

Abstract
Co(II)-based metalloradical catalysis (MRC) has been successfully applied for effective construction of the highly strained 2-sulfonyl-1,3-diazabicyclo[3.1.0]-hexane structures in high yields through intramolecular radical aziridination of allylic sulfamoyl azides. The resulting [3.1.0]-bicyclic aziridines prove to be versatile synthons for preparation of a diverse range of 1,2- and 1,3-diamine derivatives by selective ring-opening reactions in exo- and endo-fashion, respectively. As a demonstration of its application for target synthesis, the metalloradical intramolecular aziridination reaction has been incorporated as a key step for efficient synthesis of a potent neurokinin 1 (NK1) antagonist in 60% overall yield.
Keywords: intramolecular aziridination, aziridine ring-opening, neurokinin 1 (NK1) receptor, metalloradical catalysis, cobalt porphyrin
Radical chemistry has displayed vast potential in modern organic synthesis with many attractive features that are complementary to ionic chemistry.[1] To address the existing challenges in the field,[2] metalloradical catalysis (MRC) presents a new approach to achieve controllable reactivities and selectivities through catalytic generation of metal-stabilized organic radicals, such as the fundamentally new α-Co(III)-alkyl radicals and α-Co(III)-aminyl radicals.[3],[4] For example, the α-Co(III)-aminyl radicals (also known as Co(III)-nitrene radicals), which can be generated through a unique radical transfer process from the Co-centered radicals of cobalt(II) porphyrins [Co(Por)] to organic azides upon metalloradical activation,[3e]-[3g] have been demonstrated with well-controlled reactivities of both H-atom abstraction of C–H bonds and radical addition to C=C π bonds to furnish catalytic radical amination[5] and aziridination,[6] respectively, with high level of selectivities. In particular, the Co(II)-based MRC system exhibits remarkable chemoselectivity, as exemplified by the selective transformation of the sulfamoyl azide containing both N-allyl and N-bishomoallyl groups (Scheme 1A). Exclusive intramolecular 1,6-radical amination of the allylic C–H bonds was reported without observation of the potential reaction of intramolecular radical aziridination of the C=C π bonds.[5e] In the absence of the more reactive N-bishomoallyl group, however, it was unclear if intramolecular aziridination of N-allyllic sulfamoyl azides is possible in view of the high strain associated with the resulting 2-sulfonyl-1,3-diazabicyclo[3.1.0]hexane structure (Scheme 1B).[7] This type of catalytic process (Scheme 1C) would require the corresponding α-Co(III)-aminyl radical I to undergo efficient intramolecular addition to the olefin unit to generate the γ-Co(III)-alkyl radical intermediate II-a (Scheme 1C; left: 5-exo-trig cyclization) or γ-Co(III)-alkyl radical II-b (Scheme 1C; right: 6-endo-trig cyclization), followed by 3-exo-tet radical cyclization to deliver the aziridine product with regeneration of catalyst [Co(Por)]. If successful, this catalytic transformation would be highly useful as the resulting [3.1.0]-heterobicyclic structures are the key motifs in compounds of medicinal importance as well as can serve as versatile building blocks for preparation of functionalized diamines.[8]-[11]
Scheme 1.

Co(II)-based metalloradical catalysis (MRC) for intramolecular C–H amination versus intramolecular C=C aziridination of allylic sulfamoyl azides
A number of catalytic systems have been successfully developed for intramolecular aziridination of olefins.[12]-[15] While several types of [n.1.0]-heterobicyclic structures have been synthesized,[13]-[15] there is no previous report on the synthesis of [3.1.0]-bicyclic sulfamoyl aziridines via catalytic intramolecular aziridines (Scheme 1B).[7] This is presumably due to the high ring strain of the bicyclic transition state associated with the concerted asynchronous mechanism of most intramolecular catalytic systems involving metallonitrenes as the key intermediates. We envisioned that the Co(II)-based metalloradical catalysis could potentially address this challenge by constructing the bicyclic structure through a stepwise ring formation via two separate radical cyclization steps (Scheme 1C). Herein we report the development of the first catalytic system for intramolecular aziridination with allylic sulfamoyl azides via Co(II)-based metalloradical catalysis, allowing for effective construction of the highly strained 2-sulfonyl-1,3-diazabicyclo[3.1.0]hexane structures in high yields. Taking advantage of the high strain, the resulting sulfonylated [3.1.0]-bicyclic diaza aziridine products can be efficiently utilized as a new type of versatile synthons for preparation of 1,2- and 1,3-diamine derivatives by selective ring-opening reactions in exo- and endo-fashion, respectively.
