

Abstract
Naturally occurring enzyme homologs often display highly divergent activity with non-natural substrates. Exploiting this diversity with enzymes engineered for new or altered function, however, is laborious because the engineering must be replicated for each homolog. We demonstrate that a small set of mutations of the tryptophan synthase β-subunit (TrpB) from Pyrococcus furiosus, which mimic the activation afforded by binding of the α-subunit, has a similar activating effect in TrpB homologs with as little as 57% sequence identity. Kinetic and spectroscopic analyses indicate that the mutations function through the same mechanism, mimicry of α-subunit binding. From this collection of stand-alone enzymes, we identified a new catalyst that displays a remarkably broad activity profile in the synthesis of 5-substituted tryptophans, a biologically important class of compounds. This investigation demonstrates how allosteric activation can be recapitulated throughout a protein family to efficiently explore natural sequence diversity for desirable biocatalytic transformations.
Keywords: allostery, protein engineering, tryptophan synthase, non-canonical amino acids, biocatalysis
TOC image
The tryptophan synthase enzyme complex is active toward a number of indole analogs. The β-subunit (TrpB) performs the synthetically useful reaction, but requires the α-subunit to be fully active. We have transferred mutations from a re-activated TrpB variant from Pyrococcus furiosus into homologous TrpBs to generate a panel of stand-alone TrpB catalysts, one of which is especially useful for making 5-substituted tryptophans, an important biological motif.
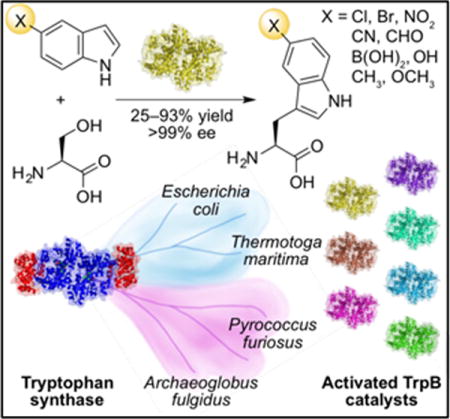
Tryptophan synthases (TrpSs) are α2β2 heterodimer complexes that catalyze the synthesis of tryptophan (1) from 3-indole-D-glycerol phosphate (IGP, 2) and L-serine (3, Figure 1a). TrpS also reacts with myriad indole analogs, providing a direct biocatalytic route to making tryptophan derivatives.[1] In such reactions, only the β-subunit (TrpB) performs catalysis (Figure 1b), but its activity is greatly diminished in the absence of the α-subunit (TrpA), limiting its utility as a biocatalyst.[2] Recently, we applied directed evolution to the β-subunit from Pyrococcus furiosus (PfTrpB) to identify mutations that emulate the effect of TrpA binding and imbue the β-subunit with high activity in isolation.[3] However, this stand-alone catalyst, PfTrpB0B2, exhibited poor levels of activity in the synthesis of 5-substituted tryptophans, a prevalent structural motif in bioactive natural products.[4] We hypothesized that if TrpB homologs could be activated by transferring the allostery-mimicking mutations identified in PfTrpB0B2, some of the resultant catalysts might have greater activity with 5-substituted indoles.
Figure 1.
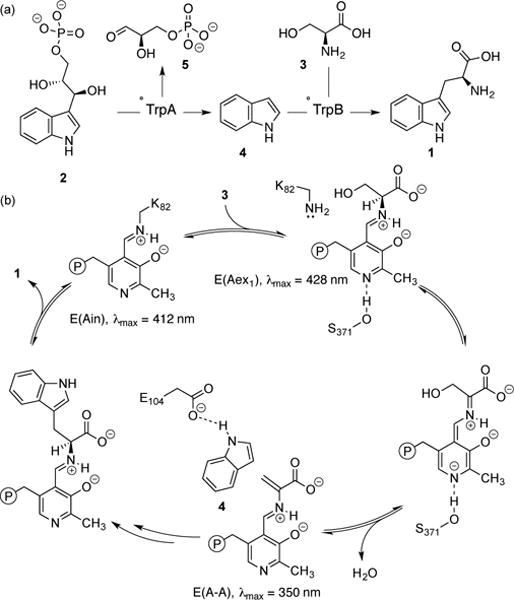
Native reaction mediated by TrpS and the catalytic cycle of TrpB. (a) TrpA degrades 2 through retro-aldol cleavage to give indole (4) and D-glyceraldehyde (5). Substrate 4 then reacts with 3 to form 1. (b) TrpB contains a pyridoxal 5′-phosphate (PLP) cofactor bound as an internal aldimine, E(Ain), which covalently binds 3 as an external aldimine, E(Aex1). Subsequent deprotonation and dehydration form the amino-acrylate, E(A-A). Finally, 4 reacts with E(A-A) to form 1.
