

Abstract
The pneumococcal autolysin LytA is a key virulence factor involved in several important functions including DNA competence, immune evasion and biofilm formation. Here, we present the 1.05 Å crystal structure of the catalytic domain of LytA in complex with a synthetic cell-wall-based peptidoglycan (PG) ligand that occupies the entire Y-shaped substrate-binding crevice. As many as twenty-one amino-acid residues are engaged in ligand interactions with a majority of these interactions directed towards the glycan strand. All saccharides are intimately bound through hydrogen bond, van der Waals and CH-π interactions. Importantly, the structure of LytA is not altered upon ligand binding, whereas the bound ligand assumes a different conformation compared to the unbound NMR-based solution structure of the same PG-fragment. Mutational study reveals that several non-catalytic glycan-interacting residues, structurally conserved in other amidases from Gram-positive Firmicutes, are pivotal for enzymatic activity. The three-dimensional structure of the LytA/PG complex provides a novel structural basis for ligand restriction by the pneumococcal autolysin, revealing for the first time an importance of the multivalent binding to PG saccharides.
Keywords: X-ray crystal structure, N-acetylmuramoyl l-alanine amidase, peptidoglycan, autolysin, LytA, cell wall hydrolase, Streptococcus pneumoniae, virulence factor
Introduction
LytA, the major autolysin of the human respiratory pathogen Streptococcus pneumoniae, is likely the most intensively studied of all peptidoglycan (PG)-degrading amidases. LytA is involved in several important processes during pneumococcal pathogenesis, including fratricidal lysis during DNA competence, immune evasion, facilitation of the release of pneumolysin and contribution to biofilm formation through the release of extracellular DNA and other structural biofilm components (Eldholm et al., 2009, Martner et al., 2008, Moscoso et al., 2006, Martner et al., 2009, Ramos-Sevillano et al., 2015). However, LytA is not essential for normal growth and lytA-deficient strains do not display any cell-morphological phenotypes besides a tendency for bacterial chain formation in certain strain backgrounds (Sanchez-Puelles et al., 1986, Meroueh et al., 2006, Lacks, 1970, Tomasz et al., 1970). Nonetheless, lytA is one of the most highly conserved pneumococcal virulence genes and is present in all clinical isolates, underscoring its essential contribution to pneumococcal colonization and virulence. Indeed, due to its high conservation, lytA is a target-gene for clinical diagnostic identification of S. pneumoniae (Rubin & Rizvi, 2004, Sheppard et al., 2004). Interestingly, LytA-mediated lysis is induced following pneumococcal treatment with cell wall-targeting antibiotics such as penicillin or vancomycin which contributes to the bactericidal effects of these drugs (Tomasz & Waks, 1975). However, it is still unclear how and why these drugs induce activation of LytA or similar autolysins in other bacteria. In fact, despite an immense interest in LytA it is far from clear how the lytic activity of this enigmatic enzyme is being regulated during normal growth and during infection. We have previously provided experimental data supporting the control of LytA lytic activity at the level of substrate recognition and the fact that protein complexes that constitute the cell-wall-synthesis machinery appear to control LytA activity through substrate (PG) sequestration (Mellroth et al., 2012).
LytA is a two-domain protein with an N-terminal catalytic N-acetylmuramoyl l-alanine amidase domain and a C-terminal choline-binding domain. LytA associates with the cell wall through its choline-binding domain by non-covalent interactions with phospho-choline molecules present within the pneumococcal teichoic acids (Fernandez-Tornero et al., 2001). The cell-wall-degrading activity of LytA is mediated by the amidase domain that specifically hydrolyzes the lactyl-amide bonds that link peptide stems to glycan chains in the PG (Howard & Gooder, 1974). We have previously reported the crystal structure of the substrate-free amidase domain of LytA, at 1.05 Å (Mellroth et al., 2014). The amidase domain comprised a large and shallow Y-shaped substrate-binding crevice with a zinc-containing catalytic center positioned at the branch point. Our biochemical in vitro data demonstrated that LytA could not cleave small muropeptides containing one or two saccharides (M5P or GM5P), but instead required a larger PG fragment named di(GM5P) spanning four contiguous saccharides, two alternating N-acetyl glucosamine (NAG) and N-acetyl muramic acid (NAM) residues (i.e. NAG1-NAM2-NAG3-NAM4), linked to the stem peptides (l-Ala-γ-d-Glu-l-Lys-d-Ala-d-Ala) via the lactyl moieties of the NAM residues (Hesek et al., 2004, Mellroth et al., 2014).
Here, we present the crystal structure of a catalytically inactive amidase domain of LytA in complex with the substrate di(GM5P), which reveals how a substrate occupies the entire binding crevice of LytA. The atomic resolution of the structure allows for precise determination of the conformation of the substrate as well as for an investigation of the role of the twenty-one substrate-interacting residues within the binding crevice. Mutational analysis demonstrates that several glycan-interacting residues located outside the catalytic site play nonetheless critical roles in the lytic activity. When bound into the binding crevice of this inactive variant of LytA, the di(GM5P) substrate adopts a different conformation compared to the solution structure of the same compound previously determined by nuclear-magnetic resonance (Meroueh et al., 2006). These findings shed light on the multivalent interactions between the catalytic domain of LytA and the PG component of the cell wall.
Results and Discussion
Di(GM5P), tightly bound on the surface of LytA, occupies the entire Y-shaped binding crevice
In order to determine the crystal structure of the N-terminal amidase domain of LytA (LytAAmi) with a PG fragment, we co-crystallized the inactive LytAAmi (C60A, H133A, C136A) variant (Mellroth et al., 2014) with the synthetic substrate di(GM5P) (Supporting Information, Fig. S1). The crystal structure of the complex was refined to 1.05Å resolution (Table I), revealing an overall excellent quality of the electron density for the entire protein chain. An unambiguous Fo-Fc electron density was found within the substrate-binding cleft after molecular replacement (Fig. 1A). The atomic resolution data allowed modeling of 105 substrate atoms, including those for all four saccharide residues NAG1-NAM2-NAG3-NAM4 and the four amino-acid residues l-Ala5-d-γ-Glu6-l-Lys7-d-Ala8 of the first peptide stem that occupies the binding crevice, as well as the two residues l-Ala9-d-γ-Glu10 of the second peptide stem that projects outside the binding cleft at the end of the glycan-binding-groove (Fig. 1A). The fifth residue of the first peptide stem (the C-terminal d-Ala) and three residues within the second peptide stem were not seen in the electron density due to their flexibility and were therefore not included in the final model. Simulated annealing omit Fo-Fc difference map allows unambiguous inclusion of all four sugars, four amino acids from the first peptide stem as well as two amino acids from the second stem into the final model (Fig.1A). Thus, the entire LytA substrate-binding crevice is occupied by a PG fragment, that comprises four contiguous saccharides and a tetrapeptide linked to the NAM2 moiety within the catalytic center.