At the outset of this project, N-benzyl-N-allyl-sulfamoyl azide 1a, which was prepared from the corresponding amine in one step,[5d] was selected as a model substrate to explore the possibility of Co(II)-MRC for intramolecular aziridination of allylic sulfamoyl azides and to establish effective catalytic conditions (Scheme 2). While the metalloradical catalyst [Co(TPP)] (TPP: tetraphenylporphyrin) was ineffective, [Co(P1)], in which the D2h-symmetric porphyrin 3,5-DitBu-IbuPhyrin P1 has amide functionalities at the ortho-positions of the meso-phenyl groups,[6b] could effectively catalyze the intramolecular aziridination reaction under mild conditions without the need of other reagents or additives, affording the desired sulfonylated [3.1.0]-bicyclic diaza aziridine 2a in 92% yield with N2 as the sole byproduct (Scheme 2). The dramatic difference in catalytic activity between [Co(TPP)] and [Co(P1)] is a remarkable demonstration of ligand-accelerated catalysis and could be rationalized as the outcome of hydrogen-bonding stabilization of Co(III)-nitrene radical intermediate (Scheme 2).[3f]
Scheme 2.
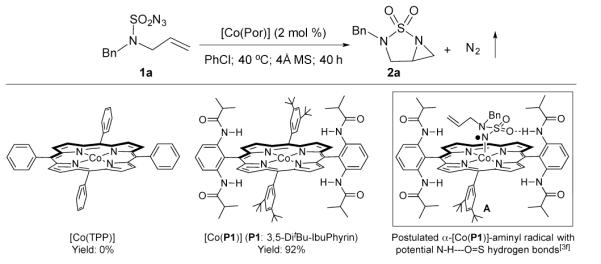
Ligand effect on [Co(Por)]-catalyzed intramolecular aziridination of allylic sulfamoyl azide: importance of potential hydrogen-bonding interaction
Under the optimized conditions (2 mol % of [Co(P1)] in PhCl at 40 °C for 40 h), the Co(II)-based intramolecular aziridination was found to have a broad substrate scope for a wide range of allylic sulfamoyl azides (Table 1). In addition to the N-benzyl group (Bn) in allylic sulfamoyl azide 1a (entry 1), the metalloradical system could tolerate a variety of N-substituted functionalities due to its neutral and nonoxidative conditions, as demonstrated by the high-yielding aziridination reactions of allylic sulfamoyl azides 1b–e that contain 4-methoxybenzyl (PMB), 4-chlorobenzyl (PCB), allyl, and propargyl groups, respectively (entries 2–5). It is worth noting that these electron-rich N-substituents could be oxidatively degraded in other catalytic systems that require the use of stoichiometric oxidant such as PhI(OAc)2 or PhI=O.[15] Similarly, allylic sulfamoyl azides bearing electron-withdrawing groups were also suitable substrates for the catalytic system. For example, azides bearing N-ethoxycarbonyl (1f) and N-Boc (1g) groups were intramolecularly aziridinated to generate the desired bicyclic aziridines 2f and 2g in 86% and 98% yields, respectively (entries 6 and 7). Besides the tolerance of various functional groups, the Co(II)-catalyzed aziridination process was shown interesting regioselectivity. For example, when azide 1h containing both internal and terminal olefins was utilized as the substrate, aziridine 2h was isolated in 70% yield as the sole product, indicating preferred reactivity of less sterically-hindered terminal olefins over internal olefins (entry 8). In the absence of terminal olefins, however, both cis- and trans-internal olefins could also be successfully aziridinated with stereospecificity (entries 9 and 10). Presumably also due to steric effect, cis-olefin-derived azide 1i (entry 9) was found to be more reactive than trans-olefin-derived azide 1j (entry 10), resulting in stereospecific formation of the corresponding aziridines 2i and 2j in 95% and 30% yields, respectively. Interestingly, it was found that allylic azides derived from disubstituted terminal olefins exhibited even higher reactivity. For example, at as low as 0.5 mol % catalyst loading and in much shorter reaction time (8 h), the 2-Ph allylic azide 1k could be effectively aziridinated by [Co(P1)], affording the desired sulfonylated [3.1.0]-bicyclic diaza aziridine 2k in nearly quantitative yield (entry 11). It is worth noting the exceedingly high strain of the 2-sulfonyl-1,3-diazabicyclo[3.1.0]hexane structure in 2k where the two bridgeheads are a quaternary carbon center and a tertiary nitrogen center, respectively. As an additional example, the intramolecular aziridination reaction of 2-Cl allylic azide 1l could be facilely catalyzed by [Co(P1)] to afford the corresponding chloro-[3.1.0]-bicyclic diaza aziridines 2l in full conversion (entry 12). Although the tertiary alkyl chloride 2l could be characterized by both 1H- and 13C-NMR (see Supp Info), it underwent subsequent ring-opening reactions during the purification, affording the ketone product 3 in nearly quantitative yield (entry 12). The ready formation of the 6-membered α,α’-diamino cyclic ketone 3 is presumably a result of the unusual kinetic susceptibility of the bridgehead quaternary amino halide units to halide substitution by water, followed by rearrangement. Similarly, 2-Br allylic azide 1m could also be converted to the same cyclic 1,3-diamino ketone 3 in an excellent yield, presumably through the initial aziridination product bromo-[3.1.0]-bicyclic diaza aziridines 2m (entry 13). Remarkably, even the cyclohexene-derived azide 1n could be successfully employed as the substrate for the Co(II)-based intramolecular aziridination, allowing for construction of the highly strained tricyclic aziridine 2n in 87% yield with excellent control of diastereoselectivity (entry 14). It is noted that 2n and related fused tricyclic aziridine compounds may serve as useful precursors for preparation of the valuable 1,2,3-cyclohexanetriamine motif that have been found in a number of biologically and pharmaceutically important compounds.[9]
Table 1.

|
Performed under N2; [azide 1]: 0.08 M.
Isolated yields.
PCB: para-chlorobenzyl.
with 30% azide recovered.
PMB: para-methoxybenzyl.
In PhCl at 80 °C; with 60% azide recovered.
0.5 mol % [Co(P1)] for 8 h.
In view of the high ring-strain associated with the 2-sulfonly-1.3-diazabicyclo[3.1.0]hexane structure, we then turned our attention to explore further transformations of the resulting bicyclic aziridine products 2 from the Co(II)-catalyzed intramolecular aziridination. In particular, we were interested in the possibility of regioselectively opening the bicyclic rings of these compounds. Using diaza aziridine 2k as a representative example, it was shown that the three-membered aziridine ring of the bicyclo[3.1.0] structure could be regioselectively opened at the exo-position by disparate nucleophiles (Scheme 3). For examples, 2k could be effectively opened by trimethylsilyl azide, thiophenol and benzylamine under mild conditions to afford the corresponding five-membered thiadiazolidine dioxide derivatives 4–6 in good to excellent yields. In addition to exo-ring opening reactions by nucleophiles, we also demonstrated the selective endo-ring opening at the bridgehead position of the bicyclo[3.1.0] structure in the presence of Lewis acids.[10] When treated with BF3•OEt2, 2k was regioselectively opened to form thiadiazine dioxide 7 in quantitative yield, presumably via a 1,3-dipole intermediate.[10] The six-membered cyclic enamine structure of 7 was confirmed by X-ray crystallographic analysis (see Supp Info). In the presence of AgSbF6, the resulting 1,3-dipole intermediate from 2k could be partially trapped by dipolarophiles such as benzonitrile and benzaldehyde to form tricyclic compounds 8 and 9, respectively, in addition to the formation of 7 (Scheme 3). X-ray crystallographic analysis (see Supp Info) established the unique [3.2.1]-tricyclic structure of 9 showing the presence of four heteroatoms with tertiary nitrogen and quaternary carbon bridgeheads.
