

Abstract
Background
Malignancy-associated hemophagocytic lymphohistiocytosis (HLH) in adults is a highly lethal disorder. Knowledge gaps have resulted in under-diagnosis or delayed diagnosis.
Patients and Methods
The University of Texas MD Anderson Cancer Center pathology database (1991–2014) was retrospectively interrogated for the keywords “hemophagocytosis” and/or “lymphohistiocytosis”. 77 adult patients were identified. All had an underlying malignancy. 16 patients with insufficient documentation were excluded.
Results
The majority of patients with pathologic evidence of hemophagocytosis/lymphohistiocytosis had incomplete work-up to confirm or refute HLH using the HLH-2004 criteria (HLH-2004 = 8 variables) and this is a common problem in adult HLH. Only 13/61 (21%) patients met the HLH-2004 diagnostic criteria based on available retrospective data. To identify potentially missed cases of HLH we reviewed published literature and selected additional variables known to be associated with adult HLH resulting in an extended diagnostic criteria of 18 variables. 35 patients met the extended criteria and 33 had follow-up data. The median OS of the 13 patients who met both the extended criteria and the HLH-2004 criteria was similar to the 20 patients who met the extended criteria but NOT the HLH-2004 criteria (1.43 vs 1.76 months, P=0.34) indicating similar underlying aggressive systemic process. 26 patients did not meet either criteria and 17 had follow-up data. The median OS of the 17 patients who had pathologic hemophagocytosis or lymphohistiocytosis but met neither criteria was significantly superior to those who met both the extended criteria and the HLH-2004 criteria or those who met the extended criteria but NOT the HLH-2004 criteria (17.27 vs 1.43 vs 1.76, P=0.002).
Conclusion
Addition of diagnostic laboratory variables that are more easily and rapidly available in smaller institutions and primary care settings than the HLH-2004 variables may be a good surrogate to raise early suspicion of malignancy-associated HLH. Prospective validation is warranted.
Keywords: hemophagocytosis, lymphohistiocytosis, ferritin, adults
BACKGROUND
HLH is characterized by severe inflammation and immune mediated organ damage. A highly stimulated and defective inflammatory response involving 3 main pathways is central to the initiation and propagation of HLH, (1) hyperactivation of CD8+ T lymphocytes and macrophages, (2) proliferation and infiltration of these cells into organs and tissues, and (3) uncontrolled hypercytokinemia1–4. Patients present with a spectrum of features of an acute illness including fever, cytopenias, spleen and/or liver enlargement, coagulopathy, encephalopathy, and transaminits3,5–7. HLH is often referred to as “primary”, assuming genetic inheritance of defects in cytotoxic immune function or “secondary”, assuming pathogenesis due to extreme or persistent antigen activation. It is becoming increasingly clear that the immune trigger and host in which HLH arise both play important roles, blurring the distinctions between “primary” and “secondary”8. In the case of malignancy-associated HLH, possible mechanisms of pathogenesis include extreme inflammation, persistent antigen stimulation by the tumor cells, and loss of immune homeostasis due to chemotherapy or hematopoietic stem cell transplant or infection. Inherited immune disorders that predispose to both HLH and malignancy (e.g. X-linked lymphoproliferative diseases) should also be considered. More recently, symptoms consistent with HLH have been described in patients receiving immunotherapies3,4,7–9.
Hemophagocytic lymphohistiocytosis (HLH) or hemophagocytic syndrome is a rare and almost universally fatal condition in adults. The true incidence remains uncertain, with diagnosis challenged both by lack of recognition of hemophagocytic syndrome and uncertain diagnostic criteria. Information specific to adult HLH is limited, and current diagnostic and treatment approaches for HLH in adults are extrapolations from retrospective databases and clinical trials in childhood HLH. These knowledge gaps have led to under-diagnosis or delayed diagnosis of HLH in adults with further worsening of outcomes5,6,10. Identification of clinical and laboratory features specific to HLH in adults may allow for early detection and intervention with improved outcomes. Improved management of adult HLH with optimal combinations of T-lympholytic and immunosuppressive agents based on prior experience gathered by our pediatric colleagues will hopefully further improve outcomes in adults and such trials are currently underway (ClinicalTrials.gov Identifier: NCT02385110).
METHODS
In this single-center retrospective study performed at the University of Texas MD Anderson Cancer Center (UT/MDACC) we designed a study to identify the frequency of HLH in adult patients with underlying malignancies and to understand the features of extreme activation in order to identify improved strategies for diagnosis and therapy for this group of patients with dismal outcomes.
The UT/MDACC pathology database (1991 – 2014) was broadly interrogated for the keywords “hemophagocytosis” and/or “lymphohistiocytosis”. 77 adult patients (>18 years) were identified. Sixteen patients had insufficient documentation (patients were referred to MDACC for pathological verification of diagnosis, but were not treated and followed up there), and were excluded from the study. For the remaining 61 patients the clinical, laboratory, demographic, treatment, disposition, and outcome information was collected and reviewed.