Table I. Data collection and refinement statistics for the crystal structure of the LytAAmi/di(GM5P) complex (PDB code 5CTV).
| Data collection | |
|---|---|
| Wavelength (Å) | 0.9724 |
| Space group | P212121 |
| a (Å), b (Å), c (Å) | 44.6 54.6 78.9 |
| Resolution (Å) | 39.44 – 1.05 (1.07 – 1.05) |
| No. of observed reflections | 531390 (21850) |
| No. of unique reflections | 90326 (4399) |
| Multiplicity | 5.9 (5.0) |
| Completeness (%) | 99.8 (98.9) |
| 1Rmerge (%) | 9.4 (65.1) |
| CC(1/2)(%) | 99.8 (77.1) |
| I/σ(I) | 10.2 (2.1) |
| Refinement statistics | |
| Resolution of Data (Å) | 39.50 – 1.05 |
| 2Rcryst (%) | 14.05 |
| 3Rfree (%) | 16.05 |
| Number of protein atoms | 1405 |
| Water molecules | 284 |
| Number of substrate atoms | 105 |
| Rmsd from ideal geometry | |
| Bond length (Å) | 0.006 |
| Bond angles (deg.) | 1.283 |
| Ramachandran Plot (%) | |
| Residues in preferred regions | 99.4 |
| Residues in allowed regions | 0.6 |
| Outliers | 0 |
| B- value (Å2) | |
| B-value from Wilson plot | 9.9 |
| Protein | 7.8 |
| Water | 22.1 |
| Substrate | 12.5 |
Values in parentheses are for the highest resolution shell
Rmerge = Σhkl Σi |Ii (hkl) - ‹I (hkl)›|/ Σhkl Σi Ii (hkl), where Ii(hkl) is the ith observation of reflection hkl and ‹I (hkl)› is the weighted average intensity for all observations i of reflection hkl.
Rcryst = Σ‖Fo| - |Fc‖/Σ|Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes of a particular reflection and the summation is over 95 % of the reflections in the specified resolution range. The remaining 5 % of the reflections were randomly selected (test set) before the structure refinement and not included in the structure refinement.
Rfree was calculated over these reflections using the same equation as for Rcryst.
Figure 1. The di(GM5P) substrate occupies the entire Y-shaped binding crevice of LytA.

A. An unambiguous positive electron density (2.5 σ) colored in green is present in the Fo-Fc electron density map calculated for the final model with omitted ligand di(GM5P) after several cycles of the simulated annealing performed in Phenix (Adams et al., 2010). The final model of di(GM5P), colored in yellow, fits perfectly within the Y-shaped substrate-binding groove. The different moieties comprising the di(GM5P) substrate are annotated in blue. The surface of the LytA amidase domain is colored white.
B. Orthogonal view of the LytAAmi/di(GM5P) complex reveals that the scissile bond formed between the lactyl moiety of NAM2 and residue L-Ala5 is buried within the active site of LytA underneath the carbohydrate chain. The LytA protein is displayed as a semitransparent white surface. The carbon atoms of the glycan chain and the peptide stems are in yellow and cyan, respectively.
When bound in the shallow binding crevice, a significant portion of the substrate is exposed to the solvent (Fig. 1A and 1B). The conformation of the bound substrate renders the position of the sugars perpendicular to the stem peptides (Fig. 1B). The N-acetyl and the lactyl moieties of NAM2 project towards the active site such that the scissile bond is buried under the saccharide rings in a conformation that is similar to that found in human PGRP-IαC (Guan et al., 2006), AmiD from Escherichia coli (Kerff et al., 2010) and AmpDh3 from Pseudomonas aeruginosa (Lee et al., 2013).
Substrate binding does not alter the conformation of the amidase domain of LytA
The overall three-dimensional structures of the substrate-free LytAAmi (Mellroth et al., 2014) and LytAAmi in complex with di(GM5P) presented here are remarkably similar. Indeed, superimposition of these crystal structures results in a root-mean-square deviation (rmsd) value of 0.34 Å and 0.42 Å for 172 Cα atoms compared to LytAAmi and full-length LytA, respectively (Li et al., 2015), demonstrating that no major conformational change is induced within the amidase structure upon substrate binding (Fig. 2). Only two LytA residues, Glu48 and Asp146, localized on different loops around the substrate-binding crevice, shift their main-chain atoms by more than 0.9 Å upon substrate binding (Fig. 2A). While Glu48 comes closer to l-Lys7 in the PG first stem peptide forming a weak salt bridge interaction, the side chain of Asp146 adopts a different conformation to interact with NAG1 (Fig. 2A). Thus, the substrate-free conformation of the amidase domain of LytA is poised to bind to its substrate. Such a structural pre-organization has also been described in other PG-binding proteins with similar fold. For example, the conformation of the Staphylococcus aureus-associated autolysin AmiA does not change upon binding to a muramyl-tetrapeptide substrate (Buttner et al., 2014). Similarly, the P. aeruginosa AmpDh2 (Martinez-Caballero et al., 2013) and AmpDh3 (Lee et al., 2013), as well as the human PG-recognition protein I alpha (Guan et al., 2006) all maintain similar structures in free form and in complex with their ligands.
Figure 2. Di(GM5P) binding does not alter the conformation of LytA.

A. Superimposition of LytA/di(GM5P) on the substrate-free LytA, in white and green, respectively, demonstrates that the amidase does not change conformation upon substrate binding. The conformation of the side chains of glutamate and aspartate residues Glu48 and Asp146 are altered upon complex generation. A violet sphere represents the catalytic Zn2+ ion in the free form of LytA. Substrate and LytA residues are labeled in blue and black, respectively.
B. Comparison of active-site residues in substrate-free LytA and the LytA-di(GM5P) complex in green and grey, respectively, reveals that the water molecule Wat1 replaces the Zn2+ ion, absent in the LytA-di(GM5P) structure. A second water molecule, Wat2, is sequestered at the location of the imidazole ring of residue His133 in the substrate free amidase. The Zn-coordinating residues Asp149 and His26 maintain their positions in both the unbound LytA and the LytA/di(GM5P) complex, whereas the side chains of the catalytic residues His147 and Glu87 are rotated upon interactions with the substrate.
Superimposition of LytAAmi and LytAAmi/di(GM5P) also demonstrates that neither mutation of the zinc-ligand His133 to an alanine nor the absence of the Zn2+ ion affect the conformation of the active site (Fig. 2B). Indeed, the conformations of the Zn-coordinating residues His26 and Asp149 are not altered in the absence of the catalytic zinc ion in the mutant protein. Two water molecules (Wat1 and Wat2) replace the catalytic Zn ion and the missing imidazole moiety of residue His133, respectively (Fig. 2B). In contrast, both catalytic residues Glu87 and His147 are more mobile and adopt different rotamer states upon substrate binding though the position of the main-chain atoms is preserved. The carboxylate of Glu87 in the LytAAmi/di(GM5P) complex forms a hydrogen bond with the lactyl oxygen of the central N-acetyl-muramoyl residue NAM2 (torsion angles χ3 of residue Glu87 are -59° and -11.7° in the unbound and complexed LytA, respectively) (Fig. 2B). However, it should be noted that in the recently determined crystal structure of the full-length unbound LytA, which crystallized in different conditions (Li et al., 2015), the conformation of Glu87 is similar to that in the LytA/di(GM5P) (χ3 = +11.4°). Thus the side chain of Glu87 is rather mobile in unbound state and can fluctuate between different rotamers.
The side chain of His147, which participates in several important interactions with the bound substrate, makes a 90° rotation positioning itself parallel to the hexose ring of NAG1 with its Nδ1 atom turned towards the scissile bond. Consequently, a hydrogen bond is formed between atoms Nε2 in His147 and O6 in NAG1 (Fig. 2B). These new orientations of the two catalytic residues Glu87 and His147 agree well with the reaction mechanism previously suggested by us and others in which Glu87 and the Zn2+ ion polarize the scissile bond (Mellroth et al., 2014, Zoll et al., 2010, Buttner et al., 2014).
Glycan strand/LytA interactions are essential for amidase lytic activity
The overall interaction between LytA and the substrate di(GM5P) is extensive with 21 LytA residues that form an intricate network of hydrogen bond, van-der-Waals and CH-π interactions with the tetrasaccharide-dipeptide motif of di(GM5P) (Fig. 3A-D and Supporting Information Fig. S2). It should be noted that each substrate peptide is composed of five residues. Interestingly, interactions between LytA and the glycan strand are far more numerous than with the peptide stem. Ten hydrogen-bond interactions are formed between the carbohydrate moieties of the substrate and LytA residues, while the peptide stem forms only four hydrogen bonds with LytA, all with the d-γ-Glu6 atoms (Supporting Information, Fig. S2). Furthermore, di(GM5P) forms five additional water-mediated hydrogen bonds, two with the peptide stem, two with NAG3 and one with NAM4.