The central challenge in enzyme engineering is to traverse the sequence space that separates a wild-type enzyme from its variant with novel functional properties. Instead of repeating the directed evolution to activate each new TrpB homolog, we decided to try to shortcut that effort by transferring beneficial mutations discovered in PfTrpB to different homologs. This would create a panel of TrpB enzymes, possibly with different substrate scopes or other useful properties for biocatalytic applications. Transfer of beneficial mutations to other, closely-related enzymes is widely used to improve properties, such as stability, but this approach assumes either that the effects of the mutations are independent and additive, as with thermostabilization by consensus design,[5] or at least that the protein context is shared (high sequence identity in the region of the mutation).[6] In an allosterically modulated enzyme such as TrpS, catalytic activity is increased by ligand (TrpA) binding at a location separate from the active site. Allosteric activation thus involves the participation of many residues, even the entire protein, as well as the surrounding solvent.[7] Furthermore, residues that contribute to allosteric signalling are poorly conserved by evolution,[8] causing homologous proteins to develop different allosteric mechanisms.[9] It was thus uncertain whether mutations that mimic allostery in TrpS from one species would be generalizable to other homologs.
We selected three phylogenetically diverse TrpBs[10] to serve as the basis for new stand-alone catalysts: the hyperthermostable enzymes from Archaeoglobus fulgidus (AfTrpB, 72% sequence identity to PfTrpB) and Thermotoga maritima (TmTrpB, 64% identity), and the mesophilic enzyme from Escherichia coli (EcTrpB, 57% identity).[11] We were especially interested in TmTrpB because its wild-type kcat is already four times that of PfTrpB.[12] Activation of EcTrpB would be desirable because it is adapted to a different temperature (37 °C versus 96 °C for PfTrpB), which may be useful for less stable substrates.
Previously, we observed that PfTrpB was a sluggish catalyst (Table 1, entry 1), but that variant PfTrpB0B2, which was engineered using three rounds of directed evolution, exhibited a 9-fold increase in kcat with an equivalent decrease in KM (Table 1, entry 2). We expressed and purified the three TrpB homologs and their corresponding 0B2 variants to test whether the activating mutations would produce a similar effect. Compared to AfTrpB (Table 1, entry 3), the variant AfTrpB0B2 has a 7-fold higher kcat and 2-fold lower KM for indole (Table 1, entry 4).
Table 1.
Kinetic parameters of the Pyrococcus furiosus and Archaeoglobus fulgidus TrpB wild-type enzymes and 0B2 variants.[a]
| Entry | Enzyme |
kcat (s−1) |
KM (μM indole) |
kcat/KM (μM−1 s−1 indole) |
|---|---|---|---|---|
| 1 | PfTrpBWT | 0.31 | 77 | 4.0 |
| 2[b] | PfTrpB0B2 | 2.9 | 9 | 300 |
| 3 | AfTrpBWT | 0.074 | 12 | 6.0 |
| 4[c] | AfTrpB0B2 | 0.51 | 4.8 | 110 |
Assays conducted in potassium phosphate buffer (pH 8) at 75 °C for Pf and 60 °C for AfTrpB. See SI section 5.4 for experimental details. Standard errors are in Table S1.
Contains the mutations P12L, E17G, I68V, F274S, T292S, and T321A.
Contains the mutations P25L, P30G, I80V, L285S, T303S, and T321A.
The absorption spectrum of PLP changes as the cofactor passes through different states of the catalytic cycle (Figure 1b).[2a] Thus, the absorption spectrum of the enzyme under reaction conditions directly reflects the steady-state distribution of catalytic intermediates. Before the addition of Ser, AfTrpS, AfTrpB, and AfTrpB0B2 all display an absorbance peak at 412 nm (Figure 2), which corresponds to the absorbance of E(Ain). When Ser is added to AfTrpS, its spectrum exhibits a new λmax at 350 nm (Figure 2a), indicating that E(A-A) is prevalent in the steady state of the catalytic cycle for this enzyme. Conversely, AfTrpB lacks a λmax at 350 nm, but instead possesses an absorbance peak at 428 nm (Figure 2b), indicating that E(Aex1) is the most prevalent intermediate. The spectrum of AfTrpB0B2, on the other hand, is almost identical to that of AfTrpS, with a prominent λmax of 350 nm (Figure 2c). This behavior matches what we observed previously[3] and provides compelling evidence that the 0B2 mutations activate AfTrpB by the same mechanism as they did in PfTrpB, namely by mimicking the allosteric activation produced by binding of TrpA to TrpB.
