

Summary
Opioids remain the standard for analgesic care, however adverse effects of systemic treatments contraindicate long-term administration. While most clinical opioids target mu opioid receptors (MOR), those that target the delta class (DOR) also demonstrate analgesic efficacy. Furthermore, peripherally-restrictive opioids represent an attractive direction for analgesia. However, opioid receptors including DOR are analgesically incompetent in the absence of inflammation. Here, we report that G Protein-Coupled Receptor Kinase 2 (GRK2) naively associates with plasma membrane DOR in peripheral sensory neurons to inhibit analgesic agonist efficacy. This interaction prevents optimal Gβ subunit association with the receptor, thereby reducing DOR activity. Importantly, bradykinin stimulates GRK2 movement away from DOR and onto Raf Kinase Inhibitory Protein (RKIP). Protein kinase C (PKC)-dependent RKIP phosphorylation induces GRK2 sequestration, restoring DOR functionality in sensory neurons. Together, these results expand the known function of GRK2, identifying a non-internalizing role to maintain peripheral DOR in an analgesically incompetent state.
Graphical abstract
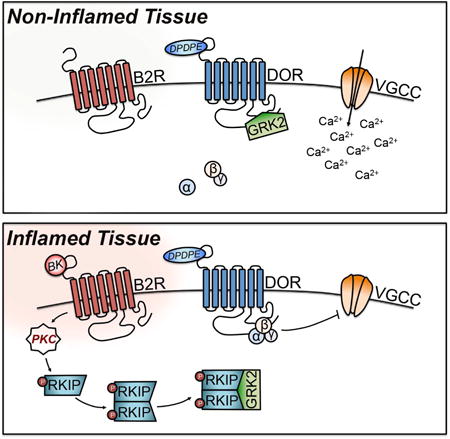
Introduction
Opioid agonists are essential therapeutic strategies in the treatment of pain. Opioids produce analgesia by activating G Protein-Coupled Receptors (GPCRs) known as mu (μ-, MOR), delta (δ-, DOR), and kappa (κ-, KOR). Traditionally, MOR analgesics are prescribed for the treatment of severe pain. However, DOR agonists have reduced side effect profiles compared to MOR agonists in rodent and non-human primate models (Vanderah TW, 2010). Like other opioid receptors, DOR primarily signals downstream through Gαi and Gβγ subunits (Alvez et al., 2004) that inhibit neuronal depolarization by decreasing cAMP activity (Law & Bergsbaken, 1995) and inhibiting voltage-gated Ca2+ channels (VGCCs) (Ford et al., 1998). In an effort to reduce systemic side effects and abuse potential, peripherally-restricted DOR agonists serve as an attractive alternative to systemic opioid therapies. However, multiple reports demonstrate that peripheral DOR analgesic competence requires an inflammatory pre-stimulus (Stein et al., 1989; Patwardhan et al., 2005; Patwardhan et al., 2006; Gaveriaux-Ruff et al. 2008; Rowan et al., 2009; Pettinger et al., 2013). Importantly, peripheral DOR incompetence is not well understood, and could provide important insight on the role of inflammatory mediators in peripheral opioid receptor regulation.
Peripheral tissues release inflammatory mediators such as bradykinin (BK) in response to injury (Levy & Zochodne, 2000). Nociceptive responses following BK administration are mediated via Gαq/11–coupled GPCRs expressed by primary afferent neurons that co-express opioid receptors (Steranka et al., 1988; Patwardhan et al., 2005; Jeske et al., 2006; Petcu et al., 2008). Importantly, BK induces rapid functional competence of DOR antinociception (Patwardhan et al., 2005; Rowan et al., 2009), indicating that peripheral DOR exists naively in a desensitized state. Inflammation-induced DOR analgesic competence at peripherally restrictive doses is known as “priming” (Patwardhan et al., 2005; Rowan et al., 2009; Pradhan et al., 2013), yet a mechanism for this phenomenon remains unknown. Recent work has identified a role for protein kinase C (PKC) (Patwardhan et al., 2005; Rowan et al., 2009), which agrees with work demonstrating that BK activation drives phospholipase C (PLC) activity to stimulate downstream PKC isoforms (Fu et al., 1989; Tippmer et al., 1994; Graness et al., 1997). Indeed, careful dissection of this mechanism would increase the application of peripherally-restrictive DOR agonists to treat pain and reduce centrally-mediated negative side effects associated with systemic MOR agonist administration.
Agonist-induced desensitization of DOR is dependent on hierarchical phosphorylation by G Protein-Coupled Receptor Kinase 2 (GRK2) (Kouhen et al., 2000; Guo et al., 2000). In contrast, the scaffolding protein Raf Kinase Inhibitory Protein (RKIP) facilitates opioid receptor activity (Kroslak et al., 2001). An important regulatory feature of RKIP modulation of GPCR activity is direct phosphorylation by PKC, which induces RKIP dimerization and subsequent sequestration of GRK2 (Lorenz et al., 2003; Deiss et al., 2012). This represents a fundamental research effort to identify that GRK2 chronically downregulates DOR antinociception. Furthermore, we provide support for the hypothesis that BK primes DOR analgesic competency in peripheral sensory neurons via RKIP sequestration of GRK2.