Figure 3. Binding of the di(GM5P) tetrasaccharide occurs through several van der Waals, CH-π and hydrogen bond interactions.

The overall structure of the LytA/di(GM5P) complex is presented in the center of the figure in the same orientation as in Figure 2. The position of the Zn2+ ion is represented by a violet sphere. Protein residues, substrate saccharides and substrate peptide stems are in grey, yellow and cyan, respectively. Regions of interest are highlighted around the overall structure.
A. The substrate NAG1 moiety forms CH-π stacking and hydrogen bonds with LytA residue His147 and are further stabilized through hydrogen bonds with Asp146 and Val148.
B. The orientation of the NAM2 and NAG3 moieties is enforced by van der Waals and hydrogen bonds with LytA residues Thr28, Gly29, Asn30, Glu38 and water molecules (red spheres) through Lys45.
C. The substrate NAM4 moiety forms CH-π stacking interactions with residue Tyr41 and hydrogen bonds with Arg44.
D. The stem peptide d-γ-Glu6 residue in di(GM5P) forms extensive interactions with LytA residues. Several LytA residues, including Asp73, Gly75, Asn79 and Ser145, interact extensively with the d-γ-Glu6 section of the peptide stem of di(GM5P).
Additionally, the pyranose rings of the two terminal sugars NAG1 and NAM4 form CH-π stacking interactions with the imidazole and phenol rings of residues His147 and Tyr41, respectively (Fig. 3A and 3D). CH-π interactions between saccharides and aromatic amino-acid residues are key for molecular recognition of carbohydrates (Chen et al., 2013). We evaluated the impact of all substrate-interacting residues by mutagenesis in a turbidometric autolysis assay where the relative lysis rate of the targeted point mutant proteins was determined in relation to the activity of wild-type LytA using stationary phase T4ΔlytA cells as a substrate (Table II and Supporting Information Fig. S3). A whole cell lysis assay was used for the comparison instead of purified peptidoglycan since LytA requires association with choline containing teichoic acids for full activity (Giudicelli & Tomasz, 1984). Comparative circular dichroism (CD) spectroscopy analysis of mutants with reduced activity was performed to verify that the introduced mutations did not affect folding nor the secondary structure of the modified proteins (Supporting Information Fig. S4). Surprisingly, our results demonstrated that several murein-saccharide interacting LytA residues localized outside the catalytic site were crucial for the autolytic activity of LytA (Table II). Indeed, mutation of Tyr41 to either alanine or arginine drastically decreased LytA activity, whereas substitution to histidine only slightly affected activity, confirming the importance of this CH-π interaction. Similarly, mutation of His147 to alanine also caused a significant reduction in enzymatic activity (Table II). The terminal substrate saccharides NAG1 and NAM4 are further stabilized by hydrogen bonds formed with the side chain of Asp146, the backbone oxygen of Val148 and the side chain of Arg44 (Fig. 3A, Fig. 3D, Supporting Information Fig. S2 and Table II).
Table II. Substrate interacting residues of LytA, their respective ligand partner and relative impact of point mutations on lytic activity of LytA.
| Liganda | LytA residue | Type of interactionb | Mutation | Activityc | Referenced | |
|---|---|---|---|---|---|---|
| Catalytic center | Zn | His26 | Co-ordinate | H26A | - | (Mellroth et al., 2014) |
|
| ||||||
| Zn | His133 | Co-ordinate | H133A | - | (Mellroth et al., 2014) | |
|
| ||||||
| Zn | Asp149 | Co-ordinate | D149A | - | (Mellroth et al., 2014) | |
|
| ||||||
| Catalytic L-Ala5 | Glu87 | H-bond | E87A | - | (Mellroth et al., 2014) This study | |
|
| ||||||
| Catalytic NAG1 | His147 | CH-π + H | H147A/K | * | (Mellroth et al., 2014) This study | |
|
| ||||||
| Glycan interaction | NAG1 | Asp146 | H × 2 | D146A/L | *** | This study |
| D146N | ***** | This study | ||||
|
| ||||||
| NAG1 | Val148 | H | V148E/K | *** | (Mellroth et al., 2014) | |
|
| ||||||
| NAM2 | Ser27 | vdW | S27A | *** | This study | |
|
| ||||||
| NAM2 | Thr28 | H+vdW | T28A/S/V | *** | This study | |
| T28I | * | This study | ||||
| T28G | - | This study | ||||
|
| ||||||
| NAM2 | Gly29 | vdW | G29A | **** | (Mellroth et al., 2014) | |
|
| ||||||
| NAM2 | Phe52 | vdW | F52A/V | ***** | (Mellroth et al., 2014) | |
|
| ||||||
| NAG3 | Asn30 | H | N30A | *** | This study | |
| N30L/Q | *** | (Mellroth et al., 2014) | ||||
|
| ||||||
| NAG3 | Ser33 | vdW | S33A | **** | (Mellroth et al., 2014) | |
| S33Q | * | (Mellroth et al., 2014) | ||||
|
| ||||||
| NAG3 | Glu38 | H | E38A/Q/R | - | This study | |
| E38D | ** | This study | ||||
|
| ||||||
| NAM4 | Tyr41 | CH-π | Y41A/R | * | (Mellroth et al., 2014) | |
| Y41H | **** | This study | ||||
|
| ||||||
| NAM4 | Arg44 | H × 2 | R44A | ***** | This study | |
| R44E/Q | *** | (Mellroth et al., 2014) | ||||
|
| ||||||
| NAM4 | Lys45 | H (via H2O) | K45A | ** | This study | |
| K45R | ***** | (Mellroth et al., 2014) | ||||
|
| ||||||
| Peptide interaction | D-Glu6 | Gly75 | H | G75A | *** | (Mellroth et al., 2014) |
|
| ||||||
| D-Glu6 | Asn79 | H | N79A | *** | (Mellroth et al., 2014) | |
|
| ||||||
| D-Glu6 | Ser145 | H | S145A | ***** | (Mellroth et al., 2014) | |
|
| ||||||
| L-Lys7 | Glu48 | Weak SB | E48A | ***** | This study | |
|
| ||||||
| L-Lys7 | Trp72 | vdW | W72A | *** | (Mellroth et al., 2014) | |
|
| ||||||
| D-Ala8 | His144 | vdW | H144A | ***** | (Mellroth et al., 2014) | |
|
| ||||||
| L-Ala9 (stem 2) | Asn37 | vdW | N37A | **** | This study | |
The co-factor or substrate residue which is ligand for the given residue of LytA.