The HLH 2004 criteria are the standard diagnostic criteria for HLH in pediatric patients10. The HLH 2004 study required patients to have five of eight features to be diagnosed with HLH. These eight features include fever, splenomegaly, cytopenias (affecting ≥ 2 of 3 lineages: hemoglobin<9.0 g/L, platelets<100 × 109/L, absolute neutrophil count (ANC)< 1.0 × 109/L), hemophagocytosis in bone marrow or spleen or lymph node, hypertriglyceridemia (fasting triglycerides ≥ 265mg/dl) and/or hypofibrinogenemia (fibrinogen ≤150mg/dL), hyperferritinemia (ferritin ≥500 microgram/L), low or absent NK cell activity, and increased soluble CD25 (IL2-receptor) (≥2,400 U/ml) as defined in the HLH-2004 criteria. On our review we noted that a majority of the patients did not have many of the requisite eight HLH 2004 variables collected making the diagnosis of HLH difficult. No patient had all 8 or 7 variables collected, 5 (8%) had 6 variables collected, 12 (20%) had 5 variables collected, and 44 (72%) had <5 variables collected. Lack of comprehensive data in these patients was likely due to under-recognition of HLH in adults resulting in an incomplete HLH work-up and difficulty in obtaining some of the variables such as NK-cell activity and soluble CD25 as these are not routinely available and require special send-out testing.
Available literature and published expert opinions suggest that a number of additional clinical and laboratory features beyond those outlined in HLH 2004 may help confirm or refute a diagnosis of HLH in children and adults3,5,6,10–15 A number of these variables are easily obtained on routine laboratory or physical examination in most community and academic institutions and do not need special lab investigation or send-out testing. These criteria if successful at identifying HLH would be a surrogate for earlier recognition of possible HLH in smaller centers and earlier referral of suspected cases to larger centers with expertise in HLH. After reviewing the available literature and experts opinions, 18 variables (clinical and laboratory characteristics) shown to be closely associated with a diagnosis of HLH were selected and data on these 18 variables was collected with an intent to identify additional patients with HLH who may have been missed by the standard HLH 2004 criteria either due to insufficient number of variables or because the disease manifestations differ between children and adults. These 18 variables included BM/lymph node/spleen hemophagocytosis per pathology evaluation, fever, splenomegaly (clinically palpable spleen), hepatomegaly (clinically palpable liver), anemia (hemoglobin<9.0 g/L), thrombocytopenia (platelets<100 × 109/L), neutropenia (absolute neutrophil count (ANC)< 1.0 × 109/L), monocytosis (absolute monocyte count (AMC)>1.0 × 109/L), renal failure (≥50% of increase of creatinine over baseline), elevation of hepatic enzymes (≥2.5 × upper limit of normal), hypofibrinogenemia (fibrinogen ≤150mg/dL), hyperferritinemia (ferritin ≥500 microgram/L), coagulopathy (PT ≥ 1.5 × upper limit of normal and/or PTT ≥ 1.5 × upper limit of normal and/or D-dimer ≥ 10.0mcg/mL), hypoalbuminemia (<3.5g/dL), elevated LDH (≥ 2.5 × upper limit of normal), hypertriglyceridemia (≥ 265 mg/dL), elevated b2-microglobulin (≥ 2mg/L), and elevated soluble IL-2 receptor (i.e. CD25) ≥ 2400 U/ml.
We reassessed our patient population using this extended 18-point diagnostic criteria. Sensitivity analysis suggested that patients meeting 5 of 18 above mentioned criteria would be considered to have a high likelihood of secondary HLH.
Kaplan-Meier method16 was used to estimate the probabilities of overall survival, and survival distributions were compared using log-rank tests. For statistical analysis IBM SPSS Statistics 20 program was used. We calculated the sensitivities by using different cutoffs in the extended 18-point diagnosis criteria. Since we are not able to calculate the specificity using the observed data (a limitation of the study), it is not possible to derive an optimal cutoff in the current study. However, as seen from the plot (Figure 1), the sensitivity will be < 50% if the cut-off value is >=6 (so 5 variables was considered a reasonable number).
Figure 1.
