapy. Forty-eight percent had self-reported asthma, 69% had allergic rhinitis and 48% had food allergies. Sixty-three and 73% of subjects had IgE testing that was positive to foods and aeroallergens, respectively. Among the paired treatment group, 13 were treated with swallowed topical swallowed budesonide and 1 was treated with topical fluticasone.
As compared with non-diseased control subjects, patients with active EoE had significantly more PAI-1 expression in the epithelium (79% versus 14% of epithelial height) (Figure 1a,b,d). In order to understand if esophageal PAI-1 expression was decreased following EoE-directed therapy, we utilized paired, well oriented biopsy specimens. Following therapy, PAI-1 expression was significantly decreased to 24% of the epithelial height (p<0.001) (Figure 1b,c,e). Since epithelial height can vary between biopsies, we utilized a second methodology to confirm that PAI-1 expression was higher in active as compared with inactive or control biopsies. We assessed the numbers of highly positive PAI-1 epithelial cells per high power field and found that this method also demonstrated that active EoE biopsies had more PAI-1 than control or inactive biopsies (see Supplemental Material and Figure E1).
Figure 1.
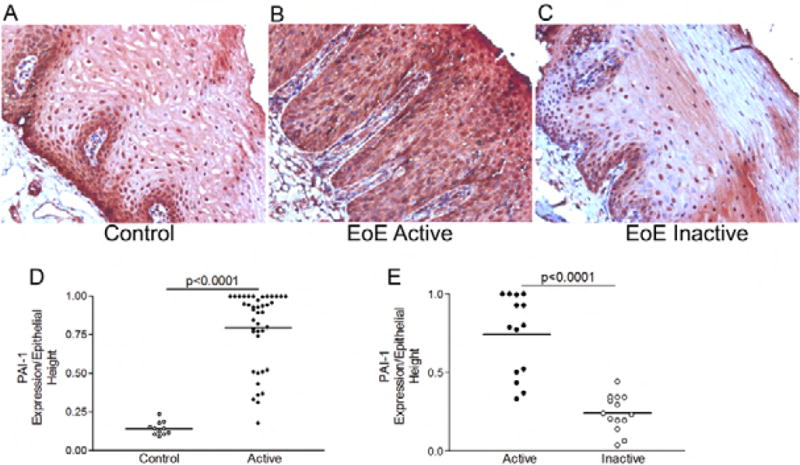
PAI-1 is expressed in the EoE esophageal epithelium. Representative image of PAI-1 immunohistochemistry on esophageal biopsy specimens from a non-diseased control (A), active EoE (B), and inactive EoE (C). Epithelial staining was quantified as a percent of the total epithelial height in control and active EoE (D) and in paired inactive and active EoE biopsies (E). Lines represent means.
PAI-1 expression in the EoE esophagus and association with epithelial features
To assess if actively proliferating epithelial cells, expressed PAI-1, we performed double immunofluorescence confocal imaging studies using the basal zone marker, cytokeratin 14, and the proliferation marker, Ki67 (Figure 2a,b). Quantification of double positive cells demonstrated that both cytokeratin 14 positive basal cells and actively proliferating cells expressed PAI-1 (Figure 2c,d). Quantification demonstrated significantly more Ki67/PAI-1 and cytokeratin 14/PAI-1 double positive cells in the basal than the non-basal zone in the active disease state (Figure 2c,d) as well as more PAI-1/Ki67 double positive cells in the basal zone of active versus inactive subjects (Figure 2d).
Figure 2.
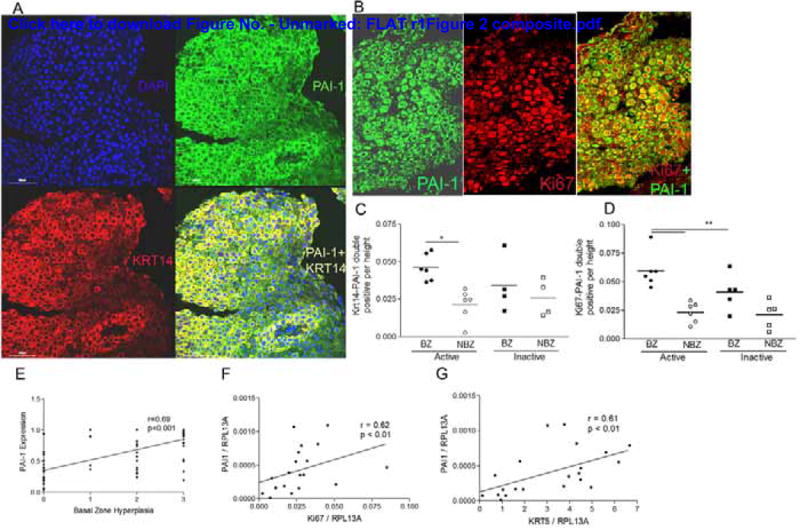
Actively proliferating cells express PAI-1. Representative images of double immunofluorescence for PAI-1 (green, A, B) and cytokeratin 14 (Krt14) (red, A) or nuclear Ki67 (red, B) shows that PAI-1 co-localizes with cytokeratin 14 (yellow, A) and Ki67 positive cells (green cytoplasm, red nucleus, B). Quantification of cells that are KRT14-PAI-1 (C) and Ki67-PAI-1 (D) double positive in the basal (BZ) and non-basal (NBZ) zones of a subset of biopsies. Epithelial PAI-1 expression correlates with the severity of basal zone hyperplasia (E). PAI-1 mRNA expression correlates with Ki67 (F) and cytokeratin 5 (Krt5) (G) in esophageal biopsies from active, inactive, and control subjects.