H = hydrogen bond, vdW = van der Waals force, CH-π interaction, SB – salt bridge
Lytic activity of mutant protein against pneumococcal cells in a turbidometric lysis assay in relation to the activity of wt-LytA protein. Stars indicate the following intervals:
(100 – 80%),
(79 – 60%);
(59 – 40%);
(39 – 20%);
(19 – 5%); - (4 – 0%)
Interactions with the central sugars NAM2 and NAG3 are also important for the catalytic activity of LytA. Both saccharides are kept tightly bound by several hydrogen bonds and van der Waals interactions with LytA residues (Fig. 3B and Supporting Information Fig. S2). The hydrogen bonds formed by main-chain oxygens of Thr28 and Glu38 to NAM2 and NAG3, respectively, appear to be essential since mutations of Thr28 or Glu38 to glycine and alanine, respectively, abolish enzymatic activity (Table II). The hexose ring of the central muramic acid NAM2 is parallel to the peptide bonds of the stretch of LytA residues Thr28-Gly29-Asn30 (Fig. 3B). Hydrogen bonds are formed on one side of the binding crevice between the nitrogen of N-acetylamino group of NAM2 and the main-chain carbonyl group of Thr28, and on the other side between the oxygen of the same N-acetylamino group of NAM2 and Lys45, via two water molecules. Also, the oxygen of the lactyl group forms a hydrogen bond with the side chain of the catalytic residue Glu87 (Fig. 3B). A similar interaction between the corresponding threonine residue and the substrate has been previously described in the S. aureus AmiA/M4P co-crystal (Buttner et al., 2014). Although the side chain of Thr28 faces the interior of the protein, away from the active site, any mutation of this residue significantly impairs autolytic activity of the amidase (Table II). Indeed, while substitution of Thr28 to a glycine yielded a fully inactive enzyme, introduction of a bulkier side chain through mutation to an isoleucine severely affected activity to below 20 % compared to wild type LytA (Table II). Even LytA variants with relatively similar residues at this position (Thr28Ala, Thr28Ser and Thr28Val) displayed reduced activity suggesting that the smallest structural perturbation alters the complementarity of the substrate-binding groove wall and the central part of the substrate, profoundly affecting catalysis.
The NAG3 moiety forms hydrogen bonds with LytA residues Asn30 and Glu38 (Fig. 3B). While mutation of Asn30 to alanine, leucine or glutamine causes a 50% decrease, substitution of Glu38 to alanine, glutamine or arginine nearly abolishes LytA lytic activity (Table II). Even the conservative substitution of Glu38 to aspartate reduces drastically lytic activity, demonstrating the importance of this fine-tuned hydrogen-bond interaction (Table II). Thus, His147, Thr28, Glu38 and Tyr41 form essential hydrogen bonds and/or CH-π interactions to each respective sugar moiety within the tetrasaccharide.
The stem peptide is loosely bound as a potential adaptation to allow for residues modifications
The electron density for the first three amino acids of the stem peptide (l-Ala5-d-γ-Glu6-l-Lys7) is of very good quality, allowing unambiguous modeling of this part of the substrate. The first residue of the peptide stem, L-Ala5 does not form any hydrogen bond with LytA (Fig.3C). We believe that absence of interactions between L-Ala5 and LytA is important, allowing for conformational rearrangement in this region of the substrate. The main anchor of the peptide stem, d-γ-Glu6 forms three hydrogen bonds with the hydroxyl of Ser145, the side chain of Asn79, a residue conserved among most Gram-positive bacterial amidases Fig. S5 and (Zoll et al., 2010), and the main-chain nitrogen atom of Gly75 in LytA (Fig. 3C). A similar role has been found for the corresponding residue in the staphylococcal amidases AmiE (Zoll et al., 2010) and AmiA (Buttner et al., 2014). It should be noted that the α-carboxylate of d-γ-Glu6 is regularly modified to a carboxamide in Gram-positive bacteria by the GatD/MurT enzyme-complex (Figueiredo et al., 2012, Munch et al., 2012). Such a modification would still be well accommodated in the binding pocket of LytA since the NH2 group of the α-carboxamide would remain solvent accessible and would not form any direct hydrogen bond with LytA residues. We have previously demonstrated that substitution of Ser145 to an alanine did not affect the activity of LytA on pneumococcal cells (that mainly contain an amidated D-γ-Glu) (Table II). Using the same di(GM5P) substrate we have also previously demonstrated that wild-type LytA cleaves this ligand in vitro (Mellroth et al., 2014). Thus interactions between Ser145 and D-γ-Glu6 are insignificant to lytic activity and our results indicate that both amidated and unmodified d-γ-Glu6 are tolerated by LytA (Table II).
In contrast to d-γ-Glu6, l-Lys7 in the stem peptide forms only moderate interactions with LytA, with its NZ atom located at 3.7 Å from the carboxylate of residue Glu48 and the hydrophobic part of its side chain forming van der Waals contacts with the indole moiety of residue Trp72 (Fig. 3C). The side chain of the l-Lys7 moiety in the substrate is frequently linked to another stem peptide via an inter-peptide bridge composed of l-Ala-l-Ser or l-Ala-l-Ala in intact pneumococcal PG (Garcia-Bustos et al., 1987).
The quality of the electron density for the terminal residue d-Ala9 of the peptide stem is poorer, abrogating reliable modeling of the terminal d-Ala. This residue is removed in the 4,3-transpeptidation or dd-carboxypeptidation reactions during cell wall crosslinking and the penultimate d-Ala8 may also be displaced in the course of 3,3-transpeptidation or ld-carboxypeptidase reactions (Hoyland et al., 2014, Fisher & Mobashery, 2015). Hence, the two terminal d-Ala residues are often leaving groups in reactions that PG undergoes and are not tightly retained by enzymes that interact with the cell wall. In mature cell walls, only a small fraction of the PG retains an unprocessed pentapeptide stem (Bui et al., 2012, Fisher & Mobashery, 2015). Thus, the low degree of LytA interaction with the peptide moiety of the substrate is likely an adaptation to allow the lysis of both unlinked and cross-linked stem peptides as well as PG with stem peptide residue modifications.
The LytA/PG complex provides first insights within the tetrahedral high-energy species of PG hydrolysis
The catalytic mechanisms of amidase-2 proteins have been studied for several PG-degrading proteins from different bacteria (Zoll et al., 2010, Kerff et al., 2010, Genereux et al., 2004), revealing similarities to thermolysin-mediated catalysis (Kester & Matthews, 1977). Structural alignment of crystallized Zn-dependent amidases demonstrated that Staphylococcus aureus AmiA and Staphylococcus epidermidis AmiE are the most closely related enzymes to LytA (Mellroth et al., 2014, Buttner et al., 2015, Zoll et al., 2010). Since these two enzymes catalyze PG at the same site as LytA, and since all the catalytic residues are conserved between LytA, AmiA and AmiE, we believe that the LytA catalysis reaction is very similar to what has been previously described (Zoll et al., 2010, Buttner et al., 2014). However, the atomic-resolution structure of the LytA/di(GM5P) complex provides additional information into these mechanisms. Indeed, the rigidity of the active site, conserved between the substrate-free and the bound forms of LytA, allows us to provide additional insights into the formation of tetrahedral high-energy species, the likely reaction transition state.
In contrast to synthetic PG samples with one or two sugar moieties (M5P and GM5P) which could not serve as substrate for LytA, the tetrasaccharide variant di(GM5P) is clearly a substrate that LytA cleaves efficiently in vitro (Mellroth et al., 2014). In order to assess how the substrate binds to the active site of LytA, we determined the crystal structure of the inactive LytA mutant His133Ala, which does not comprise Zn2+. Two water molecules are found within the space normally occupied by the Zn2+ ion and the side chain of His133 in the active LytA (Fig. 2B and 4A). Additionally, the position of the hydrolytic water molecule is occupied in the present structure by the oxygen atom of the lactyl moiety at the scissile bond (Fig. 4A). Both the Zn2+ ion and the side chain of His133 can easily be modeled in the same positions found in the substrate-free form of LytA without any sterical clashes with other LytA residues or substrate atoms (Fig. 4B). Moreover, the hydrolytic water can be modeled into the same position as in the substrate-free structure. As described above, di(GM5P) is tightly bound in the substrate-binding groove through multiple interactions including 15 hydrogen bonds and van der Waals interaction with at least eight LytA residues. The lactyl-amid bond projects inwards towards the buried active site. Since the first residue of the peptide stem, Ala5 does not form any hydrogen bonds with LytA, free available space allows for substrate rearrangement in the close vicinity of the scissile bond (Fig. 4B). Thus, it is possible to model di(GM5P) with the oxygen atom of the lactyl group forming a hydrogen bond with His147 in a similar conformation as found in the AmiA/M4P complex (Buttner et al., 2014). Similarly to AmiA, the catalytic water can become highly polarized through interactions with both Zn2+ and the side chain of Glu87, which would enable the promotion of the water molecule as the nucleophile for approach to the amide carbonyl of the scissile bond. This process leads to the transient formation of the stabilized high-energy tetrahedral species (Fig. 4C) in the reaction profile of the enzyme. This high-energy species is the likely transition state for the reaction that is being catalyzed, whose collapse leads to the formation of the two products.