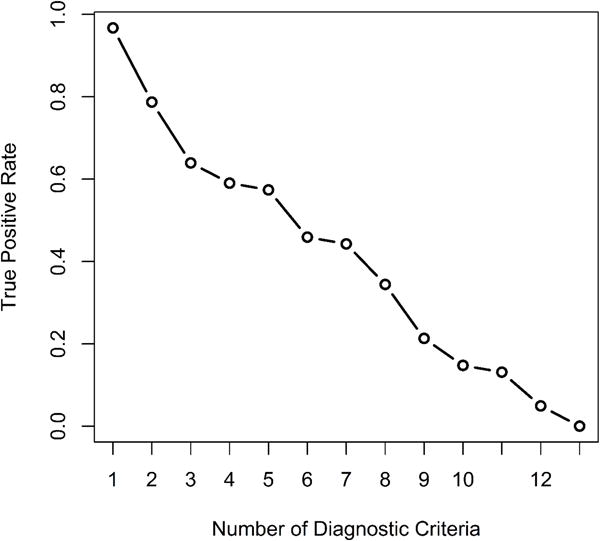
Sensitivity Plot
RESULTS
All 61 patients had pathologically confirmed presence of hemophagocytosis or lymphocytosis at UT/MDACC. The median age was 54 years (range, 18–83 years). Thirty-three (54%) were males and 42 (69%) were Caucasian, 10 (16%) Hispanics, 3 (5%) Blacks and 6 (10%) Asians. The median WBC count, hemoglobin, and platelets at diagnosis were 4.75 × 109/L [range, 100 – 65,300], 9.6 g/dL [range, 7.4 – 13.9], and 114 × 109/L [range, 6,000 – 534,000]. The most common presenting signs were fever in 42% (23/55); splenomegaly in 35% (17/49); thrombocytopenia in 48% (28/58); anemia in 39% (22/57); neutropenia in 30% (18/61); elevation of hepatic enzymes in 31% (16/51); hyperferritinemia in 88% (30/34); coagulopathy in 27% (10/37); hypoalbuminemia in 54% (26/48); high LDH in 30% (17/57); hypertriglyceridemia in 47% (8/17); high b2-microglobulinin 93% (26/28) and high soluble IL-2 receptor level in 75% (9/12). NK-cell activity test was performed in only 1 (2%) of the patients. From the all pathology specimens that described hemophagocytosis 48 were in BM (from which 3 also involved the lymph nodes, 3 liver, 2 spleen and 1 skin), 1 in lymph node and 1 in liver. As has been described by the others5, the most common underlying malignancies in our cohort of patients were AML/MDS (13 patients; 21%) and T-lymphoma (10 patients; 16%). Other commonly identified underlying malignancies included diffuse large B cell lymphoma (6 patients; 10%), Hodgkin lymphoma (6 patients; 10%), CLL (4 patients; 6.5%), CML (2 patients; 3%), follicular lymphoma (2 patient; 3.3%), one patient each with splenic marginal B-cell lymphoma, PTLD, CMML, aplastic anemia, Castleman’s disease. Solid tumors were less frequently associated with HLH with one case each associated with melanoma, breast cancer, testicular cancer, prostate cancer, squamous cell carcinoma of the neck, and a pituitary mass, respectively. Follow-up data is not available for 10 patients. The median follow-up period for the remaining 51 patients was 3.5 months (range, 0.17 months – 142.3 months). The median survival time among these 51 patients was 13.3 months. 35% (18/51) patients died within 8 weeks and 55% (28/51) patients died within 6 months from the identification of “hemophagocytosis” or “lymphohistiocytosis”.
Thirteen of the 61 patients (21%) fulfilled the HLH 2004 criteria10 (Table 1). Among the 13 patients HLH preceded the diagnosis of malignancy (AML) by 3 months in one case, HLH was noted at the same time as diagnosis of the malignancy in five cases, and HLH occurred after the diagnosis of malignancy in 7 cases. Among these 7 cases: 4 were receiving frontline therapy and 3 were receiving salvage therapy. No patients were in remission at time of HLH diagnosis. 10/13 patients received HLH directed therapy including etoposide in combination with dexamethasone in 6 (46%) patients (1 of the 6 patients additionally received ATG + cyclosporine), rituxan with dexamethasone in 1 patient, and dexamethasone + supportive care in 3 (23%) patients. Among the remaining 3 patients, one received treatment for the underlying lymphoma and the other two received supportive care. From those, who received treatment with etoposide/dexamethasone 3 had MDS/AML and 3 T-cell lymphoma. One of the MDS patients received concurrent therapy for the MDS (with clofarabine) and two of the T-ALL patients received concurrent therapy for T-ALL (with cyclophosphamide). 2/13 patients eventually proceeded to allogeneic stem cell transplant. The median follow-up for the 13 patients was 1.43 months (range, 0.17 months – 29.03 months). The median survival time among these 13 patients was 1.43 months. 54% (7/13) patients died within 8 weeks and 85% (11/13) patients died within 6 months. 2/13 (15%) patients are alive at this time.
Table 1.