We used our standardized histologic scoring tool in order to assess the relationship between the degree of PAI-1 expression and the severity of BZH. BZH is scored as 0, 1, 2, or 3 when <20%, 21–50%, 51–75%, or >75% of the epithelial height is encompassed by basal cells, respectively. The degree of PAI-1 expression correlated positively with the degree of basal zone hyperplasia on histologic scoring (p<0.001), consistent with the fact that PAI-1 was expressed in these cells (Figure 2e).
In order to understand if PAI-1 gene expression associated with the expression of epithelial proliferation markers, we ran a panel of epithelial genes including the proliferation marker, Ki67, and cytokeratin 5. These analyses demonstrated that gene expression of PAI-1 correlated positively with the expression levels of Ki67 and cytokeratin 5 (Figure 2f,g). Together these analyses show that proliferating cells express PAI-1 mRNA and protein.
PAI-1 expression relative to epithelial and fibrotic gene expression
Recent studies have demonstrated that mRNA panels can be used to gauge EoE disease activity (31). Indeed, these panels can be completed using formalin fixed paraffin embedded biopsy specimens, allowing post-biopsy analysis to occur even if a frozen/fresh sample was not procured specifically for RNA analysis at the time of EGD. We utilized frozen biopsy specimens from control (n=3), gastroesophageal reflux disease (n=1), active EoE (n=10), and inactive EoE (n=7) to determine if PAI-1 gene expression correlated with the expression of other pro-fibrotic genes and epithelial mesenchymal transformation genes that are induced by TGFβ1. Since TGFβ1 is a major regulator of EoE associated remodeling (5, 17, 32, 33), we hypothesized that PAI-1 would be elevated as part of a network of TGFβ1-induced genes. Since PAI-1 causes fibrotic remodeling, we were curious to know if the degree of epithelial PAI-1 expression would correlate with the degree of subepithelial fibrosis. Using a histologic scoring system that we have previously published, we graded fibrosis from 0–3 based on the severity of collagen accumulation and collagen bundle thickness. PAI-1 expression level associated positively with subepithelial fibrosis score (Figure 3ab).
Figure 3.
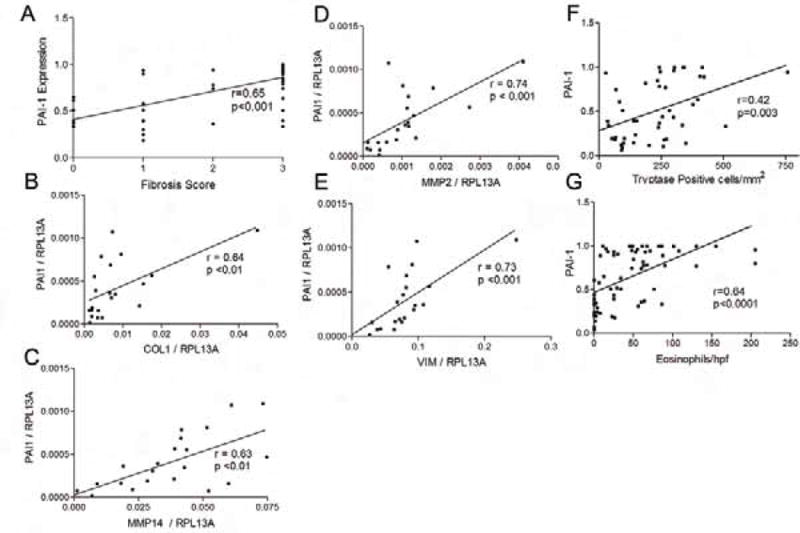
PAI-1 expression associates with fibrosis and remodeling gene expression. Epithelial expression of PAI-1 correlates with the severity of LP fibrosis (A). PAI-1 mRNA expression correlates with mRNA expression of the pro-fibrotic genes collagen I (B), MMP-14 (C), and MMP-2 (D) and the epithelial mesenchymal transformation marker vimentin (VIM) (E). PAI-1 expression correlates with the numbers of tryptase positive cells (F) and eosinophils (G).
In terms of fibrotic gene expression, we found that the degree of PAI-1 gene expression corresponded with the expression of the fibrotic genes collagen I (r=0.64, p<0.01), MMP-2 (r=0.63, p<0.01), and MMP-14 (r=0.74, p<0.001) and the TGFβ1 induced marker of epithelial mesenchymal transformation vimentin (33) (Figure 3b–e). While we did not find that PAI-1 mRNA corresponded significantly with the gene expression of either of the inflammatory markers eotaxin-3 or CPA-3 (not shown), we did find that the numbers of tryptase positive cells and eosinophils correlated positively with the degree of PAI-1 expression by immunohistochemistry in the biopsies (Figure 3f, g).