Figure 2.
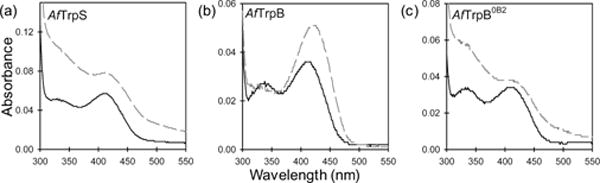
UV-vis absorption spectra of (a) AfTrpS, (b) AfTrpB, and (c) AfTrpB0B2 before (solid black) and after (dashed grey) addition of L-serine.
The TmTrpB homolog (Table 2, entry 1) already contains the residue A321 in its native sequence. However, incorporation of the remaining five mutations from PfTrpB0B2 reduced the kcat to just 10% of the wild-type activity (Table 2, entry 2). To investigate whether a subset of these mutations could still be activating, we constructed a recombination library of the 0B2 mutations in TmTrpB and screened for activity with indole. Three mutations, P19G, I69V, and T292S, increased the activity with respect to wild type. The variant having all three mutations was most active, with an 8-fold increase in kcat (Table 2, entry 3). The T292S mutation by itself was also substantially activating, producing a 4-fold increase in kcat (Table 2, entry 4).
Table 2.
Kinetic parameters of Thermotoga maritima TrpB variants and TrpS.[a]
As with AfTrpS and PfTrpS, the UV-vis absorption spectrum of TmTrpS exhibits a strong λmax at 350 nm under the reaction conditions. Once again, TmTrpB lacks this probative peak and instead exhibits the λmax at 428 nm that was observed for AfTrpB and PfTrpB. While all permutations of the three mutations (P19G, I69V, and T292S) led to improved kcat values (Figure S3), only the variants with T292S exhibited a λmax at 350 nm, equivalent to TmTrpS (Figure S4). The importance of the T292S mutation was also observed in PfTrpB, where this mutation alone restored the kcat of the isolated PfTrpB to that of the PfTrpS complex[3]. The effects of this conservative mutation were even more dramatic in TmTrpB, producing a kcat almost 3-fold higher than TmTrpS (Table 2, cf. entries 4 and 5).
Mutational activation of the most distant homolog, EcTrpB (57% identity), proved more challenging. The crucial Thr→Ser mutation was not possible for EcTrpB because EcTrpB already has Ser at this position (S297). Site-saturation mutagenesis confirmed that serine is the optimal residue at that position (Figure S5a). Unlike TmTrpB, recombination of the 0B2 mutations in EcTrpB yielded no variants with enhanced activity and only a few with activity similar to wild type (Figure S5b).
The initial screening effort with PfTrpB[3] had also identified a variant with mutations M144T and N166D that was almost as active as PfTrpBT292S (Table 3, entry 1). These two residues, unlike T292, reside in the so-called communication (COMM)[2a] domain, which interfaces with TrpA and undergoes large conformational motions during the catalytic cycle. These residues are identical in the four homologs studied here and are almost universally conserved across all TrpBs (Figure S6). We hypothesized that the effects of mutations at these sites might also be transferrable. Upon making the equivalent mutations in EcTrpB, TmTrpB, and AfTrpB, we observed activation in all variants, with approximately 2- to 5-fold increases in kcat (Table 3, entries 2 to 4).
Table 3.