Results
DOR competence in naïve and primed sensory neurons
Opioids elicit their analgesic effects, in part, via receptor-mediated inhibition of VGCCs (Stein & Zöllner, 2009). In sensory neurons, transient exposure to 50 mM KCl evokes a measureable increase in intracellular Ca2+, which is attributable to an influx of extracellular Ca2+ through VGCCs following neuronal depolarization (Kahsabova et al., 2002). Activation of DOR inhibits KCl-evoked Ca2+ influx through L-, N-, and P/Q- type VGCCs, as well as KCl-induced neurotransmitter release in DRG (Kahsabova et al., 2004). Multiple investigators have previously quantified opioid inhibition of VGCCs via KCl-evoked Ca2+ influx in cultured sensory neurons (Kahsabova et al., 2004; Pettinger et al., 2013). Thus, DOR agonist inhibition of KCl-evoked Ca2+ influx is a validated method for quantifying DOR activity in a population of sensory neurons. Given that DOR is not expressed in all peripheral sensory neurons, this method circumvents the limitation of user bias when determining whether native DOR is functionally expressed within a given cell and eliminates potential changes to receptor activity and/or biochemistry that result from receptor-fusion proteins introduced by gene targeting.
DOR activity was assessed in adult rat TG and DRG CAP-sensitive neurons (Figure 1A-1B). At doses ranging from 1 nM to 1 μM, the DOR agonist DPDPE did not significantly inhibit Ca2+ influx elicited by KCl in vehicle-treated TG or DRG neurons. However, at doses above 1 μM DPDPE efficiently inhibited KCl-evoked Ca2+ accumulation in both vehicle-treated TG and DRG. Pretreatment with BK (200 nM) significantly increased the potency of DPDPE to inhibit KCl-evoked Ca2+ influx in both populations of TG and DRG neurons at 1 μM (also known as the BK priming effect). Importantly, equimolar dose of DOR agonist DADLE has previously been shown to increase the population of TG capable of DOR-mediated inhibition of VGCCs (Pettinger et al., 2013). Thus, 1 μM DPDPE was the dose used for the remainder of this study in both cultured TG and DRG neurons. IC50 values for DPDPE were 6.91 × 10-6 M versus 5.21 × 10-6 M for vehicle-treated TG and DRG, respectively. IC50 values for DPDPE were 6.15 × 10-7 M versus 7.39 × 10-7 M for BK-treated TG and DRG, respectively. The efficacy or maximal response to DPDPE was unaltered by BK in both TG and DRG. Thus, the BK priming effect on DOR activity was indistinguishable between TG and DRG neurons.
Figure 1. Functional DOR competence in sensory neurons.
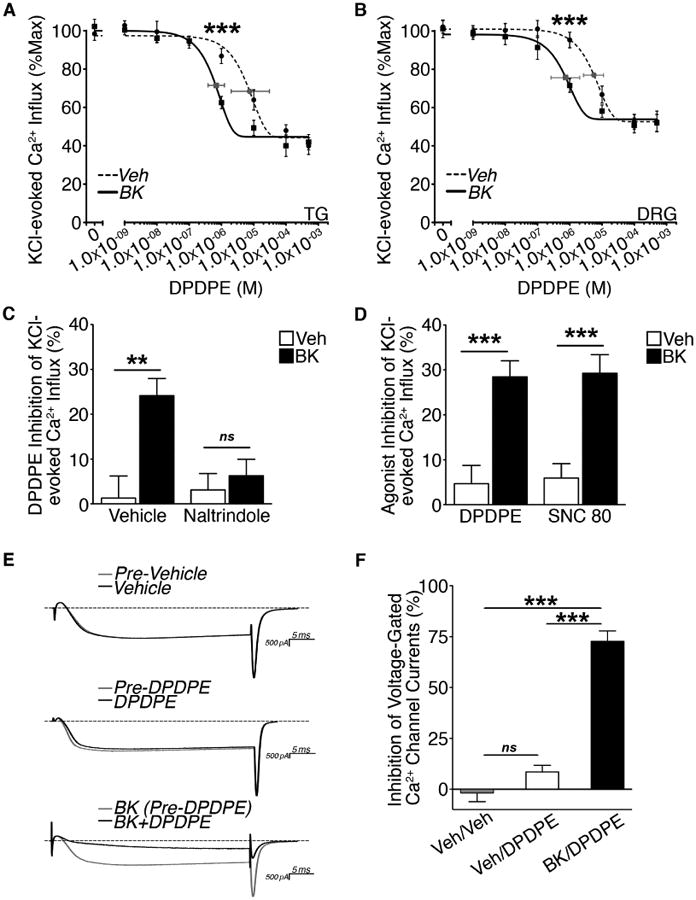
(A and B) Dose response for DPDPE inhibition of KCl (50 mM)-evoked Ca2+ influx in (A) TG or (B) DRG pretreated with vehicle or BK (200 nM, 5 min) following 2 h serum-starvation (***p<0.005 vs. vehicle; A: n=26-57 and B: n=17-39 neurons/dose; two-way ANOVA Bonferroni post-hoc; mean ± SEM). Least squares fit (best-fit) variable slope curves used to determine IC50 for vehicle (dotted line) and BK (solid line) and 95% confidence intervals (grey).
(C) Effect of naltrindole pretreatment on DPDPE (1 μM) inhibition of KCl-evoked Ca2+ influx in DRG neurons pretreated vehicle or naltrindole (10 nM, 5 min) prior to vehicle or BK (200 nM, 5 min) treatment following 2 h serum-starvation (**p<0.01; ns = no significance; n=14-28 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
(D) Comparison of DPDPE (1 μM) versus SNC 80 (1 μM) inhibition of KCl-evoked Ca2+ influx in DRG neurons pretreated with vehicle or BK (200 nM, 5 min) following 2 h serum-starvation(***p<0.005; n=24-34 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
(E and F) (E) Representative traces and (F) quantification of DPDPE (1 μM) inhibition of VGCCs in DRG neurons (20-35 pF) pretreated with vehicle or BK (200 nM, 10 min) following 2 h serum-starvation (***p<0.005; n=4-9 DRG/group; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
See also Figure S1-S2.