TGFβ1 induces PAI-1 expression in human esophageal epithelial cells and EoE fibroblasts
In order to understand if epithelial PAI-1 expression and secretion are regulated by TGFβ1 in esophageal cells, we isolated primary human epithelial cells from organ transplant donors and treated them in culture with recombinant human TGFβ1 and assayed for PAI-1 gene expression by qPCR and protein expression and secretion using immunoblotting and ELISA. TGFβ1 induced PAI-1 gene expression 11-fold in primary human esophageal epithelial cells (Figure 4a). PAI-1 protein was induced 2-fold in esophageal epithelial cell extracts (Figure 4b,d) and there was an almost 6-fold increase in PAI-1 release into the supernatant following TGFβ1 treatment (Figure 4c).
Figure 4.
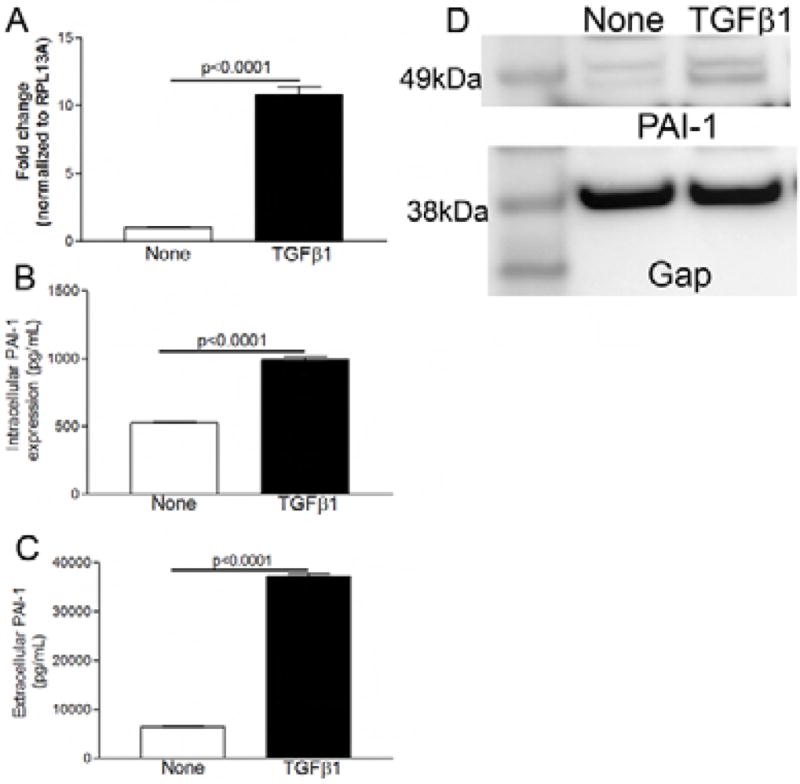
TGFβ1 induces PAI-1 expression in epithelial esophageal cells. Treatment of primary human esophageal epithelial cells with recombinant human TGFβ1 significantly induces PAI-1 gene expression (A), increases intracellular PAI-1 protein expression (B, D), and induces PAI-1 release into the supernatant (C).
Since TGFβ1 induced PAI-1 in human esophageal epithelial cells and PAI-1 was elevated in EoE, we determined whether PAI-1 and TGFβ1 expression correlated with one another. Immunohistochemistry for epithelial PAI-1 and LP TGFβ in the same biopsy specimens demonstrated that the esophageal expression of PAI-1 and TGFβ1 positively associated with one another (Figure 5a–c). In addition, TGFβ1 gene expression positively correlated with PAI-1 mRNA levels (Figure 5d).
Figure 5.