Kinetic parameters of Pyrococcus furiosus TrpBM144T N166D and its homologs.[a]
| Entry | Enzyme |
kcat (s−1) |
KM (μM indole) |
kcat/KM (μM−1 s−1 indole) |
|---|---|---|---|---|
| 1 | PfTrpBM144T N166D | 0.83 | 42 | 20 |
| 2[b] | EcTrpBM149T N171D | 0.34 | 18 | 19 |
| 3 | TmTrpBM145T N167D | 3.3 | 32 | 100 |
| 4 | AfTrpBM156T N178D | 0.34 | 11 | 31 |
We wished to verify that the proteins were still being activated by allosteric mimicry. UV-vis analysis of the steady-state distributions of intermediates upon addition of L-serine revealed that the double mutants of PfTrpB and EcTrpB still accumulated E(Aex1) rather than E(A-A) (Figure S7a and d). However, the homologous double mutants of A. fulgidus and T. maritima showed shifted spectra, in which E(A-A) predominated (Figure S7b and c). These data suggest that the double mutation is also activating the enzymes through allosteric mimicry, but that it does not quite reach the activation generated by adding TrpA. This situation is reminiscent of the earlier evolution of PfTrp0B2, wherein the lone T292S mutation was insufficient to completely shift the UV-vis spectrum to E(A-A) without at least four additional mutations.[3]
In the pantheon of tryptophan-derived natural products, one can find substitution at every position on the indole moiety (Figure 3). Position 5, for example, is chlorinated by the halogenase PyrH, en route to pyrroindomycin B (6),[4a] and mono-oxygenated by tryptophan hydroxylase in the biosynthesis of serotonin (7) and melatonin (8).[4d] Such substituents have a profound effect on biological activity because they can mask sites of metabolic degradation and change the compound’s electronic properties. This, in turn, alters properties like solubility and creates new binding interactions through effects such as π-stacking and halogen bonding.[1b, 13] Halogens can also provide handles for further diversification of biologically active compounds through cross-coupling reactions.[14] While biocatalytic routes to tryptophan derivatives tend to be inefficient and limited in substrate scope, TrpS can provide direct access to many of these products. Previously, however, 5-substituted indoles bearing anything larger than fluorine caused a substantial decrease in activity[1d, 1f, 15]. Furthermore, TrpS activity with electron-deficient indoles had not been explored.
Figure 3.
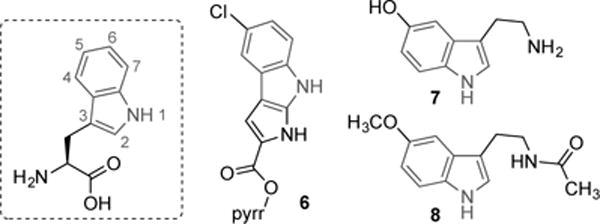
Numbering of positions on the indole moiety of tryptophan and examples of natural products bearing substitution at position 5.
One enzyme in our repertoire, TmTrpBM145T N167D, showed higher activity with 5-bromoindole than even our most optimized catalyst, PfTrpB0B2 (Figure S8). To assess whether this was a general property of the catalyst, we compared the relative rates of TmTrpBM145T N167D to PfTrpB0B2 with a set of challenging 5-substituted indoles (Table 4). We then applied TmTrpBM145T N167D in reactions that were run to higher conversion in order to isolate and characterize the products.
Table 4.
Synthesis of 5-substituted tryptophan derivatives with Thermotoga maritima TrpBM145T N167D[a]
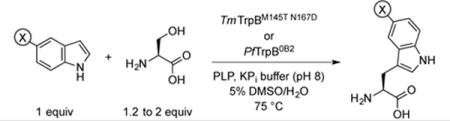
| ||||
|---|---|---|---|---|
|
| ||||
| Reaction with TmTrpBM145T N167D
[a]
|
||||
| Entry | X | Relative rate vs PfTrpB0B2 | Isolated yield (%)[b] | Total turnovers[c] |
| 1 | Cl | 3.0 | 93 | 9300 |
| 2 | Br | 5.6 | 88 | 4400 |
| 3 | NO2 | 7.5 | 25 | 1250 |
| 4 | CN | 4.5 | 49 | 2450 |
| 5 | CHO | 1.9 | 32 | 1600 |
| 6 | B(OH)2 | 1.8 | 38[d] | 1900[d] |
| 7 | OH | 1.4 | 93[d] | 9300[d] |
| 8 | CH3 | 1.4 | 91 | 9100 |
| 9 | OCH3 | 1.5 | 76 | 7600 |
Reactions used either 2 equiv (entries 1–6) or 1.2 equiv (entries 7–9) of L-serine. Products isolated by chromatography with C-18 silica.
Extrapolated from isolated yield based on maximum theoretical turnover number.
Determined by 1H NMR based on an internal standard.