The response to DPDPE (1 μM) treatment in DRG pretreated with BK (200 nM; 5 min) was blocked when co-treated with irreversible selective DOR antagonist naltrindole (10 nM, 50 X Ki; 5 min) (Figure 2C). This demonstrates that inhibition of KCl-evoked Ca2+ influx by DPDPE at the selected dose is mediated by DOR. SNC 80 was included as a more selective nonpeptide agonist for DOR, compared to peptide agonist DPDPE (Calderon et al., 1994). At a concentration of SNC 80 equal to the dose used for DPDPE and previously verified to inhibit VGCCs in sensory neurons (Rowan et al., 2014), SNC80 (1 μM) and DPDPE (1 μM) equally inhibited DOR activity in DRG when primed by BK (200 nM; 5 min) (Figure 1D). Furthermore, patch-clamp electrophysiology revealed that DPDPE (1 μM) significantly inhibited VGCCs in DRG neurons (20-35 pF) only following BK (200 nM) pretreatment (Figure 1E-1F). Collectively, these studies demonstrate that DOR is functionally incompetent unless primed by BK in peripheral sensory neurons.
Figure 2. GRK2 modulation of DOR activity.
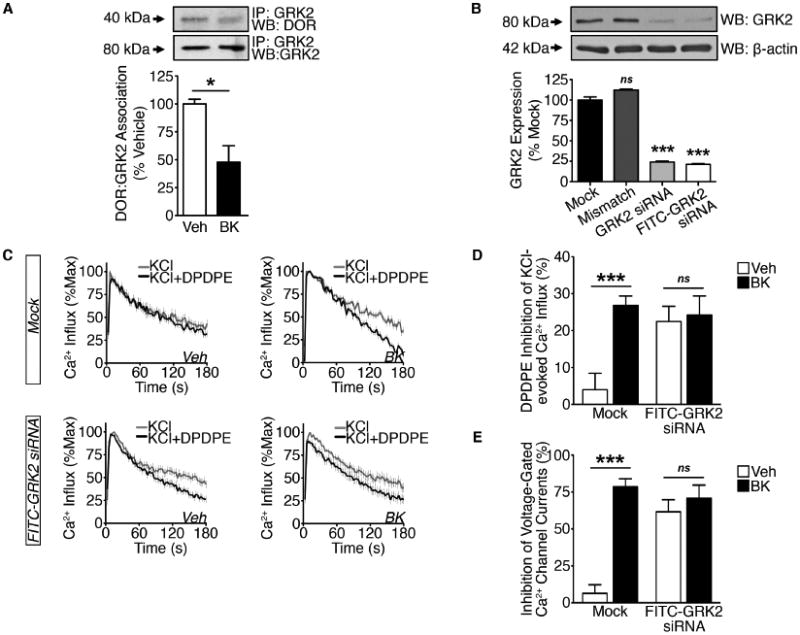
(A) Crude PM Co-IP from TG cultures serum-starved for 18 h and treated with vehicle or BK (200 nM, 5 min) (*p<0.05; n=3 independent trials; unpaired two-tailed student's t test; mean ± SEM).
(B) WCL from 2 h serum-starved TG cultures transfected in mock fashion or with mismatch, GRK2, or FITC-GRK2 siRNA (***p<0.005; ns = no significance; n=6 independent trials; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
(C and D) (C) Cumulative traces and (D) quantification of DPDPE (1 μM) inhibition of KCl (50 mM) -evoked Ca2+ influx in DRG (Mock-treated or transfected with FITC-GRK2 siRNA) pre-treated with vehicle or BK (200 nM, 5 min) following 2 h serum-starvation (***p<0.005; n=22-37 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
(E) DPDPE (1 μM) inhibition of VGCCs in DRG (Mock-treated or transfected with FITC-GRK2 siRNA) pre-treated with vehicle or BK (200 nM, 10 min) following 2 h serum-starvation (***p<0.005; n=5-8 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
See also Figure S3.
GRK2 modulation of functional DOR competence
Agonist-induced DOR activation recruits GRK2, which stimulates receptor phosphorylation that induces canonical receptor desensitization and internalization (Kouhen et al., 2000; Guo et al., 2000; Hong et al., 2009; Xu et al., 2010). Recent work in immortalized cells demonstrated that GRK2 may chronically remain associated with a GPCR in the absence of agonist stimulation, thereby reducing receptor competence (Namkung et al., 2009). To explain the chronic analgesic incompetence of peripheral DOR under naïve conditions, we first used co-immunoprecipitation analyses to investigate the possibility that GRK2 might be constitutively associated with DOR in primary neuronal culture (Figure 2A). In plasma membrane (PM) preparations from serum-starved, vehicle-treated TG cultures, DOR co-immunoprecipitates with GRK2. Notably, there is a significant reduction in GRK2 co-immunoprecipitation with DOR following treatment with BK (200 nM; 5 min). These data indicate that in the absence of agonist stimulation, GRK2 is statically bound to DOR under naïve conditions and provides support that BK induces a reduction in DOR association with GRK2 at the PM.