Figure 4. The LytA/di(GM5P) complex provides insights into the high-energy tetrahedral species for PG hydrolysis.
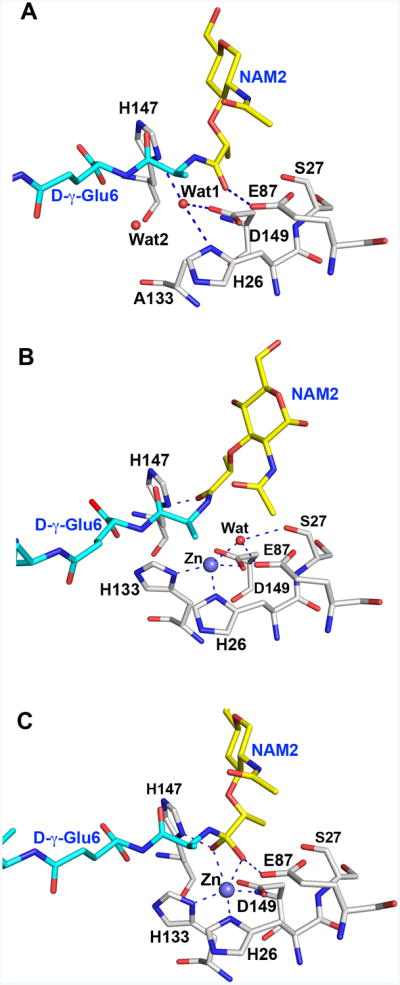
A. Active site of the LytA/di(GM5P) complex. Two water molecules Wat1 and Wat2, replace the Zn2+ ion and the side chain of His133, respectively, in the crystal structure of the LytA/di(GM5P) complex compared to the unliganded structure of LytA.
B. Both the Zn2+ ion (violet sphere) and the side chain of His133 can be easily modeled in the LytA/di(GM5P) complex. Furthermore, the catalytic water forming hydrogen bonds with both Zn2+ and Glu87 found in unbound LytA can be modeled in the active site if we hypothesize that the conformation of the stem peptide remains similar to that found in the crystal structure of AmiA/M4P complex (Buttner et al., 2014).
C. The polarized catalytic water attacks the carbonyl of the scissile bond, giving rise to the high-energy tetrahedral species that is depicted. The collapse of the tetrahedral species results in the formation of the two products of the reaction.
The substrate di(GM5P) adopts a novel conformation upon binding to LytA
The conformation of di(GM5P) in solution has previously been determined by NMR (Meroueh et al., 2006), revealing a high flexibility for the peptide stems while the conformation of the sugars, especially the core NAM2 and NAG3 units, remains stable. When the tetrasaccharide three-dimensional structure was built into a longer saccharide backbone, as found in many bacteria, the stability of the sugar backbone afforded a three-fold symmetry for the PG in a right-handed helical arrangement with a periodicity of three NAG-NAM repeat units. The crystal structure of the LytAAmi/di(GM5P) complex reveals that the conformation taken by the glycan moiety of di(GM5P) within the active site of LytA is different from the conformation it assumes in solution due to multiple interactions with 21 LytA residues (Fig. 5A). Torsion angles defining the conformation of the glycoside chains φ (O5-C1-O4-C4) and ψ C1-O4-C4-C5) are indeed different in free and bound di(GM5P) (Table III). Thus, the LytA substrate interface imposes a distinct conformation onto the PG upon binding. In this arrangement, the hexose rings of the central sugars NAM2-NAG3 align themselves in the same plane, whereas the two terminal sugars NAG1 and NAM4 turn by 45° relative to the plane of the central sugars (Fig. 5A). Although this arrangement, especially for the NAM2-NAG3 rings, requires a higher energy than in solution, the LytA substrate-binding interface would stabilize the conformation of the PG such that the energy of the complex is attainable. Furthermore, superimposition of NAG1 onto NAG3 reveals that the conformation of the saccharides of the first repeat NAG1-NAM2 does not exactly match the conformation of the second NAG3-NAM4 pair (Fig. 5B). As a consequence, the second stem peptide (l-Ala9-d-γ-Glu10; stem 2) does not share a symmetrical conformation with the first residues (l-Ala7-d-γ-Glu6; stem 1) of the first stem peptide that is bound within the binding crevice of LytA, allowing stem 2 to avoid sterical clashes with LytA and to project out of the binding crevice without forming any interaction with the enzyme. The quality of the electron density allowed us to model only the two first residues whereas the rest of peptide stem 2 is very mobile.
Figure 5. The flexible di(GM5P) substrate takes different conformations upon binding to different proteins.

A. Superimposition of the NAM2 moiety in the solution structure of di(GM5P) (in violet) on NAM2 from the LytA/di(GM5P) complex (in yellow) demonstrates that the glycan chain assumes a different conformation in the LytA binding crevice. The glycan moieties and the peptide stems of di(GM5P) bound to LytA are in yellow and cyan, respectively. Only the hexose rings of the NMR structure are displayed for clarity.
B. The NAG1-NAM2 and NAG3-NAM4 moieties take different conformations within the active site of the amidase LytA. Superposition of NAG1-NAM2-l-Ala5-d-γ-Glu6 (in orange and green for the saccharides and peptide, respectively) on NAG3-NAM4-l-Ala9-d-γ-Glu10 (in yellow and cyan for saccharides and peptide, respectively) demonstrates that the second peptide stem (stem 2) takes a different conformation compared to the first peptide stem (stem 1).
C. The conformation of the carbohydrate moiety of di(GM5P) is very similar when bound to either LytA (yellow) or the E. coli transglycosidase MltC (green).
D. The conformation of the carbohydrate moiety of di(GM5p) is very different in LytA (yellow) compared to AmpDh2 from P. aeruginosa (magenta).
Table III. Torsion angles of the carbohydrate moiety of di(GM5P).
| Torsion angle | sugars | NMR | LytA |
|---|---|---|---|
| O5-C1-O4-C4 | NAG1-NAM2 | -55 | -43 |
| C1-O4-C4-C5 | NAG1-NAM2 | -115 | -64 |
| O5-C1-O4-C4 | NAM2-NAG3 | -47 | -88 |
| C1-O4-C4-C5 | NAM2-NAG3 | -108 | -142 |
| O5-C1-O4-C4 | NAG3-NAM4 | -47 | -93 |
| C1-O4-C4-C5 | NAG3-NAM4 | -92 | -99 |
The crystal structures of several other enzymes in complex with PG tetrasaccharides have been recently determined (Lee et al., 2013, Martinez-Caballero et al., 2013, Artola-Recolons et al., 2014). Comparison of the bound LytA with these other structures reveals that the conformation of the sugars can be rather different. Interestingly, the most similar conformation of the PG sugar chain compared to LytA was found in an absolutely unrelated protein, the membrane bound lytic transglycosylase MltC from E. coli (Fig. 5C). The folds of LytA and MltC are radically different and these two enzymes catalyze different reactions but both enzymes works on intact polymeric peptidoglycan (Artola-Recolons et al., 2014). In contrast, the same PG substrate takes very different conformations within the binding pocket of the N-acetylmuramoyl l-alanine amidase AmpDh2 from P. aeruginosa (Martinez-Caballero et al., 2013), which is a similar amidase as LytA but with a less extended glycan interaction interface typical for Gram negative amidases (Fig. 5D). In conclusion, the flexibility of the PG chain allows its accommodation in the active site of very different proteins.