Scheme 3.

Regioselective ring-opening reactions of [3.1.0]-bicyclic aziridine 2k by disparate reagents.
Considerable efforts have been dedicated toward the development of efficient methods for the preparation of diamines, which are prevalent motifs in biologically active molecules and can also function as chelating ligands for complexing transition metals.[11] Given that cyclic sulfamides can serve as precursors of diamines,[16] we showed that the resulting N-sulfonyl [3.1.0]-bicyclic diaza aziridines 2 from the [Co(P1)]-catalyzed aziridination could be efficiently desulfonylated for selective formation of either 1,2-diamines or 1,3-diamines, depending on the specific reaction conditions, as demonstrated with the reactions of diaza aziridine 2k (Scheme 4). When treated with LiAlH4 in THF, the aziridine structure of 2k was selectively opened at the exo-position through facile removal of the sulfonyl group, producing 1,2-diamine 10 containing a quaternary chiral carbon-center in 99% yield. Alternatively, 2k could be selectively converted to 1,3-diamine 11 in 90% yield, when the aziridine ring was first endo-opened under H2/Pd/C reduction condition, followed by desulfonylation via transamination with 1,3-diaminopropane (DAP).
Scheme 4.

Selective reductive desulfonylation of bicyclic aziridine structure.
To showcase the application of [Co(P1)]-catalyzed intramolecular aziridination of allylic sulfamoyl azides and subsequent transformations of the resulting sulfonylated [3.1.0]-bicyclic diaza aziridines for target synthesis, we developed a concise and high-yielding synthetic route to the bioactive compound 13 (Scheme 5),[17] one member of a family of potent Neurokinin 1 (NK1) receptor antagonists.[18] Compound 13 consists of a core structure of N-methylimidazolidin-2-one with a quaternary carbon-center bearing phenyl and (3,5-bis(trifluoromethyl)benzyloxy)methyl groups. Starting from N-methyl-2-phenylallylamine, allylic sulfamoyl azide 1o was readily prepared in 91% yield. The catalytic intramolecular aziridination of 1o proceeded smoothly in the presence of only 0.5 mol % of [Co(P1)], affording the desired [3.1.0]-bicyclic sulfamoyl aziridine 2o in 99% yield on a gram scale. Upon treatment with bis(trifluoromethyl)benzyl alcohol (Ar'FCH2OH) in the presence of NaH, aziridine 2o was regioselectively opened to give compound 12 in 86% yield. Cyclic sulfamide 12 was successfully transformed to the final cyclic urea analogue 13 through straightforward desulfonylation with 1,3-diaminopropane (DAP) in 91% yield, followed by carbonylation with 1,1’-carbonyldiimidazole (CDI) in 84% yield. The five-step synthesis of NK1 antagonist 13 was achieved in an overall yield of 60%.[19]
Scheme 5.

Application of Co(II)-catalyzed intramolecular aziridination for efficient synthesis of potent NK1 antagonist 13.
In summary, we have developed the first synthetic tool for efficient construction of the highly strained 2-sulfonaly-1,3-diazabicyclo[3.1.0]hexane structures through intramolecular radical aziridination via Co(II)-based metalloradical catalysis (MRC). The [Co(P1)]-catalyzed aziridination system, which operates under neutral and nonoxidative conditions, is generally applicable to a wide range of allylic sulfamoyl azides, allowing for high-yielding synthesis of the bicyclic aziridine molecules with various substitution patterns. Taking advantage of the high strain associated with the unique heterobicyclic structure, we have demonstrated a series of synthetic applications of the resulting [3.1.0]-bicyclic diaza aziridines as intermediates for preparation of valuable 1,2- and 1,3-diamines and related derivatives. The catalytic method was also showcased as a key step in the high-yielding new synthesis of the NK1 antagonist 13. Efforts are underway to study the detailed catalytic mechanism of this Co(II)-based radical intramolecular aziridination as well as to develop its asymmetric variant.