To evaluate the role of GRK2 in functional DOR competence under naïve and primed conditions, we employed siRNA-mediated knockdown of GRK2 expression and assessed DOR activity using Ca2+ imaging and patch-clamp electrophysiology. To demonstrate the specificity of this molecular approach, we first assessed the efficiency of GRK2 knockdown relative to β-actin, and found a 76% reduction in normalized GRK2 protein expression one day post-transfection (Figure 2B). Next, we determined DOR competency in the neuronal population of primary DRG following siRNA-mediated knockdown of GRK2 using Ca2+ imaging. BK pretreatment (200 nM; 5 min) significantly increased DPDPE inhibition of KCl-evoked Ca2+ influx in mock-transfected DRG neurons (Figure 2C-2D). However, in DRG cultures transfected with FITC-GRK2 siRNA, the increase in DPDPE inhibition of KCl-evoked Ca2+ influx was independent of BK pretreatment. Similarly, DPDPE significantly inhibited VGCCs only following priming by BK (200 nM) in mock-treated small to medium DRG neurons (Figure 2F). Importantly, small to medium DRG neurons transfected with FITC-GRK2 siRNA did not require BK pretreatment to evoke significant DPDPE inhibition of VGCCs. These data indicate that GRK2 participates in functional DOR incompetence in cultured DRG neurons.
GRK2 kinase activity regulates homologous desensitization of many GPCRs through receptor-G protein uncoupling and receptor internalization following phosphorylation (Premont RT et al., 1995). Following DOR agonist stimulation, GRK2 hierarchically phosphorylates DOR first at Ser363 to mediate receptor desensitization through G protein uncoupling, and then at Thr358 to initiate internalization of the receptor to further attenuate signaling (Guo et al., 2000; Kouhen et al., 2000; Xu et al., 2010). To determine whether BK affects GRK2 phosphorylation of DOR, we used a phosphorylation site-specific antibody for DOR at Ser363 (Figure 3A). In PM preparations from serum-starved, vehicle-treated TG cultures, DOR is phosphorylated at Ser363. Interestingly, DOR phosphorylation at Ser363 remains unchanged following pretreatment with BK (200 nM; 5 min). These data demonstrate that BK does not affect GRK2-mediated phosphorylation of DOR at its primary desensitization site.
Figure 3. Constitutive DOR incompetence is independent of GRK2 kinase activity.
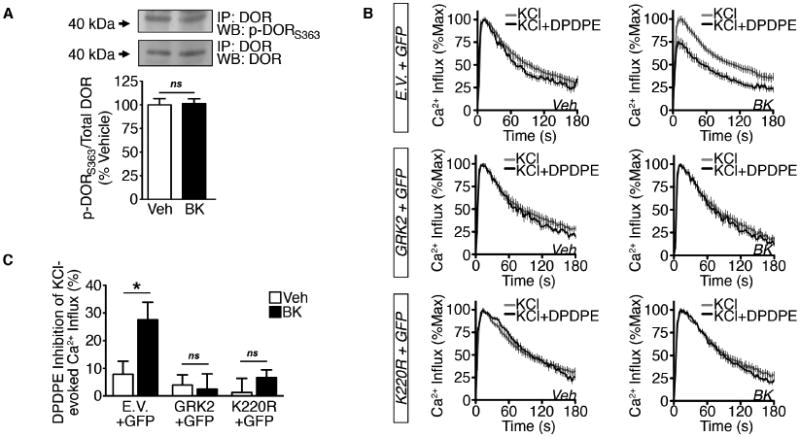
(A) DOR phosphorylation at Ser 363 in crude PM immunoprecipitates from TG cultures serum-starved for 18 h and treated with vehicle or BK (200 nM, 5 min) (ns = no significance; n=3 independent trials; unpaired two-tailed student's t test; mean ± SEM).
(B and C) (B) Cumulative traces and (C) quantification of DPDPE (1 μM) inhibition of KCl (50 mM)-evoked Ca2+ influx in nucleofected DRG (Overexpression: Empty Vector (E.V.) + GFP, GRK2 + GFP, K220R + GFP cDNAs) pretreated with vehicle or BK (200 nM, 5 min) following 2 h serum-starvation (*p<0.05; n=20-31 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
To assess whether GRK2 kinase activity supports functional DOR incompetence in primary DRG cultures, we overexpressed GRK2 or a kinase-inactive mutant that maintains the ability to interact with GPCRs (K220R, Kong G et al., 1994) and measured DOR inhibition of KCl-evoked Ca+2 influx in vehicle-treated and BK-treated conditions. BK pretreatment (200 nM; 5 min) significantly increased DPDPE inhibition of KCl-evoked Ca2+ influx in GFP-positive DRG nucleofected with empty vector (E.V.) (Figure 3B-3C). However, in DRG nucleofected with GRK2 or K220R, BK was unable to induce DPDPE inhibition of KCl-evoked Ca2+ influx over vehicle-treated DRG. These data demonstrate that GRK2 kinase activity is not required for GRK2 modulation of functional DOR incompetence in primary sensory neurons.
GRK2 modulation of DOR analgesic competence in vivo
After we identified a role for GRK2 modulation of functional DOR incompetence in sensory neurons in vitro, we measured physiologic peripheral DOR analgesic incompetence in vivo. In this study, we employed antisense-oligodeoxynucleotides (AS-ODN) against GRK2 mRNA to knock down GRK2 expression in a model of BK priming of peripheral DOR. Intrathecal (i.t.) injections of GRK2 AS-ODN over 3 days significantly reduced GRK2 expression in peripheral sensory nerves (Ferrari et al., 2012). Utilizing the same AS-ODN in this study, daily i.t. injections of GRK2 AS-ODN (30 μg/day) over 3 days nearly ablated GRK2 protein expression in both ipsilateral and contralateral DRG (Figure 4A-4B). To assess whether GRK2 knockdown affects DOR analgesic competence, we assessed DPDPE inhibition of PGE2-induced mechanical and thermal allodynia following BK priming in MM- and AS-ODN-treated rats (Figure 4C-4H). In MM-ODN-treated animals, injection of a peripherally restrictive dose of DPDPE (20 μg; Rowan et al., 2009) into the hindpaw did not block PGE2 (0.3 μg)-induced mechanical or thermal allodynia unless primed by BK (25 μg). Similar to functional data, GRK2 knockdown eliminated the BK priming requirement for functional competence of the DOR in vivo. Surprisingly, both BK-induced mechanical and thermal allodynia remain unchanged with GRK2 knockdown. Although GRK2 is reduced in both ipsilateral and contralateral DRG, contralateral PWTs and PWLs remained unchanged from BL. These data suggest that GRK2 impairs peripheral DOR analgesic competence in vivo.