A common substrate interface among Gram-positive Firmicute amidases
The crystal structures of several bacterial amidases have been determined, revealing clear similarities and differences (Buttner et al., 2015). According to the DALI server (Holm et al., 2008) and despite a sequence identity of only 21%, the amidase domain of the autolysin AmiA from S. aureus in complex with the ligand NAM-tetrapeptide (M4P) (Buttner et al., 2014) is most structurally similar to LytA/di(GM5P), with an rmsd of 1.76 Å for 161 amino-acid residues. The active-site residues of these two amidases are conserved and the conformations of the bound substrate peptide stems within the active sites are at first sight similar. However, the central N-acetyl muramic acid in the AmiA/M4P complex, corresponding to NAM2 in the LytA/di(GM5P) complex is positioned differently. While residues 2-4 of the peptide stem (d- iGln-l-Lys-d-Ala) in M4P align well with the corresponding residues in di(GM5P), with a displacement of 0.6-1.2Å, the l-Ala and the N-acetyl muramic acid are shifted by 2.3-4.0 Å in relation to corresponding residues in the LytA complex (Fig. 6A). Despite the good resolution (1.55 Å) of the crystal structure of the AmiA/M4P complex, the electron density of the muramic acid remains poor, which makes it difficult to assess whether the observed movement is due to differences between the two proteins or in the bound substrate. Most likely, a larger muropeptide structure such as di(GM5P) if bound into the binding crevice of AmiA could influence the relative positioning of the central NAM residue. Indeed, structural superimposition of LytA with AmiA reveals that all glycan-interacting residues we identified as critical for di(GM5P) binding to LytA (Thr28, Glu38, Tyr41 and His147) are conserved between the two amidases and take similar positions (Fig. 6B). The extended glycan interaction interface appears to be structurally conserved among Gram-positive Firmicute amidases (Mellroth et al., 2014) which has been confirmed by structural sequence alignment of amidases with known three-dimensional structures (Buttner et al., 2015).
Figure 6. Key LytA residues essential for saccharide binding are structurally conserved in Gram-positive Firmicute amidases.

A. At first sight, the substrate peptide stems of M4P and di(GM5P) bind similarly to AmiA and LytA, respectively. However, while residues 2-4 of the peptide stem (d-iGln-l-Lys) in M4P align well with the corresponding residues in di(GM5P), with a displacement of 0.6-1.2Å, the position of l-Ala5 and the N-acetyl muramic acid NAM2 differ between 2.3-4.0 Å. Thus the central N-acetyl muramic acid in the AmiA/M-4P complex, corresponding to NAM2 in the LytA/di(GM5P) structure is positioned differently.
B. Structural alignment of the amidases AmiA, AmiE and PlyL in violet, magenta and green, respectively, on LytA in grey demonstrates that the position and conformation of the key residues Thr28, Glu38, Tyr41 and His147 are conserved except that a lysine residue replaces His147 in the bacteriophage amidase PlyL. Glycan moieties and peptide stems of the di(GM5P) substrate are in yellow and cyan, respectively.
Multi-sequence alignment of Firmicute amidases displays the conservation of the substrate interacting residues (Supporting Information Fig. S5). The four key glycan-interacting residues His147, Thr28, Glu38, and Tyr41 are highly conserved among the species. His147 that forms CH-π interaction with NAM1 is also involved in catalysis and therefore fully conserved. Thr28 is sometimes replaced by a serine residue but otherwise conserved. Interestingly, Glu38 is conserved in most species but replaced with an isoleucine residue in B. subtilis and B. cereus. It has been observed that S. aureus AmiA cannot digest PG from B. subtilis and thus suggested that differences in the peptide unit (meso- diaminopimelic acid instead of L-Lys and direct linkage instead of penta-glycine interpeptide bridge) could explain the lack of amidase activity (Buttner et al., 2014). Thus, the absence of a conserved glutamic acid residue is a further indication that the cell wall of most Bacillus species may have different structural features than other Firmicutes. In fact, Bacillus cecembensis, one of the few Bacillus species that comprises an L-Lys residue and a bridging D-Glu, also shares the conserved Glu38 (Reddy et al., 2008). Also Tyr41 that forms a CH-π interaction with NAM4 is highly conserved but with a Phe residue that can form similar interactions in S. epidermidis. The glycan interacting residues Asn30 and Asp146 are also conserved in most non-Bacilli Firmicute amidases. The extensive glycan interface of Firmicutes and the glycan interacting network described within this study has partly been highlighted in previous studies (Buttner et al., 2014, Mellroth et al., 2014, Buttner et al., 2015). Finally, the sequence alignment also confirms that peptide-interacting residues Trp72, Gly75 and Asn79, which all display reduced activity in the corresponding alanine mutation background (Table II), are all highly conserved.
The results presented herein imply that LytA and amidases from most Firmicute species share common structural features including an extended Y-shaped binding crevice and conserved key amino acid residues that are central for positioning the glycan backbone and required for degradation of the thick Gram-positive bacterial cell wall. Our study provides the first detailed structural framework underlying how lytic amidases interact with their complex substrate.
Concluding remarks
Structural studies on PG-interacting proteins are often limited by the complexity of the substrate. The crystal structure of the major pneumococcal autolysin LytA in complex with a large synthetic PG fragment, determined at 1.05 Å, reveals that the ligand occupies the entire binding crevice and allows for a study of the complete protein-substrate interface into the finest detail. Our data demonstrated that as many as twenty-one LytA amino-acid residues interact with the substrate-motif. Interestingly, the majority of these interactions are directed towards the glycan backbone, where all four saccharide units are intimately bound through key hydrogen bond and CH-π stacking interactions. Mutational analysis of substrate-interacting residues defined their relative impact for the lytic activity of LytA and revealed that several non-catalytic glycan-interacting residues were pivotal for enzymatic activity. Some of these residues are not localized within the immediate vicinity of the catalytic site and are most likely important for stringent positioning of the PG substrate in relation to the active site to facilitate cleavage. These results are fully in line with our previous in vitro enzymatic assays, which demonstrated that LytA required a large muropeptide substrate comprising more than two saccharides for cleavage (Mellroth et al., 2014). Previous studies also strongly indicated that the lytic activity of LytA is regulated at the level of substrate recognition and that an inactive form of LytA displayed affinity for a substrate within the nascent PG (Mellroth et al., 2012). Our results demonstrate that the flexible di(GM5P) substrate adopts a distinct three-dimensional conformation upon binding into the binding crevice of the pneumococcal amidase. We propose that a PG epitope with a similar conformation is present within the nascent peptidoglycan.
Experimental Procedures
Production and isolation of the mutated LytAAmi variants
A mutated variant of the amidase domain of the major autolysin LytAAmi, in which the three residues C60, H133 and C136 were substituted by alanine, was produced for the structural determination of the complex with the PG derivative. We have recently determined the crystal structure of LytAAmi in which both cysteine residues were replaced by alanine (Mellroth et al., 2014). To prevent turnover of the bound substrate by LytAAmi, we produced an enzymatically inactive form of LytA, introducing an additional mutation in the zinc-ligand residue H133 that was substituted to an alanine (H133A). The H133A mutation was introduced using the QuickChange II mutagenesis kit (Agilent Technologies) with the pET21d/lytAAmi-C60A-C136A(C-termHis6) vector (Mellroth et al., 2014) as template and using primers that replaced the 133 codon CAT(His) with a GCT(Ala) codon. The same approach was used to introduce mutations in the full-length LytA protein using the pET21d-lytA vector as a template and primers with degenerate sequences at the site of the target codon. All mutations were confirmed by DNA sequencing. Expression and purification of all recombinant proteins were performed as previously described (Mellroth et al., 2012, Mellroth et al., 2014).