Supplementary Material
Acknowledgements
We are grateful for financial support by NIH (R01-GM102554) and in part by NSF (CHE-1624216).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Quiclet-Sire B, Zard SZ. Pure. Appl. Chem. 2011;83:519–551. [Google Scholar]; b) Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; c) Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Du J, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392–396. doi: 10.1126/science.1251511. For recent successful examples, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hoyt JM, Schmidt VA, Tondreau AM, Chirik PJ. Science. 2015;349:960–963. doi: 10.1126/science.aac7440. [DOI] [PubMed] [Google Scholar]; c) Jeffrey JL, Terrett JA, MacMillan DWC. Science. 2015;349:1532–1536. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J. Am. Chem. Soc. 2010;132:10891–10902. doi: 10.1021/ja103768r. For detailed studies on the involvement of α-Co(III)-alkyl radical intermediates (also known as Co(III)-carbene radicals) for catalytic cyclopropanation and C.H alkylation, see: [DOI] [PubMed] [Google Scholar]; b) Belof JL, Cioce CR, Xu X, Zhang XP, Space B, Woodcock HL. Organometallics. 2011;30:2739–2746. doi: 10.1021/om2001348. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lu H, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J. Am. Chem. Soc. 2011;133:8518–8521. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]; d) Cui X, Xu X, Jin L-M, Wojtas L, Zhang XP. Chem. Sci. 2015;6:1219–1224. doi: 10.1039/c4sc02610a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J. Am. Chem. Soc. 2011;133:12264–12273. doi: 10.1021/ja204800a. For detailed studies on the involvement of related α-Co(III)-aminyl radical intermediates (also known as Co(III)-nitrene radicals) for catalytic aziridination and C.H amination, see: [DOI] [PubMed] [Google Scholar]; f) O. Suarez AI, Jiang H, Zhang XP, de Bruin B. Dalton Trans. 2011;40:5697–5705. doi: 10.1039/c1dt10027k. [DOI] [PubMed] [Google Scholar]; g) Goswami M, Lyaskovskyy V, Domingos SR, Buma WJ, Woutersen S, Troeppner O, Ivanović-Burmazović I, Lu H, Cui X, Zhang XP, Reijerse EJ, DeBeer S, van Schooneveld MM, Pfaff FF, Ray K, de Bruin B. J. Am. Chem. Soc. 2015;137:5468–5479. doi: 10.1021/jacs.5b01197. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Suarez AIO, Lyaskovskyy V, Reek JNH, van der Vlugt JI, de Bruin B. Angew. Chem., Int. Ed. Angew. Chem. 2013;2013;52125:12510–12529. 12740–12760. doi: 10.1002/anie.201301487. For a recent review on nitrogen-centered radical ligands, see: [DOI] [PubMed] [Google Scholar]
- [4].a) Gansäuer A, Hildebrandt S, Vogelsang E, Flowers RA., II Dalton Trans. 2016;45:448–452. doi: 10.1039/c5dt03891j. For a recent review on Ti(III)-based MRC see: [DOI] [PubMed] [Google Scholar]; b) Nugent WA, RajanBabu TV. J. Am. Chem. Soc. 1988;110:8561–8562. For the first demonstration of Ti(III)-based metalloradical non-catalytic process, see: [Google Scholar]; c) Gansäuer A, Rinker B, Pierobon M, Grimme S, Gerenkamp M, Mück-Lichtenfeld C. Angew. Chem., Int. Ed. Angew. Chem. 2003;2003;42115:3687–3690. 