Figure 4. GRK2 modulation of DOR-mediated antinociception.
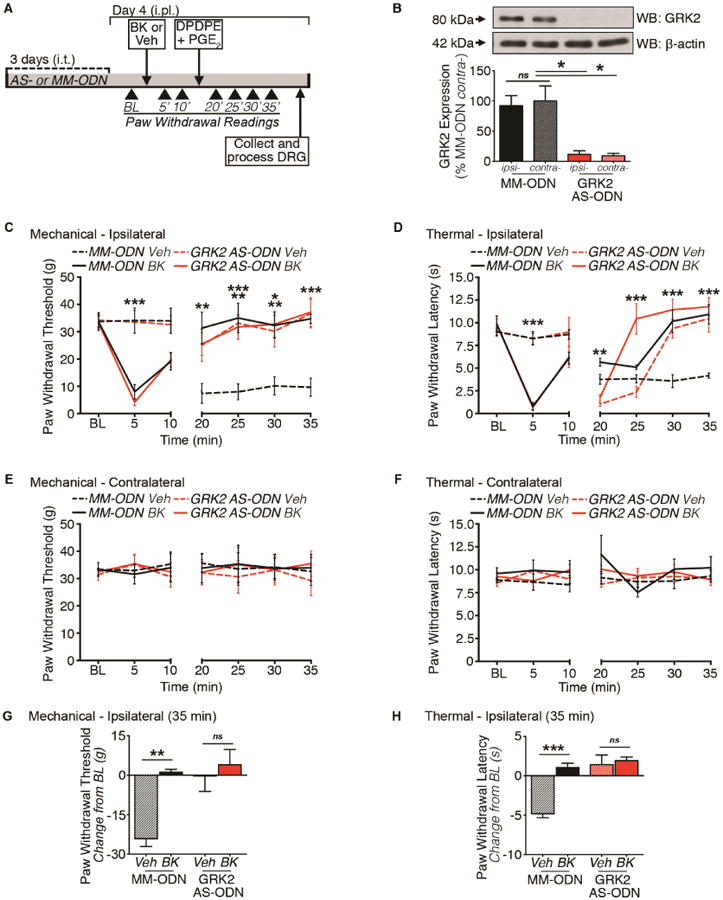
(A) Timeline for ODN injections and rat behavior protocol for BK priming of peripheral DOR antinociception.
(B) WCL from ipsilateral (ipsi-) and contralateral (contra-) DRG of rats treated with MM-ODN or GRK2 AS-ODN (*p<0.05; ns = no significance; n=3 independent trials; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
(C-F) Time course for DPDPE inhibition of PGE2-induced allodynia in ipsilateral (C, mechanical; D, thermal) and contralateral (E, mechanical; F, thermal) hindpaws. Paw withdrawal readings were measured 5 min and 10 min post-intraplantar (i.pl.) injection (Vehicle or BK (25 μg), and 5 min intervals for 20 min following second i.pl. injection (co-injection DPDPE (20 μg)/PGE2 (0.3 μg) (BK-induced allodynia: Mechanical and Thermal - p***<0.005 vs. vehicle-treated groups; DPDPE inhibition of PGE2-induced allodynia: Mechanical - 20 min, *p<0.05 (MM-ODN/BK); 25 min, **p<0.01 (AS-ODN/BK), ***p<0.005 (AS-ODN/Veh, MM-ODN/BK); 30 min, *p<0.05 (AS-ODN/Veh), **p<0.01 (AS-ODN/BK, MM-ODN/BK); 35 min, ***p<0.005 (MM-ODN/BK, AS-ODN/Veh, AS-ODN/BK) vs. MM-ODN/Veh; Thermal - **p<0.01 MM-ODN/BK vs. AS-ODN/Veh; 25-35, ***p<0.005 vs. groups below baseline (BL) readings; n=6 rats per group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
(G and H) Quantified antinociceptive effect of DPDPE at 35 min for (G) mechanical and (H) thermal readings (***p<0.005; ns = no significance; n=6 rats per group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
BK activates PLC-PKC pathway to modulate DOR
BK stimulation of B2R activates downstream PLC or cAMP signaling, and leads to PKC or PKA activation, respectively (Liebmann & Bohmer, 2000). To determine whether kinases downstream of BK signaling were involved in BK-mediated GRK2 dissociation from DOR, we employed inhibitors of PLC (U73122, 10 μM), PKC (GF 109203X (GFX), 10 μM), and PKA (H-89 20 μM) (Figure S3). BK-induced GRK2 dissociation from DOR was reversed by inhibitors for PLC and PKC, but not PKA. These results demonstrate that a PLC-PKC-dependent pathway is involved in BK-mediated GRK2 dissociation from DOR in cultured TG neurons.
BK-induced functional DOR competence is mediated by PKC both in vitro and in vivo (Patwardhan et al., 2005; Rowan et al., 2009). However, whether other second messengers downstream of BK-receptor activation mediate DOR priming remains unknown. Thus, we tested whether inhibitors for PLC (U73122, 10 μM) or PKA (H-89 20 μM) could block the BK effect on DPDPE inhibition of KCl-evoked Ca2+ influx in CAP-sensitive DRG neurons, using the PKC inhibitor (GFX, 10 μM) as a positive control. Inhibition of PLC and PKC, but not PKA, blocked BK priming of functional DOR competence in DRG (Figure S3). In agreement with GRK2 dissociation from DOR, these data indicate that BK priming of DOR competence in peripheral sensory neurons is mediated by the PLC-PKC pathway and not PKA.