Baseline characteristics of 13 patients fulfilled HLH-2004 criteria
| Variable | N | % |
|---|---|---|
| Age | ||
| Median [range] | 46 [18–77] | |
| Gender | ||
| Male/Female | 10/3 | 77/23 |
| Fever | 10/13 | 77 |
| Splenomegaly | 8/12 | 66.7 |
| Bi- or pancytopenia | 12/13 | 92.3 |
| Ferritin (ng/mL), Median [range] | 5,124 [665 – 60,761] | |
| >10,000 | 8/13 | 61.5 |
| >50,000 | 6/13 | 46.15 |
| Coagulopathy | 5/12 | 41.7 |
| LDH (U/L), Median [range] | 760 [325 – 19,841] | |
| >2.5 times | 3/13 | 23.1 |
| Fibrinogen (mg/dL), Median [range] | 294.5 [78 – 616] | |
| <150 | 3/12 | 25 |
| Triglycerides (mg/dL), Median [range] | 334.5 [105–599] | |
| >268 | 8/12 | 66.7 |
| Soluble IL-2 receptor, (pg/mL), Median [range] | 2891 [35 – 39,100] | |
| ≥2400 | 6/8 | 75 |
35 (57%) out of 61 patients had 5 or more variables when evaluated using the extended HLH criteria. Their information is summarized in Figure 3. Only 15/35 (43%) of these patients with high likelihood of secondary HLH received HLH-directed therapy. These 35 patients included the 13 definitive HLH-2004 patients mentioned above and an additional 22 patients who did not meet the HLH-2004 but met the extended HLH criteria. 9/13 of the definitive HLH patients received HLH-directed therapy as discussed above. Thus, only 6/22 (27%) of the patients who did not meet the definitive HLH but met the extended HLH criteria received HLH directed therapies included dexamethasone + etoposide based in 4 patients, alemtuzumab + steroid in 1, and steroids alone in 1 patient. Only 1/22 of the patients eventually went for stem cell transplant. The remaining 16 patients received therapy directed to the underlying malignancy. The low percentage of HLH directed therapy in this group is not surprising as these patients were not at that time considered to have HLH by their treating physicians highlighting the historical problem of poor awareness of this entity in adults. Follow-up data is not available for 2 patients. The median follow-up for the remaining 33 patients was 1.5 months (range, 0.17 months – 142.3 months). Among the remaining 33 patients 51.5% (17/33) died within 8-weeks from diagnosis and 69.7% (23/33) of patients within 6 months from diagnosis. The median survival for the 33 patients with follow-up was 1.50 months. This is very similar to the median survival of 1.43 months among the 13 patients who met the HLH-2004 criteria suggesting that the extended criteria may be an effective surrogate for identifying adult patients with malignancy-associated secondary HLH and dismal outcomes. The median survival was 1.98 months among patients treated with HLH directed therapy as compared to 2.03 months among those not treated with HLH directed therapies.
Figure 3. Hemophagocytic histiocytosis characteristics.
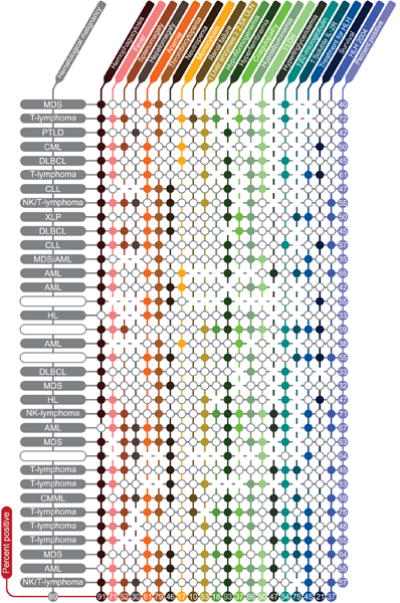
Each closed circle (○) represents a negative result. Blanks represent missing information. Each row is one patient and each column is a variable. Numbers show percent positive patients (for each characteristic) or percent positive characteristics (for each patient) excluding missing information. Variables evaluated (listed in order of columns) included BM/lymph node/spleen hemophagocytosis per pathology evaluation, fever, splenomegaly (clinically palpable spleen), hepatomegaly (clinically palpable liver), anemia (hemoglobin < 9.0 g/L), thrombocytopenia (platelets < 100 × 109/L), neutropenia (absolute neutrophil count (ANC) < 1.0 × 109/L), monocytosis (absolute monocyte count (AMC) > 1.0 × 109/L), renal failure (≥ 50% increase in creatinine over baseline), elevation of hepatic enzymes (≥ 2.5× upper limit of normal), hypofibrinogenemia (fibrinogen ≤ 150mg/dL), hyperferritinemia (ferritin ≥ 500micrograms/L), coagulopathy (PT ≥ 1.5× upper limit of normal and/or PTT ≥ 1.5× upper limit of normal and/or D-dimer ≥ 10.0mcg/mL), hypoalbuminemia (< 3.5g/dL), elevated LDH (≥ 2.5× upper limit of normal), hypertriglyceridemia (≥ 265 mg/dL), elevated b2-microglobulin (≥ 2mg/L), and elevated soluble IL-2 receptor (CD25) ≥ 2400U/mL. Abbreviations: Hb, hemoglobin; ULN, upper limit of normal; LDH, lactate dehydrogenase; HLH 2004, Hemophagocytic lymphohistiocytosis 2004 diagnostic criteria; MDS, myelodysplastic syndromes; PTLD, post-transplant lymphoproliferative disorder; CML, chronic myeloid leukemia; DLBCL, diffuse large B-cell lymphoma; CLL, chronic lymphocytic leukemia; XLP, X-linked lymphoproliferative disease; AML, acute myeloid leukemia; HL, Hodgkin’s lymphoma.