With 5-chloroindole, the Tm variant exhibits a 3-fold rate enhancement compared to PfTrpB0B2 (Table 4, entry 1). Despite this improvement, the reaction still appeared to stall at about 85% conversion when the substrates were used in equal amounts, possibly due to competing decomposition of serine.[16] We overcame this limitation by using a small excess of serine, allowing us to obtain 5-chlorotryptophan in 94% isolated yield. PfTrpB0B2 has even greater difficulty with 5-bromoindole, but the Tm variant is almost six times as fast for this substrate, allowing us to obtain 5-bromotryptophan in 88% isolated yield (Table 4, entry 2). These results compare favorably with previous reports, in which TrpS from Salmonella enterica was shown to form 5-chloro and 5-bromotryptophan in 61% and 26% yield, respectively[1f].
TmTrpBM145T N167D exhibits substantially faster rates, ranging from 2- to over 7-fold, with substrates that bear electron-withdrawing groups, such as nitro, cyano, formyl, and even boronate (Table 4, entries 3–6), representing a new substrate class in the TrpS literature. The reaction with 5-boronoindole is particularly interesting, as the catalyst must contend with competing proto-deborylation. Boronic acids can serve as handles for bio-orthogonal conjugation,[17] pH-sensitive delivery of therapeutics in vivo,[18] and substrates for cross-coupling, complementing 5-halotryptophans, for which electron-deficient coupling partners lead to reduced yields.[19] Although the reactivity with these new substrates remains low, we believe that TmTrpBM145T N167D, which only has two mutations compared to six in PfTrpB0B2, is the ideal parent for further optimization.
PfTrpB0B2 already has excellent activity with substrates that bear electron-donating groups at the 5-position (e.g., hydroxy, methyl, and methoxy); nonetheless, TmTrpBM145T N167D delivers a 1.5-fold faster rate (Table 4, entries 7–9), which is helpful because such electron-rich substrates are prone to aerobic oxidation. Thus, TmTrpBM145T N167D performs almost 10,000 turnovers in just two hours, allowing the products to be isolated in high yield without the need for oxygen-free conditions.
By mining the wealth of activating mutations in PfTrpB, we have identified subsets that retain their effects when transferred into related enzymes, including those from different domains of life (archaea and bacteria). Importantly, spectroscopic data indicate that the homologs are activated through the same mechanism as the PfTrpB variants, namely mimicking the effects of TrpA binding. By screening the resulting panel of activated TrpB homologs, we have identified a variant with broadly improved activity toward 5-substituted indoles, a substrate class that had proven problematic for all previous catalysts. The strategy used here could be applied to other TrpBs and exemplifies how the transfer of activating mutations to homologous enzymes can rapidly expand activity with non-native substrates.
Supplementary Material
Acknowledgments
The authors thank Dr. Jackson Cahn for the data on frequency of amino acids in each position of TrpB and Dr. Jennifer Kan for helpful discussions and comments on the manuscript. J.M.-C. gratefully acknowledges support from the Alfonso Martín Escudero Foundation. This work was funded through the Jacobs Institute for Molecular Engineering for Medicine and Ruth Kirschstein NIH Postdoctoral Fellowships F32GM117635 (to D.K.R) and F32G110851 (to A.R.B.).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Ferrari D, Yang LH, Miles EW, Dunn MF. Biochem. 2001;40:7421–7432. doi: 10.1021/bi002892l. [DOI] [PubMed] [Google Scholar]; b) Goss RJM, Newill PLA. Chem Comm. 2006:4924–4025. doi: 10.1039/b611929h. [DOI] [PubMed] [Google Scholar]; c) Winn M, Roy AD, Grüschow S, Parameswaran RS, Goss RJM. Bioorg Med Chem Lett. 2008;18:4508–4510. doi: 10.1016/j.bmcl.2008.07.053. [DOI] [PubMed] [Google Scholar]; d) Tsoligkas AN, Winn M, Bowen J, Overton TW, Simmons MJH, Goss RJM. ChemBioChem. 2011;12:1391–1395. doi: 10.1002/cbic.201100200. [DOI] [PubMed] [Google Scholar]; e) Perni S, Hackett L, Goss RJ, Simmons MJ, Overton TW. AMB Express. 2013;3:66–75. doi: 10.1186/2191-0855-3-66. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Smith DRM, Willemse T, Gkotsi DS, Schepens W, Maes BUW, Ballet S, Goss RJM. Org Lett. 2014;16:2622–2625. doi: 10.1021/ol5007746. [DOI] [PubMed] [Google Scholar]; g) Corr MJ, Smith DRM, Goss RJM. Tetrahedron. 2016 doi: 10.1016/j.tet.2016.02.016. [DOI] [Google Scholar]