Modulation of RKIP in sensory neurons
RKIP is an important signal modifier of GPCR signaling. In sensory neurons, BK evokes rapid PKC activation (Delmas et al., 2002; Cesare et al., 1999). Studies in immortalized cells have demonstrated that agonist-induced PKC phosphorylation of RKIP facilitates its self-dimerization, which is crucial for RKIP association with GRK2 (Corbit et al., 2003; Lorenz et al., 2003; Deiss et al., 2012). Dimerized RKIP can then sequester GRK2 and hinder GRK2-mediated receptor desensitization. Given its expression in intact TG and DRG tissue (Figure 5A) and sensitivity to PKC, we hypothesized that BK stimulates PKC-dependent modulation of RKIP in primary sensory neurons.
Figure 5. BK modulation of RKIP signaling in sensory neurons.
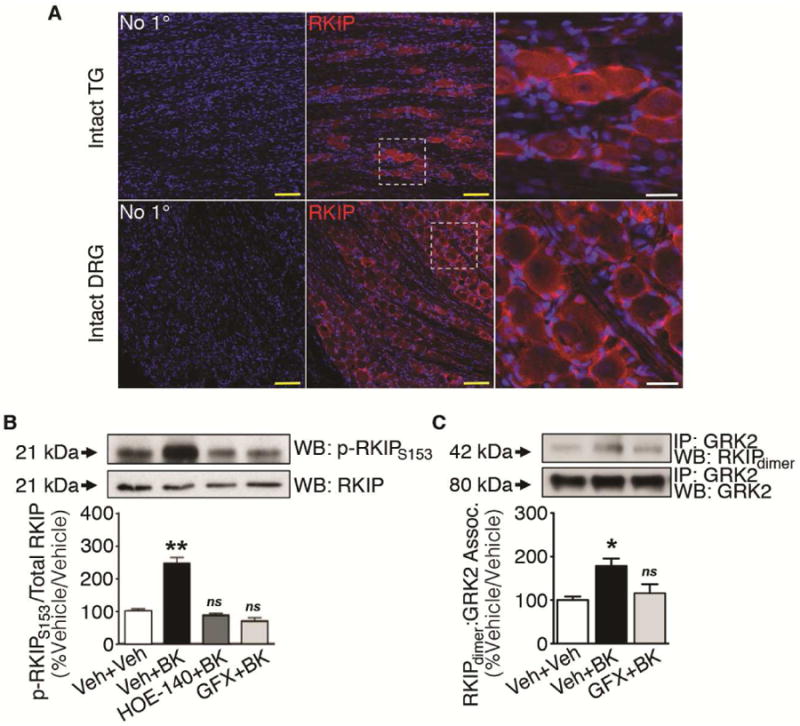
(A) Immunohistochemical expression of RKIP (red) in rat TG and DRG with Topro (blue) to identify nuclei. Scale bars: yellow = 50 μm; white = 15 μm. Confocal images are representative of 4 independent trials.
(B) Cytosolic lysates from TG cultures serum-starved for 18 h and treated with vehicle (5 min)/vehicle (5 min), BK (200 nM, 5 min)/vehicle, BK/HOE-140 (10 μM, 5 min), or BK/GFX (GF 109203X, 10 μM, 5 min) (**p<0.01 vs vehicle/vehicle, ns = no significance; n=3 independent trials; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
(C) Cytosolic co-IP from TG cultures serum-starved for 18 h and treated with vehicle (5 min)/vehicle (5 min), vehicle/BK (200 nM, 5 min), or BK/GFX (GF 109203X, 10 μM, 5 min) (*p<0.05; ns = no significance; n=3 independent trials; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
See also Figure S4.
Previous studies have demonstrated that activation of Gαq–coupled GPCRs results in PKC phosphorylation of RKIP at Ser153, followed by RKIP self-dimerization and recruitment of GRK2 in multiple cell lines (Corbit et al., 2003; Lorenz et al., 2003; Deiss et al., 2012). We sought to determine whether this mechanism occurs in sensory neurons. To determine whether BK activation of B2R leads to PKC phosphorylation of RKIP, we utilized a phosphorylation site-specific antibody for RKIP at Ser153 (Figure 5B). BK (200 nM; 5 min) treatment nearly triples RKIP phosphorylation at Ser153 in serum-starved TG, which was blocked by pretreatment with a B2R antagonist (HOE-140; 10 μM; 5 min) or PKC inhibitor (GFX, 10 μM; 5 min). These data demonstrate that BK stimulation of B2R results in PKC phosphorylation of RKIP at Ser153 in sensory neurons.
We next sought to determine whether BK-induced PKC stimulation also directs RKIP dimerization and association with GRK2, using Co-IP analyses (Figure 5C). BK (200 nM; 5 min) induced GRK2 co-immunoprecipitation with the RKIP dimer in cytosolic lysates from serum-starved TG cultures, in a manner sensitive to PKC inhibition (GFX, 10 μM; 5 min). These data indicate that BK-induced PKC activation stimulates RKIP self-dimerization and association with GRK2 in sensory neurons, supporting PKC-dependent RKIP sequestration of GRK2.