We then evaluated the remaining 26 of 61 (43%) patients who had a pathologic evidence of hemophagocytosis/lymphohistiocytosis on tissue biopsy (bone marrow or lymph node) but did not meet the extended 18-point diagnostic criteria. Follow-up data was not available for 9 patients. The median follow-up among the remaining 17 patients was 17.27 months (range, 2.9 – 82 months). The median survival among these 17 patients has not been reached. We plotted the overall survival curves by patients who met the expanded HLH diagnostic criteria and patients who did not meet the expanded HLH diagnostic criteria (Figure 2). Log-rank test suggested that the patients who did not meet the expanded HLH-criteria had significantly better overall survival rate that those who met the expanded HLH-criteria (p=0.001). The 1-year and 3-year survival rates of patients who did not meet the expanded diagnostic criteria were 0.875 (95% CI: 0.727, 1.000) and 0.624(95% CI: 0.406, 0.960), respectively. The 1-year and 3-year survival rates of patients who met the expanded HLH diagnostic criteria were 0.294 (95% CI: 0.172, 0.503) and0.224 (95% CI: 0.116, 0.432).
Figure 2A.
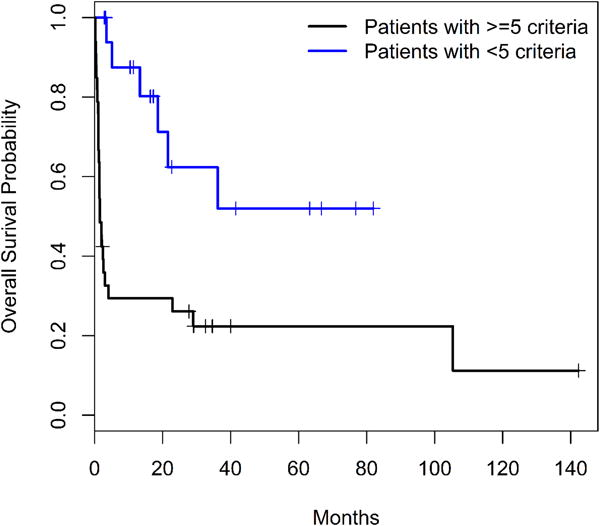
Overall survival of patients classified by the extended HLH criteria
In addition, as the 33 patients who met the expanded criteria included the 13 patients who met the HLH-2004 criteria and it is possible that the 13 patients who met the HLH 2004 diagnostic criteria negatively impacted the survival in the entire cohort of 33 patients. To overcome this, the 33 patients who met the expanded HLH criteria were analyzed into two subgroups: those who met the HLH 2004 criteria versus the others who didn’t meet the HLH 2004 criteria. The median OS of the 13 patients who met the extended criteria and the HLH-2004 criteria is 1.43 (95% CI: 0.73, infinity). The median OS of the 20 patients who met the extended criteria but not the HLH-2004 criteria is 1.76 (95% CI: 1.43, infinity). The p-value of long-rank test for the survival comparison between the two subgroups is 0.34 (not significant). When patients with follow-up data in all three subgroups were compared: 13 patients who met HLH-2004 and extended HLH criteria versus 20 patients who did not meet HLH-2004 but met extended HLH criteria and the 17 patients who met neither criteria log-rank test suggested that there were significant differences in overall survival rate among the three subgroups (p=0.002). The 1-year survival rates of these three subgroups were 0.277 (95% CI: 0.110, 0.699), 0.300 (95% CI: 0.154, 0.586) and 0.875 (95% CI: 0.727, 1.000), respectively (Figure 2B). Herein, we only report the 1-year overall survival as the three years overall survival is not estimable for all subgroups.
Figure 2B.
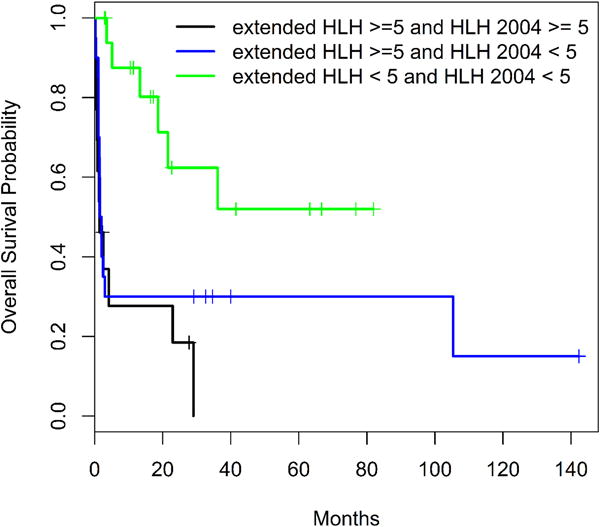
Overall survival of patients classified by both the extended HLH and HLH 2004 criteria
DISCUSSION
In patients with malignancy associated HLH, the prognosis is dismal. In a recent study from Sweden, all patients with secondary HLH due to malignancy died17. The median time from HLH diagnosis to death was 22 days (range 0–108 days). In our study 35 of 61 patients had possible malignancy associated secondary HLH (based on ≥ 5 features from among 18 adult HLH associated variables evaluated in the extended criteria), and only 7 of them survived. The median survival among the 33 patients who had follow-up data in this group was a very dismal 1.5 months.