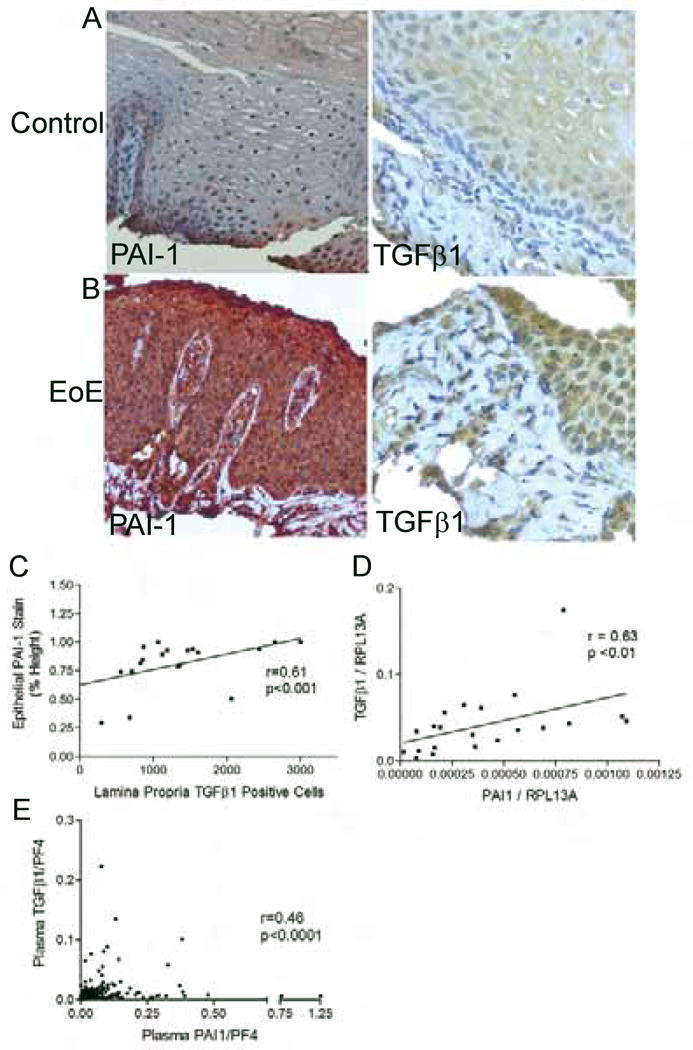
PAI-1 expression associates with the degree of TGFβ1 expression. Representative images of epithelial PAI-1 expression (left) and LP TGFβ1 positive cells (right) in control (A) and EoE (B). Epithelial PAI-1 expression correlates with the numbers LP TGFβ1 positive cells (C). mRNA expression of PAI-1 correlates with TGFβ1 in esophageal biopsies (D). Plasma PAI-1 levels correlate with plasma TGFβ1 (E).
Both PAI-1 and TGFβ1 have been reported to be detectable in the peripheral blood. We utilized stored, platelet depleted plasma samples from children with active and inactive EoE and from controls in order to explore if we could detect TGFβ1 or PAI-1 in the peripheral blood. Both PAI-1 and TGFβ1 were normalized to PF4 levels of both factors to control for platelet activation. Using 488 samples from 203 subjects, we found that, consistent with the esophageal data, the peripheral plasma levels of PAI-1 and TGFβ1 correlated with one another (Figure 5e) but the plasma levels of neither PAI-1 nor TGFβ1 correlated with esophageal disease activity or severity.
TGFβ1 induced PAI-1 is involved in TGFβ1-induced gene expression
To understand if induced PAI-1 was in its active or inactive conformation we treated EoE fibroblasts with TGFβ1 and assayed for PAI-1 activity. EoE fibroblasts released active PAI-1 at baseline and this was augmented in the presence of TGFβ1 (data not shown). We utilized the specific PAI-1 inhibitor TM5275 to assess if PAI-1 was involved in fibrotic gene expression. Treatment with TM5275 significantly decreased the baseline gene expression of collagen I and periostin in EoE fibroblasts (Figure 6a). In addition, PAI-1 inhibitor treatment significantly decreased TGFβ1-induced expression of α-smooth muscle actin (αSMA) and phospholamban (Figure 6b). It has been reported that PAI-1 can alter cellular function independently of its enzymatic activity via the low density lipoprotein receptor-related protein 1 (LRP1)/alpha-2 macroglobulin receptor 2 (35–37). Using immunocytochemistry we found that LRP1 was expressed in the LP of active EoE biopsies and on esophageal fibroblasts (Figure 6c,d), supporting the ability of PAI-1 to mediate pro-fibrotic effects either dependently or independently of its enzymatic activity in EoE.
Figure 6.
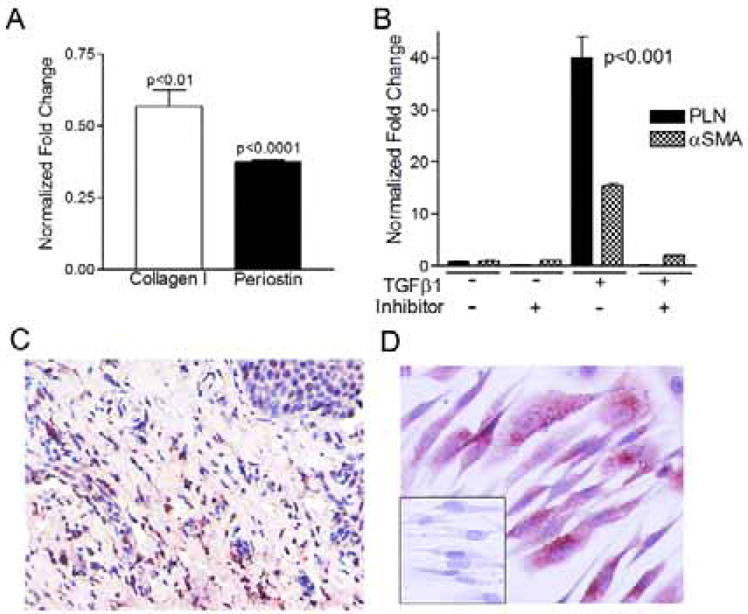
PAI-1 inhibition decreases gene expression in EoE fibroblasts. Treatment with the PAI-1 inhibitor TM5275 decreases baseline (A) and TGFβ1-induced (B) expression of pro-fibrotic and myofibroblast genes. Active EoE biopsy (C) and primary human esophageal fibroblasts (D) shows presence of low density lipoprotein like receptor-1.