RKIP sequestration of GRK2 modulates BK priming of DOR
In immortalized cells, RKIP facilitates DOR signaling (Kroslak et al., 2001). Given that BK activation of PKC induces functional DOR competence (Figure S3, Patwardhan et al., 2005), GRK2 dissociation from PM DOR (Figure 2A), and RKIP phosphorylation and self-dimerization resulting in association with GRK2 (Figure 5B-5C), we hypothesized that RKIP sequestration of GRK2 governs BK priming of DOR in sensory neurons.
We employed siRNA-mediated knockdown of RKIP expression in sensory neuron cultures to evaluate the role of RKIP in BK priming of DOR. To demonstrate the specificity of this molecular approach in TG cultures, we assessed the efficiency of RKIP knockdown relative to β-actin, and found a 65% reduction in normalized RKIP protein expression one day post-transfection (Figure 6A).
Figure 6. RKIP modulation of functional DOR competence.
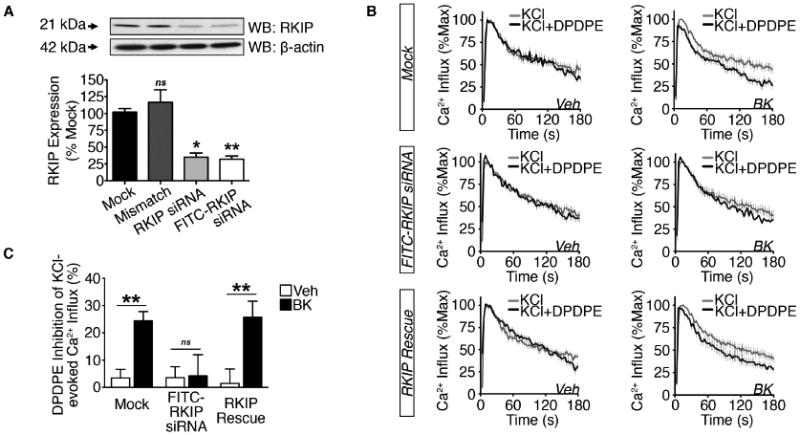
(A) WCL from 2 h serum-starved TG cultures transfected in mock fashion or with mismatch, RKIP, or FITC-RKIP siRNA (*p<0.05, **p<0.01, ns = no significance vs. mock; n=3 independent trials; one-way ANOVA Bonferroni post-hoc; mean ± SEM).
(B and C) (B) Cumulative traces and (C) quantification of DPDPE (1 μM) inhibition of KCl (50 mM)-evoked Ca2+ influx in DRG [Mock-treated, transfected with FITC-RKIP siRNA, or FITC-RKIP siRNA followed by nucleofection RKIP and GFP cDNAs (RKIP Rescue)] pretreated with vehicle or BK (200 nM, 5 min) following 2 h serum-starvation (**p<0.01; ns = no significance; n=20-26 DRG/group; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
Next, we sought to determine whether BK priming of DOR functional competence remained intact in DRG following siRNA-mediated knockdown of RKIP protein expression. BK pretreatment (200 nM; 5 min) significantly increased DPDPE inhibition of KCl-evoked Ca2+ influx in mock-transfected DRG (Figure 6B-6C). However, in DRG transfected with FITC-RKIP siRNA, there was no longer a BK-induced increase in DPDPE inhibition of KCl-evoked Ca2+ influx. Furthermore, when we re-introduced RKIP into FITC-RKIP siRNA-treated DRG (RKIP Rescue), the effect on DPDPE-inhibition of KCl-evoked Ca2+ influx was restored. These data indicate that RKIP expression is required for BK priming of DOR functional competence.
To determine whether PKC phosphorylation of RKIP is necessary for BK priming of functional DOR competence in cultured DRG, we overexpressed RKIP or a phospho-deficient mutant (RKIP-S153A) and measured DOR activity in vehicle- and BK-treated conditions. BK pretreatment (200 nM; 5 min) significantly increased DPDPE inhibition of KCl-evoked Ca2+ influx in GFP-positive DRG neurons nucleofected with empty vector (E.V.) or RKIP (Figure S4). However, in GFP-positive DRG cultures nucleofected with RKIP-S153A BK was unable to induce DPDPE inhibition of KCl-evoked Ca2+ influx. These data demonstrate that PKC phosphorylation of RKIP is required for BK-induced functional DOR competence in sensory neurons.
GRK2 modulation of DOR G protein coupling
The primary signaling event in GPCR activation involves G protein interaction (Rodbell et al., 1971). Following agonist stimulation, DOR interacts with a heterotrimer G protein complex comprised of an αi subunit and βγ dimer (Alvez et al., 2004). Upon dissociation from the receptor, Gαi and Gβγ signal as downstream effectors. Gβ subunits function to inhibit VGCCs (Herlitze et al., 1996; Ford et al., 1998). Given that BK enhances the potency of DPDPE-mediated inhibition of VGCCs in sensory neurons (Figure 1A-1B), we investigated the possibility that BK might influence interactions between Gβ and DOR using Co-IP analyses. In PM preparations from serum-starved, vehicle-treated TG cultures, Gβ co-immunoprecipitates with DOR (Figure 7A). Notably, there is an increase in Gβ co-immunoprecipitation with DOR following treatment with BK (200 nM; 5 min). Conversely, there is a decrease in DOR-bound Gβ in TG cultures treated with DPDPE (1 μM; 15 min) following initial BK pretreatment (Figure 7B). These data indicate that prior to ligand binding, BK facilitates the coupling of Gβ to DOR. Taken together with our Ca2+imaging data, these findings also indicate that Gβ is cooperatively released to potentially act on second order targets when DPDPE activates primed DOR.
Figure 7. GRK2 hinders peripheral DOR G protein coupling.