A greater awareness of the disease among adult oncologists, initiation of a comprehensive laboratory and clinical work-up for HLH in suspected adult patients, and development and implementation of an adult specific HLH treatment program may improve the outcome in adult patients with HLH. Recently published data suggests that HLH is a more common entity in adults than previously thought6. It is well known that the presence of hemophagocytosis or lymphohistiocytosis alone is insufficient to confirm or refute a diagnosis of HLH. We reviewed the published literature and expert opinions and identified 18 criteria that were demonstrated to be closely associated with HLH in adults or children in these peer-reviewed manuscripts. We then attempted to select patients who had a higher likelihood of systemic HLH from among the 61 patients who had a confirmed pathology of hemophagocytosis or lymphohistiocytosis using the identified 18 variables. 35 of the patients had 5 or more additional criteria beyond hemophagocytosis or lymphohistiocytosis and were more likely in our opinion to have a systemic HLH process rather than a reactive hemophagocytosis or lymphohistiocytosis. There is no significant difference in overall survival among the 13 patients who met the HLH-2004 criteria (median overall survival = 1.43 months) and the 20 patients who did not meet the HLH-2004 criteria but met our extended 18-point HLH criteria (median overall survival = 1.76) (P=0.34). The inferior survival in both these groups of patients suggests that these patients likely had a more aggressive systemic process rather than just reactive hemophagocytosis/lymphohistiocytosis on pathology. Further in support of this point is the significantly improved survival (median overall survival = 17.2 months) among the 26 patients who had hemophagocytosis /lymphohistiocytosis on pathology but met neither the HLH-2004 nor the expanded HLH criteria (Figure 2B). Furthermore, the similar survival among patients who met the HLH-2004 and those who did not meet the HLH-2004 but met the extended HLH criteria suggests that the extended criteria may adequately and potentially more expediently identify malignancy-associated secondary HLH in adult patients triggering early referral to tertiary centers of expertise for HLH-specific standard or clinical trial based therapies. Unfortunately, only 45.5% (15/33) of these patients received HLH targeted therapies suggesting significant under diagnosis of HLH in adults with malignancies.
As the suspicion index goes up we anticipate more oncologists to initiate a work-up for HLH. However data suggests that the HLH-2004 criteria developed for children may be inadequate for accurately diagnosing HLH in adults3,18,19. This brings us to the second major hurdle that must be resolved to enable accurate identification of this entity in adults, namely the development of adult HLH diagnostic criteria. By applying extended diagnostic criteria that included 18 variables compiled from previously published literature on adult HLH we were able to identify features highly suggestive of HLH in 57% as compared to 21% (with the standard HLH-2004 criteria) of our patients who had pathologic evidence of “hemophagocytosis” or “”lymphocytosis” on tissue biopsy. Schram and colleagues recently reported that hyperferritinemia does not clearly predict for the HLH in adult population20. In our small cohort of HLH patients most presented with hyperferritinemia at diagnosis and/or during course of the disease (88%). Although hyperferritinemia may not be specific or sufficient for diagnosis of HLH in adults as suggested by Schram et al, it appears that increased ferritin levels frequently accompany adult HLH and is an important variable in the HLH diagnostic work-up.
This suggests that identification of HLH may be more difficult in adults and that inclusion of additional diagnostic variables, especially variables that are easily obtained on routine laboratory or physical examination may improve our sensitivity to diagnose HLH and allow us to work-up cases that may have been previously missed. This manuscript is not intended to replace or refute the HLH-2004 criteria but rather to suggest that more readily and quickly available variables may be efficient surrogates in raising suspicion for this entity at an earlier time-point. The HLH-2004 criteria requires 5 of 8 criteria to be met including two criteria (soluble IL2r and NK-cell activity) that are difficult to obtain in smaller institutions and may take up to 1 week to be reported even in larger institutions such as ours. Non-availability or delayed availability waiting for these variables to be reported may delay identification and referral to a tertiary care facility with inferior outcomes. As a referral center we face this issue repeatedly and this served as the impetus to identify more easily available variables until formal adult HLH criteria can be developed. With greater awareness and initiation of a comprehensive work-up for HLH we anticipate that a number of patients who would have been diagnosed with conditions such as hepatic or renal failure of unknown etiology, sudden onset multi-organ failure, culture-negative sepsis, encephalopathy of unknown etiology may be identified to have had HLH and would be candidates for HLH directed therapy. Comparison of these expanded HLH diagnostic criteria to the standard HLH-2004 diagnostic criteria in a prospective manner in adults will allow us to determine whether these expanded criteria are more accurate not only at the diagnosis of adult secondary HLH, but also at prognostication and identification of patients most likely to respond to immunomodulatory combination therapy versus those needing allogeneic stem cell transplant. These efforts are underway and exhaustive laboratory evaluation has been incorporated in our ongoing prospective clinical trial for adults with HLH (ClinicalTrials.gov Identifier: NCT02385110).