- 2.a) Dunn MF. Arch Biochem Bioph. 2012;519:154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Biochem. 2013;52:6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buller AR, Brinkmann-Chen S, Romney DK, Herger M, Murciano-Calles J, Arnold FH. Proc Nat Acad Sci. 2015;112:14599–14604. doi: 10.1073/pnas.1516401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Zehner S, Kotzsch A, Bister B, Süssmuth RD, Méndez C, Salas JA, van Pée K-H. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]; b) Zhang P, Sun X, Xu B, Bijian K, Wan S, Li G, Alaoui-Jamali M, Jiang T. Eur J Med Chem. 2011;46:6089–6097. doi: 10.1016/j.ejmech.2011.10.036. [DOI] [PubMed] [Google Scholar]; c) Wang H, Reisman SE. Angew Chem, Int Ed. 2014;53:6206–6210. doi: 10.1002/anie.201402571. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang J, Wu C, Sheng J, Feng X. Mol Biosyst. 2016;12:1432–1435. doi: 10.1039/c5mb00888c. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann M, Wyss M. Curr Opin Biotech. 2001;12:371–375. doi: 10.1016/s0958-1669(00)00229-9. [DOI] [PubMed] [Google Scholar]
- 6.a) Brinkmann-Chen S, Flock T, Cahn JKB, Snow CD, Brustad EM, McIntosh JA, Meinhold P, Zhang L, Arnold FH. Proc Nat Acad Sci. 2013;110:10946–10951. doi: 10.1073/pnas.1306073110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heel T, McIntosh JA, Dodani SC, Meyerowitz JT, Arnold FH. ChemBioChem. 2014;15:2556–2562. doi: 10.1002/cbic.201402286. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Khanal A, Yu McLoughlin S, Kershner JP, Copley SD. Mol Biol Evol. 2015;32:100–108. doi: 10.1093/molbev/msu271. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dunn MR, Otto C, Fenton KE, Chaput JC. ACS Chem Biol. 2016;11:1210–1219. doi: 10.1021/acschembio.5b00949. [DOI] [PubMed] [Google Scholar]
- 7.Dokholyan NV. Chem Rev. 2016;116:6463–6487. doi: 10.1021/acs.chemrev.5b00544. [DOI] [PubMed] [Google Scholar]
- 8.Nussinov R, Tsai CJ, Csermely P. Trends Pharm Sci. 2011;32:686–693. doi: 10.1016/j.tips.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuriyan J, Eisenberg D. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 10.Merkl R. BMC Evol Biol. 2007;7 doi: 10.1186/1471-2148-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For sequence and structure alignments, see supporting information.
- 12.Hettwer S, Sterner R. J Biol Chem. 2002;277:8194–8201. doi: 10.1074/jbc.M111541200. [DOI] [PubMed] [Google Scholar]
- 13.Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 14.a) Roy AD, Grüschow S, Cairns N, Goss RJM. J Am Chem Soc. 2010;132:12243–12245. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]; b) Pathak TP, Miller SJ. J Am Chem Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Durak LJ, Payne JT, Lewis JC. ACS Catal. 2016;6:1451–1454. doi: 10.1021/acscatal.5b02558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaser G, Sanderson JM, Batsanov AS, Howard J. Tet Lett. 2008;49:2795–2798. [Google Scholar]
- 16.Crawford IP, Ito J. Proc Nat Acad Sci. 1964;51:390–397. doi: 10.1073/pnas.51.3.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akgun B, Hall DG. Angew Chem, Int Ed. 2016;55:3909–3913. doi: 10.1002/anie.201510321. [DOI] [PubMed] [Google Scholar]
- 18.a) Han H, Davis ME. Bioconjugate Chem. 2013;24:669–677. doi: 10.1021/bc300640j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pan DW, Davis ME. Bioconjugate Chem. 2015;26:1791–1803. doi: 10.1021/acs.bioconjchem.5b00324. [DOI] [PubMed] [Google Scholar]
- 19.Roy AD, Goss RJM, Wagner GK, Winn M. Chem Comm. 2008:4831–4833. doi: 10.1039/b807512c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.