Circular dichroism analysis
Circular dichroism (CD) spectra of wild-type and mutated LytA proteins with reduced enzymatic activity were performed to verify that the substitutions did not affect folding. Far UV CD measurements were performed using a J-810 spectro-polarimeter (Jasco, Easton, MD). Purified wild-type and mutated LytA proteins were dialyzed against 10 mM sodium phosphate buffer, pH 7.3, 5 mM choline chloride and corrected to 2 mg/ ml. Prior to analysis proteins were further diluted 50-fold in 10 mM sodium phosphate buffer to give an experimental concentration of 40 μg/ ml. CD spectra were recorded from 195 to 260 nm at 20°C with 0.5 nm steps and a total of eight scans were averaged for each protein. The optical activity of the buffer was subtracted from the protein spectra.
Turbidometric amidase activity assay
Amidase activity was performed previously described (Mellroth et al., 2014). Briefly, pneumococcal T4ΔlytA cells in PBS at OD600 = 1.0 were treated with 10 μg/ml of wt or mutant forms of recombinant LytA and the decrease in turbidity was recorded at 10 minutes intervals. The relative activities of each mutant variant were determined in relation to that of wt-LytA using average of three independent reactions.
Crystallization and data collection of the LytAAmi/di(GM-5P) complex
Crystals of the LytAAmi/di(GM5P) complex were obtained by co-crystallization. An aqueous solution of di(GM5P) in five-fold molar excess was added to 12 mg/mL solution of LytAAmi (C60A, H133A, C136A) in 0.02 M Tris HCl pH 7.5 and 140 mM NaCl. The protein-ligand solution (2 μL) was mixed with an equal volume of crystallization solution (9 % PEG6K, 1 M LiCl, 0.1 M Tris HCl pH 6.2) and equilibrated against 1 mL of the crystallization solution. A single protein crystal appeared in a week and grew within about a month to 0.3 × 0.2 × 0.8 mm in size. The crystal was soaked in crystallization solution supplemented with 25% glycerol, before freezing in liquid nitrogen. Data collection was performed on beamline ID23-1 at ESRF (Grenoble, France). Data from a total of 1200 images was processed and merged with XDS (Kabsch, 2010) and Aimless (Evans, 2006), respectively. The crystals, which belong to space group P212121 with unit cell parameters a = 44.57 Å, b = 54.62 Å, c = 78.88 Å, diffracted to at least 1.05 Å. Data collection statistics are presented in Table I.
Determination of the crystal structure of the LytAAmi/di(GM5P) complex
Since the space group of the LytAAmi/di(GM5P)-crystal was different from the crystal of substrate-free LytAAmi (Mellroth et al., 2014), the three-dimensional structure of the di(GM5P)-LytAAmi complex was determined by molecular replacement using the program PHASER (McCoy et al., 2007) with the coordinates of the monomer LytAAmi (PDB code 4IVV) as a search model. Calculation of solvent contents suggested one LytAmi/di(GM5P) molecule in the asymmetric unit with 50 % solvent. The single solution suggested by the program Phaser had an LLG value of 1977. Refinement was performed with Refmac5 (Murshudov et al., 2011) for the dataset scaled to 1.05 Å. All rebuilding and the addition of both the PG substrate and water molecules were performed with COOT (Emsley et al., 2010). The final model comprises 176 LytA residues, 105 substrate atoms and 248 water molecules. The N-LytA construct comprised LytA residues 1-180, a C-terminal His-tag and two amino acids at the N-terminus (Ala-Ser) as a cloning artifact. The electron density allowed unambiguous modeling of residues Ala(-1) to Ile174. 12 C-terminal residues including the His6-tag are not included in the final model. Refinement statistics are presented in Table I. All structural figures were created with PyMol (PyMol Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC). The schematic view of the interactions between the ligand and protein was created using LigPlot+ (Laskowski & Swindells, 2011).
Supplementary Material
Acknowledgments
We gratefully acknowledge access to the synchrotron beam line ID23-1 at ESRF (Grenoble, France) and to the crystallization facility within the Protein Science Facility, Karolinska Institutet, (http://psf.ki.se/). This work was supported by grants from the Swedish Research Council (AA, PM, BHN), the Swedish Foundation for Strategic Research (AA, BHN), the Knut and Alice Wallenberg Foundation (AA, BHN) and the National Institute of Health, USA (AI090348 and GM61629; SM).
Abbreviations
- NAM
N-acetylmuramic acid
- NAG
N-acetylglucosamine
- M5P
NAM-l-Ala-d-γ-Glu-l-Lys-d-Ala-d-Ala
- GM5P
NAG-NAM-l-Ala-d-γ-Glu-l-Lys-d-Ala-d-Ala
- Di(GM5P)
four connected saccharides with two pentapeptides linked to the NAM residues
- PG
peptidoglycan
- Dgl
D-γ-glutamic acid
Footnotes
Accession numbers: The three-dimensional coordinates and structural factors for the LytAmi/di(GM5P) complex have been deposited to the Protein Data Bank under accession number 5CTV.
The authors declare no conflicts of interest.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, Mc Coy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola-Recolons C, Lee M, Bernardo-Garcia N, Blazquez B, Hesek D, Bartual SG, Mahasenan KV, Lastochkin E, Pi H, Boggess B, Meindl K, Uson I, Fisher JF, Mobashery S, Hermoso JA. Structure and cell wall cleavage by modular lytic transglycosylase MltC of Escherichia coli. ACS Chem Biol. 2014;9:2058–2066. doi: 10.1021/cb500439c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, Simorre JP, Vollmer W. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal Biochem. 2012;421:657–666. doi: 10.1016/j.ab.2011.11.026. [DOI] [PubMed] [Google Scholar]
- Buttner FM, Renner-Schneck M, Stehle T. X-ray crystallography and its impact on understanding bacterial cell wall remodeling processes. Int J Med Microbiol. 2015;305:209–216. doi: 10.1016/j.ijmm.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Buttner FM, Zoll S, Nega M, Gotz F, Stehle T. Structure-function analysis of Staphylococcus aureus amidase reveals the determinants of peptidoglycan recognition and cleavage. J Biol Chem. 2014;289:11083–11094. doi: 10.1074/jbc.M114.557306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Enck S, Price JL, Powers DL, Powers ET, Wong CH, Dyson HJ, Kelly JW. Structural and energetic basis of carbohydrate-aromatic packing interactions in proteins. J Am Chem Soc. 2013;135:9877–9884. doi: 10.1021/ja4040472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldholm V, Johnsborg O, Haugen K, Ohnstad HS, Havarstein LS. Fratricide in Streptococcus pneumoniae: contributions and role of the cell wall hydrolases CbpD, LytA and LytC. Microbiology. 2009;155:2223–2234. doi: 10.1099/mic.0.026328-0. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Fernandez-Tornero C, Lopez R, Garcia E, Gimenez-Gallego G, Romero A. A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat Struct Biol. 2001;8:1020–1024. doi: 10.1038/nsb724. [DOI] [PubMed] [Google Scholar]
- Figueiredo TA, Sobral RG, Ludovice AM, Almeida JM, Bui NK, Vollmer W, de Lencastre H, Tomasz A. Identification of genetic determinants and enzymes involved with the amidation of glutamic acid residues in the peptidoglycan of Staphylococcus aureus. PLoS Pathog. 2012;8:e1002508. doi: 10.1371/journal.ppat.1002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JF, Mobashery S. Practical handbook of microbiology. CRC Press; Boca Raton: 2015. [Google Scholar]