Discussion
In this manuscript we document a number of novel findings regarding the TGFβ1-induced serine protease inhibitor, PAI-1, in pediatric EoE. We show that PAI-1 is significantly elevated in the active EoE esophagus as compared with non-diseased esophagi and that EoE-directed therapy reduces the degree of PAI-1 expression. We further demonstrate that TGFβ1 increases PAI-1 in primary human esophageal epithelial cells and EoE fibroblasts. The levels of PAI-1 correlate with histologic features of epithelial and subepithelial esophageal remodeling and fibrotic gene expression. The induction of a subset of genes by TGFβ1 was significantly decreased by pharmacologic PAI-1 inhibition and genes involved in myofibroblast formation such as αSMA were decreased by PAI-1 inhibition even in the absence of TGFβ1. Together these data suggest that PAI-1 is part of a pathogenic fibrotic network that can be initiated by TGFβ1.
To our knowledge, this is the first report of the potential role of PAI-1 in EoE. In asthma, PAI-1’s induction by allergen challenge and its role in fibrosis and airway remodeling have been studied for a number of years. In sensitized mice and humans, ovalbumin and house dust mite challenge, respectively, result in elevated pulmonary PAI-1 levels. Animals that are deficient in PAI-1 due to gene deletion or treatment with the PAI-1 inhibitor, tixplatinin, are protected from fibrin deposition and airway fibrosis and a murine model of TGFβ1-induced pulmonary fibrosis shows decreased myofibroblast formation and collagen deposition in the airways following treatment with oral TM5275 (23, 24, 38). In addition, rats treated with small interfering PAI-1 RNA have protection from bleomycin induced lung fibrosis and depletion of PAI-1 can improve hepatic fibrosis (39, 40). Our data suggests that PAI-1 is involved in EoE pathogenesis that also includes fibrosis. Since pharmacologic inhibition of PAI-1 decreased the baseline expression of the fibrosis genes collagen I and periostin, it is possible the use of PAI-1 blockade could be of utility in fibrotic EoE. Consistent with its role in TGFβ1-induced pulmonary fibrosis, PAI-1 was part of the pathway for increases in αSMA expression. The ability of TM2575 to reduce TGFβ1-induction of both αSMA and phospholamban may reflect the role of PAI-1 in myofibroblast formation (38). Since LRP1 is expressed in the EoE esophagus, PAI-1 may act independently of its enzymatic activity to promote esophageal fibrosis (35–37).
While all esophageal biopsies contain adequate epithelial tissue for analysis, at our center, 50% and 15% of biopsies obtained during routine clinical care have LP and muscularis mucosa, respectively. For this reason, despite the hypothesis that substantial esophageal remodeling occurs beneath the superficial surface, this tissue is not always available for analysis of fibrotic severity. Currently, other than invasive procedures that can gauge trans-mural esophageal rigidity in adults, there is no manner in which to sample esophageal rigidity. Indeed, even the relationship between histologic fibrosis and progression to strictures is not yet clear, although a logical assumption is that increasing esophageal fibrosis is one of the main underlying mechanisms for esophageal rigidity and dysfunction. Since LP is not always available for assessment, an epithelial marker that accurately reflects the severity of LP fibrosis could be of substantial utility for evaluating biopsies. Since it is possible that LP fibrosis is the closest gauge to esophageal compliance that we have currently available in children, and since epithelium is always present in the biopsy specimens, an epithelial marker of fibrosis could potentially alter clinical interventional strategies. For example, in subjects with less, absent, or minimal fibrosis, less intervention may be required to prevent remodeling, strictures, and food impactions while in patients with high levels of fibrosis, more aggressive therapy may be warranted. In this context, epithelial PAI-1 staining, which is straightforward and easily performed, could potentially serve as an epithelial marker of fibrotic severity, at least in children, and may have the potential to change EoE management.
We have previously reported that MMP-14 is also increased in the epithelium of pediatric EoE subjects (16). Like PAI-1, MMP-14 expression and release is increased in epithelial cells by TGFβ1 (16). Indeed, PAI-1 gene expression correlates positively with MMP-14 gene expression as well as with the expression of a number of other remodeling genes including collagen I, MMP-2, Ki67, vimentin, and TGFβ1. In addition PAI-1 levels correlated with the proliferation and epithelial genes Ki67 and cytokeratin 5. The fact that PAI-1 expression correlates with basal zone hyperplasia and fibrosis scores, both of which are histologic markers of remodeling, suggests that it is indeed a good epithelial marker of esophageal remodeling.