It should also be emphasized that from the results of our study – the extended diagnostic criteria are applicable to malignancy associated secondary HLH only. While they may be applied to other causes of secondary HLH (including infectious and autoimmune), and may very well be better than HLH-04 criteria in this group of patients, the data presented in this study did not include such patients.
The third major hurdle to successful management of HLH in adults is the development and validation of an effective treatment strategy. Less than 50% of the patients in our study received HLH-directed therapy. There are several reasons for this. One of the major hurdles to effective therapy in adult HLH is the delay in recognition of this entity and presentation at an advanced stage when it is not feasible to initiate effective therapy. The other major reason for <50% of patients receiving effective therapy is that patients with malignancy-associated HLH are already immunocompromised and addition of further cytotoxic therapy with etoposide or ATG-based regimens is not an appealing option. Furthermore it is difficult to abort therapy of the underlying malignancy and switch to therapy of HLH. At the same time we have noted a very aggressive and rapid progression of HLH in our patients with malignancy associated HLH making with a majority of patients dying from the HLH process in spite of continued treatment of the underlying malignancy. In our opinion the secondary but aggressive HLH process as well as the underlying malignancy must be simultaneously addressed making this a difficult situation. The advent of novel non-cytotoxic agents that do not add to cumulative toxicity to chemotherapy for the underlying malignancy may allow concurrent treatment of the malignancy and the HLH process. One such agent that is in clinic is the interferon-gamma inhibitor NI-0501 that was recently approved breakthrough designation for the treatment of primary HLH but may also prove to be a major milestone in the therapy of secondary and malignancy-associated HLH21. This agent does not cause myelosuppression and may be potentially given concomitant with anti-tumor therapy.
Current therapy of adult HLH is based on pediatric data from HLH-94 and HLH-200410,22. No adult specific frontline HLH prospective trials have been conducted in the United States. A recent report from China, described the use of combination chemotherapy with DEP (doxorubicin, etoposide, methylprednisolone) regimen as a salvage therapy for adult patients with refractory HLH. The DEP regimen showed promising results with a complete remission in 27% and partial remission in 49% of the 63 refractory HLH patients treated23. Furthermore, even within the pediatric population the outcomes of HLH-94 remain less than ideal with only approximately half of patients experiencing complete resolution of disease, with a significant early mortality rates22,24,25. The outcomes of HLH-2004 are awaited. Recently, it was shown, that alemtuzumab is an effective salvage agent for refractory HLH, leading to improved response rates and transition to allogeneic stem cell transplant in pediatric patients26. Alemtuzumab up-front in combination with dexamethasone and etoposide may afford the best combination to target HLH. A clinical trial to evaluate this approach is currently being developed by the pediatric HLH consortium. With input from our pediatric colleagues at Texas Children’s Hospital a similar IRB-approved study has opened and is accruing at UT/MDACC to treat adult patients with HLH (Figure 4) (NCT02385110). This is the only prospective clinical trial for adults with HLH in the United States and will likely help to answer many questions regarding the ideal diagnostic work-up, the optimal therapy, the need for allogeneic stem cell transplant versus maintenance approaches, and the long-term outcomes in adult patients with HLH.
Figure 4.
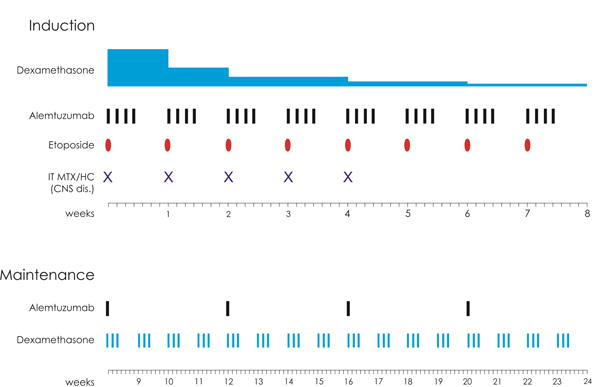
Alemtuzumab in combination with Etoposide and Dexamethasone for the treatment of adult patients with Hemophagocytic Lymphohistiocytosis
Acknowledgments
None
Funding Source: This study was conducted following the guidelines of The University of Texas MD Anderson Cancer Centre after local IRB approval. It was supported in part by the MD Anderson Cancer Centre Support Grant (CCSG) CA016672
Footnotes
P30 CA016672.
Conflict of Interest: No relevant COI to disclose.