- Garcia-Bustos JF, Chait BT, Tomasz A. Structure of the peptide network of pneumococcal peptidoglycan. J Biol Chem. 1987;262:15400–15405. [PubMed] [Google Scholar]
- Genereux C, Dehareng D, Devreese B, Van Beeumen J, Frere JM, Joris B. Mutational analysis of the catalytic centre of the Citrobacter freundii AmpD N-acetylmuramyl-L-alanine amidase. Biochem J. 2004;377:111–120. doi: 10.1042/BJ20030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli S, Tomasz A. Attachment of pneumococcal autolysin to wall teichoic acids, an essential step in enzymatic wall degradation. J Bacteriol. 1984;158:1188–1190. doi: 10.1128/jb.158.3.1188-1190.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Brown PH, Swaminathan CP, Roychowdhury A, Boons GJ, Mariuzza RA. Crystal structure of human peptidoglycan recognition protein I alpha bound to a muramyl pentapeptide from Gram-positive bacteria. Protein Sci. 2006;15:1199–1206. doi: 10.1110/ps.062077606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesek D, Lee M, Morio K, Mobashery S. Synthesis of a fragment of bacterial cell wall. J Org Chem. 2004;69:2137–2146. doi: 10.1021/jo035583k. [DOI] [PubMed] [Google Scholar]
- Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard LV, Gooder H. Specificity of the autolysin of Streptococcus (Diplococcus) pneumoniae. J Bacteriol. 1974;117:796–804. doi: 10.1128/jb.117.2.796-804.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyland CN, Aldridge C, Cleverley RM, Duchene MC, Minasov G, Onopriyenko O, Sidiq K, Stogios PJ, Anderson WF, Daniel RA, Savchenko A, Vollmer W, Lewis RJ. Structure of the LdcB LD-carboxypeptidase reveals the molecular basis of peptidoglycan recognition. Structure. 2014;22:949–960. doi: 10.1016/j.str.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallographica Section D-Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerff F, Petrella S, Mercier F, Sauvage E, Herman R, Pennartz A, Zervosen A, Luxen A, Frere JM, Joris B, Charlier P. Specific structural features of the N-acetylmuramoyl-L-alanine amidase AmiD from Escherichia coli and mechanistic implications for enzymes of this family. J Mol Biol. 2010;397:249–259. doi: 10.1016/j.jmb.2009.12.038. [DOI] [PubMed] [Google Scholar]
- Kester WR, Matthews BW. Comparison of the structures of carboxypeptidase A and thermolysin. J Biol Chem. 1977;252:7704–7710. [PubMed] [Google Scholar]
- Lacks S. Mutants of Diplococcus pneumoniae that lack deoxyribonucleases and other activities possibly pertinent to genetic transformation. J Bacteriol. 1970;101:373–383. doi: 10.1128/jb.101.2.373-383.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lee M, Artola-Recolons C, Carrasco-Lopez C, Martinez-Caballero S, Hesek D, Spink E, Lastochkin E, Zhang W, Hellman LM, Boggess B, Hermoso JA, Mobashery S. Cell-wall remodeling by the zinc-protease AmpDh3 from Pseudomonas aeruginosa. J Am Chem Soc. 2013;135:12604–12607. doi: 10.1021/ja407445x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Cheng W, Morlot C, Bai XH, Jiang YL, Wang W, Roper DI, Vernet T, Dong YH, Chen Y, Zhou CZ. Full-length structure of the major autolysin LytA. Acta Crystallogr D Biol Crystallogr. 2015;71:1373–1381. doi: 10.1107/S1399004715007403. [DOI] [PubMed] [Google Scholar]
- Martinez-Caballero S, Lee M, Artola-Recolons C, Carrasco-Lopez C, Hesek D, Spink E, Lastochkin E, Zhang W, Hellman LM, Boggess B, Mobashery S, Hermoso JA. Reaction products and the X-ray structure of AmpDh2, a virulence determinant of Pseudomonas aeruginosa. J Am Chem Soc. 2013;135:10318–10321. doi: 10.1021/ja405464b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martner A, Dahlgren C, Paton JC, Wold AE. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect Immun. 2008;76:4079–4087. doi: 10.1128/IAI.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martner A, Skovbjerg S, Paton JC, Wold AE. Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect Immun. 2009;77:3826–3837. doi: 10.1128/IAI.00290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Coy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellroth P, Daniels R, Eberhardt A, Ronnlund D, Blom H, Widengren J, Normark S, Henriques-Normark B. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J Biol Chem. 2012;287:11018–11029. doi: 10.1074/jbc.M111.318584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M, Mobashery S, Normark S, Svergun D, Henriques-Normark B, Achour A. Structural and functional insights into peptidoglycan access for the lytic amidase LytA of Streptococcus pneumoniae. MBio. 2014;5:e01120–01113. doi: 10.1128/mBio.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc Natl Acad Sci U S A. 2006;103:4404–4409. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscoso M, Garcia E, Lopez R. Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J Bacteriol. 2006;188:7785–7795. doi: 10.1128/JB.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch D, Roemer T, Lee SH, Engeser M, Sahl HG, Schneider T. Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PLoS Pathog. 2012;8:e1002509. doi: 10.1371/journal.ppat.1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Sevillano E, Urzainqui A, Campuzano S, Moscoso M, Gonzalez-Camacho F, Domenech M, Rodriguez de Cordoba S, Sanchez-Madrid F, Brown JS, Garcia E, Yuste J. Pleiotropic effects of cell wall amidase LytA on Streptococcus pneumoniae sensitivity to the host immune response. Infect Immun. 2015;83:591–603. doi: 10.1128/IAI.02811-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GS, Uttam A, Shivaji S. Bacillus cecembensis sp. nov., isolated from the Pindari glacier of the Indian Himalayas. Int J Syst Evol Microbiol. 2008;58:2330–2335. doi: 10.1099/ijs.0.65515-0. [DOI] [PubMed] [Google Scholar]
- Rubin LG, Rizvi A. PCR-based assays for detection of Streptococcus pneumoniae serotypes 3, 14, 19F and 23F in respiratory specimens. J Med Microbiol. 2004;53:595–602. doi: 10.1099/jmm.0.45550-0. [DOI] [PubMed] [Google Scholar]
- Sanchez-Puelles JM, Ronda C, Garcia JL, Garcia P, Lopez R, Garcia E. Searching for autolysin functions. Characterization of a pneumococcal mutant deleted in the lytA gene. Eur J Biochem. 1986;158:289–293. doi: 10.1111/j.1432-1033.1986.tb09749.x. [DOI] [PubMed] [Google Scholar]
- Sheppard CL, Harrison TG, Morris R, Hogan A, George RC. Autolysin-targeted LightCycler assay including internal process control for detection of Streptococcus pneumoniae DNA in clinical samples. J Med Microbiol. 2004;53:189–195. doi: 10.1099/jmm.0.05460-0. [DOI] [PubMed] [Google Scholar]
- Tomasz A, Albino A, Zanati E. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature. 1970;227:138–140. doi: 10.1038/227138a0. [DOI] [PubMed] [Google Scholar]
- Tomasz A, Waks S. Mechanism of action of penicillin: triggering of the pneumococcal autolytic enzyme by inhibitors of cell wall synthesis. Proc Natl Acad Sci U S A. 1975;72:4162–4166. doi: 10.1073/pnas.72.10.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll S, Patzold B, Schlag M, Gotz F, Kalbacher H, Stehle T. Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog. 2010;6:e1000807. doi: 10.1371/journal.ppat.1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.