Unlike the correlation of PAI-1 with other remodeling genes, its mRNA level did not correspond to gene expression of inflammatory markers such as eotaxin-3 and CPA-3. Given that we see PAI-1 expression largely in structural epithelial cells in EoE, it is possible that, in chronic EoE, PAI-1 may be part of a pro-fibrotic network that can function independently or in the presence of lower levels of inflammation. This is potentially important in the context of the inflammatory burn out and progressive fibrosis that is seen in EoE adults with long disease durations (7). In murine asthma, PAI-1 deficiency substantially reduces fibrosis but does not robustly decrease inflammation, consistent with the concept that PAI-1 has functions that are independent of inflammation (24). These observations have therapeutic implications in EoE since there are no agents that specifically target fibrosis and interventional strategies can have variable effects on remodeling. It is possible that the use of agents such as inhibitors of PAI-1 may be of utility in subjects with fibrotic EoE. In addition, since PAI-1 can be made by mast cells where it is induced by chymase-activated TGFβ1 (22), it will be of interest to see what role inflammation does play in PAI-1 expression in EoE.
PAI-1 can be detected in the peripheral blood of asthmatic subjects and can assoicate with asthma severity (21). In order to understand if we could detect PAI-1 in the periphery of EoE subjects, we utilized stored plasma samples. Since PAI-1 is released by platelets, it is important to control for the presence and degranulation of platelets. We found that plasma PAI-1 associated positively with peripheral TGFβ1 levels, supporting the concept that TGFβ1 is integral to PAI-1 expression.
In conclusion, we have demonstrated a novel potential role of PAI-1 in EoE as an epithelial marker of esophageal remodeling that is induced by TGFβ1. Since PAI-1 inhibition decreased the effects of TGFβ1 on gene expression, it is possible to that PAI-1 could be a new therapeutic target in EoE. It will be of interest to validate and extend our findings in a larger EoE population in order to better understand the role of PAI-1 in EoE pathogenesis.
Supplementary Material
Clinical Implications.
Since PAI-1 is induced by TGFβ1, expressed in EoE epithelial cells, correlates positively with fibrosis and its inhibition decreases TGFβ1-induced fibrotic gene expression, PAI-1 may be as an epithelial marker of esophageal remodeling and serve as a new therapeutic target.
Acknowledgments
We thank Elizabeth La and Loan Duong for technical assistance with cell culture. Donor tissue was obtained from NDRI and ARORA. CEGIR (U54 AI117804) is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS, and is funded through collaboration between NIAID, NIDDK, and NCATS. The UCSD/RCHSD database is supported by the NIH grant UL1TR000100 of CTSA funding prior to August 13, 2015 and grant UL1TR001442 of CTSA funding beginning August 13, 2015 and beyond. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Funding Sources: NIH/NIAID AI 092135 (S.A), ART/APFED HOPE Award (S.A.), DOD FA100044 (D.H.B, S.A.), NIH/NIAID AI 107779 (D.H.B.), AI 70535 (D.H.B.), AI 72115 (D.H.B.), NIH/NCRR/NCATS UL1TR000039 (R.K.), Hearst Foundation (R.D.), ORDR/NCATS/NIAID/NIDDK CEGIR U54 AI117804 (S.A), NIH UL1TR000100 CTSA (S.A.)
Abbreviations
- CPA-3
Carboxypeptidase A3
- EoE
Eosinophilic esophagitis
- PAI-1
Plasminogen activator inhibitor-1
- TGFβ1
Transforming growth factor beta-1
- BZH
Basal zone hyperplasia
- LP
Lamina propria
- IHC
Immunohistochemistry
- IF
Immunofluorescence
- HPF
high power field
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts: Conflicts: SA and RD are co-inventors of oral viscous budesonide, patented by UCSD and hold stock options in Meritage Pharmaceutical
References
- 1.Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. The Journal of allergy and clinical immunology. 2011 Jul;128(1):3–20. e6. doi: 10.1016/j.jaci.2011.02.040. quiz 1–2. Epub 2011/04/12. eng. [DOI] [PubMed] [Google Scholar]
- 2.Aceves SS, Ackerman SJ. Relationships between eosinophilic inflammation, tissue remodeling, and fibrosis in eosinophilic esophagitis. Immunology and allergy clinics of North America. 2009 Feb;29(1):197–211. xiii–xiv. doi: 10.1016/j.iac.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoepfer AM, Safroneeva E, Bussmann C, Kuchen T, Portmann S, Simon HU, et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology. 2013 Dec;145(6):1230–6. e1–2. doi: 10.1053/j.gastro.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Dellon ES, Kim HP, Sperry SL, Rybnicek DA, Woosley JT, Shaheen NJ. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointestinal endoscopy. 2014 Apr;79(4):577–85. e4. doi: 10.1016/j.gie.2013.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. The Journal of allergy and clinical immunology. 2007 Jan;119(1):206–12. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Aceves SS. Tissue remodeling in patients with eosinophilic esophagitis: what lies beneath the surface? The Journal of allergy and clinical immunology. 2011 Nov;128(5):1047–9. doi: 10.1016/j.jaci.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 7.Straumann A, Spichtin HP, Grize L, Bucher KA, Beglinger C, Simon HU. Natural history of primary eosinophilic esophagitis: a follow-up of 30 adult patients for up to 11.5 years. Gastroenterology. 2003 Dec;125(6):1660–9. doi: 10.1053/j.gastro.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 8.Lucendo AJ, Arias A, De Rezende LC, Yague-Compadre JL, Mota-Huertas T, Gonzalez-Castillo S, et al. Subepithelial collagen deposition, profibrogenic cytokine gene expression, and changes after prolonged fluticasone propionate treatment in adult eosinophilic esophagitis: a prospective study. The Journal of allergy and clinical immunology. 2011 Nov;128(5):1037–46. doi: 10.1016/j.jaci.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Aceves SS, Newbury RO, Chen D, Mueller J, Dohil R, Hoffman H, et al. Resolution of remodeling in eosinophilic esophagitis correlates with epithelial response to topical corticosteroids. Allergy. 2010 Jan;65(1):109–16. doi: 10.1111/j.1398-9995.2009.02142.x. Epub 2009/10/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dohil R, Newbury R, Fox L, Bastian J, Aceves S. Oral viscous budesonide is effective in children with eosinophilic esophagitis in a randomized, placebo-controlled trial. Gastroenterology. 2010 Aug;139(2):418–29. doi: 10.1053/j.gastro.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Straumann A, Conus S, Degen L, Frei C, Bussmann C, Beglinger C, et al. Long-term budesonide maintenance treatment is partially effective for patients with eosinophilic esophagitis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2011 May;9(5):400–9. e1. doi: 10.1016/j.cgh.2011.01.017. Epub 2011/02/01. eng. [DOI] [PubMed] [Google Scholar]
- 12.Lieberman JA, Morotti RA, Konstantinou GN, Yershov O, Chehade M. Dietary therapy can reverse esophageal subepithelial fibrosis in patients with eosinophilic esophagitis: a historical cohort. Allergy. 2012 Oct;67(10):1299–307. doi: 10.1111/j.1398-9995.2012.02881.x. [DOI] [PubMed] [Google Scholar]
- 13.Abu-Sultaneh SM, Durst P, Maynard V, Elitsur Y. Fluticasone and food allergen elimination reverse sub-epithelial fibrosis in children with eosinophilic esophagitis. Digestive diseases and sciences. 2011 Jan;56(1):97–102. doi: 10.1007/s10620-010-1259-5. Epub 2010/05/12. eng. [DOI] [PubMed] [Google Scholar]
- 14.Assa’ad AH, Gupta SK, Collins MH, Thomson M, Heath AT, Smith DA, et al. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology. 2011 Nov;141(5):1593–604. doi: 10.1053/j.gastro.2011.07.044. [DOI] [PubMed] [Google Scholar]
- 15.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. The Journal of allergy and clinical immunology. 2010 Dec;126(6):1198–204. e4. doi: 10.1016/j.jaci.2010.08.050. Epub 2010/11/05. eng. [DOI] [PubMed] [Google Scholar]
- 16.Beppu L, Yang T, Luk M, Newbury RO, Palmquist J, Dohil R, et al. MMPs-2 and -14 are Elevated in Eosinophilic Esophagitis and Reduced Following Topical Corticosteroid Therapy. Journal of pediatric gastroenterology and nutrition. 2014 Dec 23; doi: 10.1097/MPG.0000000000000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beppu LY, Anilkumar AA, Newbury R, Dohil R, Broide D, Aceves S. TGF-b1–induced phospholamban expression alters esophageal smooth muscle cell contraction in patients with eosinophilic esophagitis. The Journal of allergy and clinical immunology. 2014 doi: 10.1016/j.jaci.2014.04.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, Chang G, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal immunology. 2008 Jul;1(4):289–96. doi: 10.1038/mi.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abonia JP, Wen T, Stucke EM, Grotjan T, Griffith MS, Kemme KA, et al. High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. The Journal of allergy and clinical immunology. 2013 Aug;132(2):378–86. doi: 10.1016/j.jaci.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra A, Wang M, Pemmaraju VR, Collins MH, Fulkerson PC, Abonia JP, et al. Esophageal remodeling develops as a consequence of tissue specific IL-5-induced eosinophilia. Gastroenterology. 2008 Jan;134(1):204–14. doi: 10.1053/j.gastro.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho S, Kang J, Lyttle C, Harris K, Daley B, Grammer L, et al. Association of elevated plasminogen activator inhibitor 1 levels with diminished lung function in patients with asthma. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2011 May;106(5):371–7. doi: 10.1016/j.anai.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho SH, Lee SH, Kato A, Takabayashi T, Kulka M, Shin SC, et al. Cross-talk between human mast cells and bronchial epithelial cells in plasminogen activator inhibitor-1 production via transforming growth factor-beta1. American journal of respiratory cell and molecular biology. 2015 Jan;52(1):88–95. doi: 10.1165/rcmb.2013-0399OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SH, Eren M, Vaughan DE, Schleimer RP, Cho SH. A plasminogen activator inhibitor-1 inhibitor reduces airway remodeling in a murine model of chronic asthma. American journal of respiratory cell and molecular biology. 2012 Jun;46(6):842–6. doi: 10.1165/rcmb.2011-0369OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh CK, Ariue B, Alban RF, Shaw B, Cho SH. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochemical and biophysical research communications. 2002 Jun 28;294(5):1155–60. doi: 10.1016/S0006-291X(02)00577-6. [DOI] [PubMed] [Google Scholar]