3815–3818. doi: 10.1002/anie.200351240. For select examples of catalytic processes via Ti(III)-based MRC. [DOI] [PubMed] [Google Scholar]; d) Gansäuer A, Fan C-A, Keller F, Keil J. J. Am. Chem. Soc. 2007;129:3484–3485. doi: 10.1021/ja0686211. [DOI] [PubMed] [Google Scholar]; e) Gansäuer A, Fleckhaus A, Lafont MA, Okkel A, Kotsis K, Anoop A, Neese F. J. Am. Chem. Soc. 2009;131:16989–16999. doi: 10.1021/ja907817y. [DOI] [PubMed] [Google Scholar]; f) Gansäuer A, Hildebrandt S, Michelmann A, Dahmen T, von Laufenberg D, Kube C, Fianu GD, Flowers RA., II Angew. Chem., Int. Ed. Angew. Chem. 2015;2015;54127:7003–7006. 7109–7112. doi: 10.1002/anie.201501955. [DOI] [PubMed] [Google Scholar]
- [5].a) Cenini S, Gallo E, Penoni A, Ragaini F, Tollari S. Chem. Commun. 2000:2265–2266. For select examples of C–H amination via Co(II)-based MRC, see: [Google Scholar]; b) Harden JD, Ruppel JV, Gao G-Y, Zhang XP. Chem. Commun. 2007:4644–4646. doi: 10.1039/b710677g. [DOI] [PubMed] [Google Scholar]; c) Lu H, Tao J, Jones JE, Wojtas L, Zhang XP. Org. Lett. 2010;12:1248–1251. doi: 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]; d) Lu H, Jiang H, Wojtas L, Zhang XP. Angew. Chem., Int. Ed. 2010;49:10192–10196. doi: 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010;122:10390–10394. [Google Scholar]; e) Lu H, Jiang H, Hu Y, Wojtas L, Zhang XP. Chem. Sci. 2011;2:2361–2366. [Google Scholar]; f) Lu H, Hu Y, Jiang H, Wojtas L, Zhang XP. Org. Lett. 2012;14:5158–5161. doi: 10.1021/ol302511f. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Jin L-M, Lu H, Cui Y, Lizardi CL, Arzua TN, Wojtas L, Cui X, Zhang XP. Chem. Sci. 2014;5:2422–2427. doi: 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Lu H, Li C, Jiang H, Lizardi CL, Zhang XP. Angew. Chem., Int. Ed. 2014;53:7028–7032. doi: 10.1002/anie.201400557. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014;126:7148–7152. [Google Scholar]; i) Lu H, Zhang XP. Chem. Soc. Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. For a review on C.H amination via Co(II)-based MRC, see: [DOI] [PubMed] [Google Scholar]
- [6].a) Jones JE, Ruppel JV, Gao G-Y, Moore TM, Zhang XP. J. Org. Chem. 2008;73:7260–7265. doi: 10.1021/jo801151x. Co(II)-based MRC has been applied for catalytic intermolecular aziridination with different organic azides. For phosphoryl azides, see: [DOI] [PubMed] [Google Scholar]; b) Tao JR, Jin L-M, Zhang XP. Beilstein J. Org. Chem. 2014;10:1282–1289. doi: 10.3762/bjoc.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org Lett. 2008;10:1995–1998. doi: 10.1021/ol800588p. For arylsulfonyl azides, see: [DOI] [PubMed] [Google Scholar]; d) Subbarayan V, Ruppel JV, Zhu S, Perman JA, Zhang XP. Chem Commun. 2009:4266–4268. doi: 10.1039/b905727g. [DOI] [PubMed] [Google Scholar]; e) Jin L-M, Xu X, Lu H, Cui X, Wojtas L, Zhang XP. Angew. Chem. Int. Ed. 2013;52:5309–5313. doi: 10.1002/anie.201209599. For fluoroaryl azides, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125:5417–5421. [Google Scholar]; f) Subbarayan V, Jin L-M, Cui X, Zhang XP. Tetrahedron Lett. 2015;56:3431–3434. doi: 10.1016/j.tetlet.2015.01.186. For aryloxysulfonyl azides, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Khettache N, Berredjem M, Régaïnia Z, Aouf NE. Heterocycles. 2008;75:2243–2250. Only one example was reported for synthesis of 2-sulfonyl-1,3-diazabucyclo[3.1.0]hexane structure via non-catalytic process, see: [Google Scholar]; b) Duran F, Leman L, Ghini A, Burton G, Dauban P, Dodd RH. Org. Lett. 2002;4:2481–2483. doi: 10.1021/ol0200899. The related intramolecular aziridination of sulfamate derived from an allyl alcohol failed to give the [3.1.0]-bicyclic aziridine product presumably due to the high ring strain, see: [DOI] [PubMed] [Google Scholar]