Author Contributions:
Gevorg Tamamyan, Jing Ning, Preetesh Jain, Koji Sasaki, Waleed Abdelall, Sergej Konoplev and Naval Daver collected and analyzed the data and wrote the paper. All other authors: Hagop Kantarjian, Kenneth L. McClain, Carl E. Allen, Sherry Pierce, Jorge Cortes, Farhad Ravandi, Marina Konopleva, Guillermo Garcia-Manero, Christopher B. Benton, Dai Chihara, Michael Rytting, Sa Wang participated in the discussion, have reviewed and approved the current version of the manuscript.
Naval Daver is responsible for the overall content as guarantor.
References
- 1.Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139(6):713–727. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6(1):137–154. doi: 10.1586/eci.09.58. [DOI] [PubMed] [Google Scholar]
- 3.Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015;125(19):2908–2914. doi: 10.1182/blood-2015-01-551622. [DOI] [PubMed] [Google Scholar]
- 4.Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160(3):275–287. doi: 10.1111/bjh.12138. [DOI] [PubMed] [Google Scholar]
- 5.Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89(4):484–492. doi: 10.1016/j.mayocp.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90(3):220–224. doi: 10.1002/ajh.23911. [DOI] [PubMed] [Google Scholar]
- 7.Shabbir M, Lucas J, Lazarchick J, Shirai K. Secondary hemophagocytic syndrome in adults: a case series of 18 patients in a single institution and a review of literature. Hematol Oncol. 2011;29(2):100–106. doi: 10.1002/hon.960. [DOI] [PubMed] [Google Scholar]
- 8.Allen CE, McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2015;2015(1):177–182. doi: 10.1182/asheducation-2015.1.177. [DOI] [PubMed] [Google Scholar]
- 9.Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–619. doi: 10.3109/10428194.2010.551153. [DOI] [PubMed] [Google Scholar]
- 10.Henter J-I, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 11.Hejblum G, Lambotte O, Galicier L, et al. A web-based delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One. 2014;9(4):e94024. doi: 10.1371/journal.pone.0094024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang S-C, Chen J-S, Cheng C-N, Yang Y-J. Hypoalbuminaemia is an independent predictor for hemophagocytic lymphohistiocytosis in childhood Epstein-Barr virus-associated infectious mononucleosis. Eur J Haematol. 2012;89(5):417–422. doi: 10.1111/ejh.12006. [DOI] [PubMed] [Google Scholar]
- 13.Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet (London, England) 2014;383(9927):1503–1516. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 14.Hibi S, Ikushima S, Fujiwara F, et al. Serum and urine beta-2-microglobulin in hemophagocytic syndrome. Cancer. 1995;75(7):1700–1705. doi: 10.1002/1097-0142(19950401)75:7<1700::aid-cncr2820750722>3.0.co;2-d. http://www.ncbi.nlm.nih.gov/pubmed/8826930. Accessed March 28, 2016. [DOI] [PubMed] [Google Scholar]
- 15.Emminger W, Zlabinger GJ, Fritsch G, Urbanek R. CD14(dim)/CD16(bright) monocytes in hemophagocytic lymphohistiocytosis. Eur J Immunol. 2001;31(6):1716–1719. doi: 10.1002/1521-4141(200106)31:6<1716::AID-IMMU1716>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 16.Bland JM, Altman DG. Survival probabilities (the Kaplan-Meier method) BMJ. 1998;317(7172):1572. doi: 10.1136/bmj.317.7172.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsson T. Secondary haemophagocytic lymphohistiocytosis: Experience from the Uppsala University Hospital. Ups J Med Sci. 2015 Jul;:1–6. doi: 10.3109/03009734.2015.1064500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raschke RA, Garcia-Orr R. Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest. 2011;140(4):933–938. doi: 10.1378/chest.11-0619. [DOI] [PubMed] [Google Scholar]
- 19.Padhi S, Varghese RGB, Ramdas A, Phansalkar MD, Sarangi R. Hemophagocytic lymphohistiocytosis: critical reappraisal of a potentially under-recognized condition. Front Med. 2013;7(4):492–498. doi: 10.1007/s11684-013-0292-0. [DOI] [PubMed] [Google Scholar]
- 20.Schram AM, Campigotto F, Mullally A, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood. 2015;125(10):1548–1552. doi: 10.1182/blood-2014-10-602607. [DOI] [PubMed] [Google Scholar]
- 21.Prof FL, Allen C, De Benedetti F, et al. A Novel Targeted Approach to the Treatment of Hemophagocytic Lymphohistiocytosis (HLH) with an Anti-Interferon Gamma (IFNγ) Monoclonal Antibody (mAb), NI-0501: First Results from a Pilot Phase 2 Study in Children with Primary HLH. Blood. 2015;126(23):LBA – 3. [Google Scholar]
- 22.Henter JI, Aricò M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997;28(5):342–347. doi: 10.1002/(sici)1096-911x(199705)28:5<342::aid-mpo3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Huang W, Hu L, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood. 2015;126(19):2186–2192. doi: 10.1182/blood-2015-05-644914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henter J-I, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 25.Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–109. doi: 10.1002/pbc.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]