- 25.Cho SH, Hong SJ, Chen H, Habib A, Cho D, Lee SH, et al. Plasminogen activator inhibitor-1 in sputum and nasal lavage fluids increases in asthmatic patients during common colds. The Journal of allergy and clinical immunology. 2014 May;133(5):1465–7. 7 e1–2. doi: 10.1016/j.jaci.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kowal K, Bodzenta-Lukaszyk A, Pampuch A, Szmitkowski M, Donati MB, Iacoviello L. Plasminogen activator inhibitor-1 plasma concentration in allergic asthma patients during allergen challenge. International archives of allergy and immunology. 2007;144(3):240–6. doi: 10.1159/000103998. [DOI] [PubMed] [Google Scholar]
- 27.Nie W, Li B, Xiu QY. The -675 4G/5G polymorphism in plasminogen activator inhibitor-1 gene is associated with risk of asthma: a meta-analysis. PloS one. 2012;7(3):e34385. doi: 10.1371/journal.pone.0034385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pampuch A, Kowal K, Bodzenta-Lukaszyk A, Di Castelnuovo A, Chyczewski L, Donati MB, et al. The -675 4G/5G plasminogen activator inhibitor-1 promoter polymorphism in house dust mite-sensitive allergic asthma patients. Allergy. 2006 Feb;61(2):234–8. doi: 10.1111/j.1398-9995.2005.00948.x. [DOI] [PubMed] [Google Scholar]
- 29.Schuliga M, Westall G, Xia Y, Stewart AG. The plasminogen activation system: new targets in lung inflammation and remodeling. Current opinion in pharmacology. 2013 Jun;13(3):386–93. doi: 10.1016/j.coph.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Aceves SS, Newbury RO, Dohil MA, Bastian JF, Dohil R. A symptom scoring tool for identifying pediatric patients with eosinophilic esophagitis and correlating symptoms with inflammation. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2009 Nov;103(5):401–6. doi: 10.1016/S1081-1206(10)60359-6. [DOI] [PubMed] [Google Scholar]
- 31.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, Putnam PE, et al. Molecular Diagnosis of Eosinophilic Esophagitis by Gene. Expression Profiling Gastroenterology. 2013 Aug 23; doi: 10.1053/j.gastro.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abonia JP, Franciosi JP, Rothenberg ME. TGF-beta1: Mediator of a feedback loop in eosinophilic esophagitis–or should we really say mastocytic esophagitis? The Journal of allergy and clinical immunology. 2010 Dec;126(6):1205–7. doi: 10.1016/j.jaci.2010.10.031. Epub 2010/12/08. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, Mukkada V, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. The Journal of allergy and clinical immunology. 2012 May;129(5):1387–96. e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdulnour-Nakhoul SM, Al-Tawil Y, Gyftopoulos AA, Brown KL, Hansen M, Butcher KF, et al. Alterations in junctional proteins, inflammatory mediators and extracellular matrix molecules in eosinophilic esophagitis. Clinical immunology. 2013 Aug;148(2):265–78. doi: 10.1016/j.clim.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. The Journal of biological chemistry. 2004 May 21;279(21):22595–604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- 36.Czekay RP, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM, Klein RM, et al. PAI-1: An Integrator of Cell Signaling and Migration. International journal of cell biology. 2011;2011:562481. doi: 10.1155/2011/562481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garg N, Goyal N, Strawn TL, Wu J, Mann KM, Lawrence DA, et al. Plasminogen activator inhibitor-1 and vitronectin expression level and stoichiometry regulate vascular smooth muscle cell migration through physiological collagen matrices. J Thromb Haemost. 2010 Aug;8(8):1847–54. doi: 10.1111/j.1538-7836.2010.03907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang WT, Vayalil PK, Miyata T, Hagood J, Liu RM. Therapeutic value of small molecule inhibitor to plasminogen activator inhibitor-1 for lung fibrosis. American journal of respiratory cell and molecular biology. 2012 Jan;46(1):87–95. doi: 10.1165/rcmb.2011-0139OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang YP, Li WB, Wang WL, Liu J, Song SX, Bai LL, et al. siRNA against plasminogen activator inhibitor-1 ameliorates bleomycin-induced lung fibrosis in rats. Acta pharmacologica Sinica. 2012 Jul;33(7):897–908. doi: 10.1038/aps.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. Journal of cellular physiology. 2012 Feb;227(2):493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.