- [8].a) Wehn PM, Du Bois J. Angew. Chem. Int. Ed. Angew. Chem., 2009;2009;48121:3802–3805. 3860–3863. doi: 10.1002/anie.200806292. For synthetic application for aziridination, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Möϐner C, Bolm C. Catalyzed Asymmetric Aziridination in Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals (Second Edition) Wiley-VCH Verlag GmbH; Weinheim, Germany: 2008. [Google Scholar]
- [9].b) Robert C. Patent. 2008 WO02060859 (A2) [Google Scholar]; b) Wuensch B, Schepmann D, Bourgeois C. Patent. 2009 WO2009080745 (A2) [Google Scholar]
- [10].b) Wender PA, Strand D. J. Am. Chem. Soc. 2009;131:7528–7529. doi: 10.1021/ja901799s. [DOI] [PubMed] [Google Scholar]; b) Dauban P, Malik G. Angew. Chem. Int. Ed. 2009;48:9026–9029. doi: 10.1002/anie.200904941. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121:9188–9191. [Google Scholar]
- [11].Liew SK, He Z, Denis JD, St., Yudin AK. J. Org. Chem. 2013;78:11637–11645. doi: 10.1021/jo401489q. [DOI] [PubMed] [Google Scholar]
- [12].a) Müller P, Fruit C. Chem. Rev. 2003;103:2905–2920. doi: 10.1021/cr020043t. For select reviews on aziridination via transition metal catalysis, see: [DOI] [PubMed] [Google Scholar]; b) Fantauzzi S, Caselli A, Gallo E. Dalton Trans. 2009:5434–5443. doi: 10.1039/b902929j. [DOI] [PubMed] [Google Scholar]; c) Pellissier H. Tetrahedron. 2010;66:1509–1555. [Google Scholar]; d) Jiang H, Zhang XP. In: Oxidation: C.N Bond Formation by Oxidation (Aziridines) Carreira EM, Yamamoto H, editors. Vol. 5. Elsevier; Amsterdam: 2012. pp. 168–182. Comprehensive Chirality. [Google Scholar]
- [13].a) Müller P, Baud C, Jacquier Y. Can. J. Chem. 1998;76:738–750. For the synthesis of [3.1.0]- and [4.1.0]-bicyclic sulfonyl aziridines via catalytic intramolecular aziridination, see: [Google Scholar]; b) Dauban P, Dodd RH. Org. Lett. 2000;2:2327–2329. doi: 10.1021/ol000130c. [DOI] [PubMed] [Google Scholar]; c) Dauban P, Saniere L, Tarrade A, Dodd RH. J. Am. Chem. Soc. 2001;123:7707–7708. doi: 10.1021/ja010968a. [DOI] [PubMed] [Google Scholar]; d) Liang JL, Yuan SX, Chan PWH, Che C-M. Org. Lett. 2002;4:4507–4510. doi: 10.1021/ol0270475. [DOI] [PubMed] [Google Scholar]; e) Liang JL, Yuan SX, Chan PWH, Che C-M. Tetrahedron Lett. 2003;44:5917–5920. [Google Scholar]; f) Liang JL, Yuan SX, Huang JS, Che C-M. J. Org. Chem. 2004;69:3610–3619. doi: 10.1021/jo0358877. [DOI] [PubMed] [Google Scholar]; g) Padwa A, Flick AC, Leverett CA, Stengel T. J. Org. Chem. 2004;69:6377. doi: 10.1021/jo048990k. [DOI] [PubMed] [Google Scholar]; h) Liu P, Wong EL-M, Yuen AW-H, Che C-M. Org. Lett. 2008;10:3275–3278. doi: 10.1021/ol801157m. [DOI] [PubMed] [Google Scholar]; i) Kiefer L, Gorojankina T, Dauban P, Faure H, Ruat M, Dodd RH. Bioorg. Med. Chem. Lett. 2010;20:7483–7487. doi: 10.1016/j.bmcl.2010.10.006. [DOI] [PubMed] [Google Scholar]
- [14].a) Guthikonda K, Du Bois J. J. Am. Chem. Soc. 2002;124:13672–13673. doi: 10.1021/ja028253a. For the synthesis of [4.1.0]- and [5.1.0]-bicyclic sulfonated aziridines via catalytic intramolecular aziridination. [DOI] [PubMed] [Google Scholar]; b) Wehn PM, Lee JH, Du Bois J. Org. Lett. 2003;5:4823–4826. doi: 10.1021/ol035776u. [DOI] [PubMed] [Google Scholar]; c) Guthikonda K, Wehn PM, Caliando BJ, Du Bois J. Tetrahedron. 2006;62:11331–11342. [Google Scholar]; d) Esteoule A, Duran F, Retailleau P, Dodd RH, Dauban P. Synthesis. 2007:1251–1260. [Google Scholar]; e) Harvey ME, Musaev DG, Du Bois J. J. Am. Chem. Soc. 2011;133:17207–17216. doi: 10.1021/ja203576p. [DOI] [PubMed] [Google Scholar]; f) Malik G, Esteoule A, Retailleau P, Dauban P. J. Org. Chem. 2011;76:7438–7448. doi: 10.1021/jo201209x. [DOI] [PubMed] [Google Scholar]; g) Valle MS, Saraiva MF, Retailleau P, de Almeida MV, Dodd RH. J. Org. Chem. 2012;77:5592–5599. doi: 10.1021/jo300468j. [DOI] [PubMed] [Google Scholar]
- [15].Kurokawa T, Kim M, Du Bois J. Angew. Chem. Int. Ed. 2009;48:2777–2779. doi: 10.1002/anie.200806192. For the synthesis of [4.1.0]- and [5.1.0]-bicyclic sulfamoyl aziridines via catalytic intramolecular aziridination. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009;121:2815–2817. [Google Scholar]
- [16].b) de Figueiredo RM. Angew. Chem. Int. Ed. 2009;48:1190–1193. doi: 10.1002/anie.200804362. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121:1212–1215. [Google Scholar]; b) Muñniz K, Streuff J, Hövelmann CH, Núñnez A. Angew. Chem. Int. Ed. 2007;46:7125–7127. doi: 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007;119:7255–7258. [Google Scholar]; c) McDonald RI, Stahl SS. Angew. Chem. Int. Ed. 2010;49:5529–5532. doi: 10.1002/anie.200906342. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010;122:5661–5664. doi: 10.1002/ange.200906342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].b) Shue H-J, Chen X, Shih N-Y, Blythin DJ, Paliwal S, Lin L, Gu D, Schwerdt JH, Shah S, Reichard GA, Piwinski JJ, Duffy RA, Lachowicz JE, Coffin VL, Liu F, Nomeir AA, Morgan CA, Varty GB. Bioorg. Med. Chem. Lett. 2005;15:3896–3901. doi: 10.1016/j.bmcl.2005.05.111. [DOI] [PubMed] [Google Scholar]; b) Shue H-J, Chen X, Schwerdt JH, Paliwal S, Blythin DJ, Lin L, Gu D, Wang C, Reichard GA, Wang H, Piwinski JJ, Duffy RA, Lachowicz JE, Coffin VL, Nomeir AA, Morgan CA, Varty GB, Shih N-Y. Bioorg. Med. Chem. Lett. 2006;16:1065–1069. doi: 10.1016/j.bmcl.2005.10.072. [DOI] [PubMed] [Google Scholar]
- [18].b) Varty GB, Cohen-Williams ME, Hunter JC. Behav. Pharmacol. 2003;14:87–95. doi: 10.1097/00008877-200302000-00009. [DOI] [PubMed] [Google Scholar]; b) Varty GB, Cohen-Williams ME, Morgan CA, Pylak U, Duffy RA, Lachowicz JE, Carey GJ, Coffin VL. Neuropsychopharmacol. 2002;27:371–379. doi: 10.1016/S0893-133X(02)00313-5. [DOI] [PubMed] [Google Scholar]; c) Hesketh PJ. Support. Care Cancer. 2004;12:550–554. doi: 10.1007/s00520-004-0651-0. [DOI] [PubMed] [Google Scholar]
- [19].Wen Y, Zhao B, Shi Y. Org. Lett. 2009;11:2365–2368. doi: 10.1021/ol900808z. For a recent synthesis of analogous compounds via Cu(I)-catalyzed diamination, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.