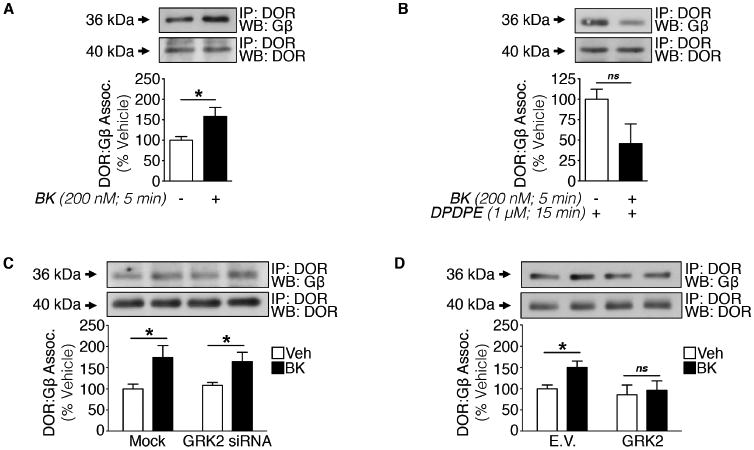
(A and B) Crude PM Co-IP from TG cultures serum-starved for 18 h and treated with (A) vehicle or BK (200 nM, 5 min) or (B) vehicle or BK (200 nM, 5 min) then DPDPE (1 μM, 15 min) (A: *p<0.05; ns = no significance; n=3 independent trials; unpaired two-tailed student's t test; mean ± SEM).
(C) Crude PM Co-IP from TG cultures serum-starved for 2 h following Mock- or GRK2-siRNA nucleofection and treated with vehicle or BK (200 nM, 5 min) (*p<0.05; n=3 independent trials; two-way ANOVA Bonferroni post-hoc; mean ± SEM).
(D) Crude PM Co-IP from TG cultures co-treated for 18 h with empty vector (E.V.) or GRK2 cDNA nucleofection (overexpression) in serum starved media followed by treatment with vehicle or BK (200 nM, 5 min) (*p<0.05; ns = no significance; n=3 independent trials; unpaired two-tailed student's t test; mean ± SEM).
Next, we hypothesized that the constitutive interaction between GRK2 and DOR may block the coupling of DOR to Gβ. For this, we utilized siRNA and Co-IP techniques in TG cultures to validate the role of GRK2 in DOR coupling to Gβ. BK pretreatment (200 nM; 5 min) increased DOR-bound Gβ in serum-starved mock- and GRK2 siRNA-treated TG culture PM lysates (Figure 7C). These data indicate that the absence of GRK2 alone is not sufficient to recruit Gβ to the receptor. To investigate whether GRK2 overexpression impairs DOR coupling to Gβ, PM Co-IPs were conducted from TG cultures nucleofected with GRK2 or E.V., serum-starved, and treated with vehicle or BK (200 nM; 5 min). As expected, Gβ co-immunoprecipitated with DOR in vehicle-treated TG cultures nucleofected with E.V., while BK treatment increased Gβ co-immunoprecipitation with DOR. GRK2 overexpression blocked the BK-dependent increase in Gβ association with DOR (Figure 7D). Thus, the constitutive association between GRK2 and DOR attenuates Gβ:receptor coupling in sensory neurons. Together with our DOR-GRK2 association and functional imaging studies, these findings suggest that GRK2 hinders receptor coupling to Gβ, such that Gβ may not inhibit VGCCs unless primed first by BK.
Discussion
The phenomenon of peripheral DOR incompetence in the periphery has been observed in vitro (Patwardhan et al., 2005; Patwardhan et al., 2006) and in vivo (Stein et al., 1989; Rowan et al., 2009), and pretreatment with an inflammatory stimulus is required for DOR activation, thereby, promoting analgesia. However, a major gap in knowledge existed concerning 1) why peripheral DOR remains functionally incompetent under naïve conditions and 2) a mechanism for DOR priming beyond PKC-dependence. This study identifies two important conclusions that fill this gap. First, our data illustrate that DOR responsiveness to agonist stimulation in naïve afferent terminals is impaired by a constitutive interaction with GRK2 at the PM that prevents receptor coupling to Gβ, which subsequently prohibits VGCC inhibition. Second, when peripheral sensory neurons undergo BK activation of B2R, PKC directly phosphorylates RKIP. This initiates RKIP self-dimerization and sequestration of GRK2. Consequently, DOR can couple to Gβ to inhibit VGCCs, which results in antinociception. Within this framework we have identified pharmaceutical targets that may enhance DOR-mediated analgesia.
Numerous reports in immortalized cell lines indicate that GRK2 phosphorylation of DOR occurs following stimulation by highly efficacious agonists such as DPDPE (Guo et al., 2000; Kouhen et al., 2000; Marie et al., 2008; Bradbury et al., 2009), deltorphin II (Bradbury et al., 2009) and (+)BW373U86, as well as SNC 80 in hippocampal lysates (Pradhan et al., 2009). In response to agonist stimulation, GRK2 hierarchically phosphorylates DOR for phosphorylation site-specific receptor regulation (Kouhen et al. 2000). The initial site phosphorylation, Ser363, promotes uncoupling of activated DOR from G proteins and desensitizes PM DOR, whereas the second phosphorylation site, Thr358, regulates receptor internalization. Although BK elicits an increase in DOR competence (Figures 1A-1B, 4C-4D, and 4G-4H), it does not affect GRK2 phosphorylation of DOR at Ser363 (Figure 3A). With no commercially available phospho-specific antibody for DOR Thr358, we could not evaluate GRK2 phosphorylation of the internalization residue. β-arrestin-2 mediates internalization of DOR following GRK2 phosphorylation at Ser363 (Bradbury et al., 2009). We report here that DPDPE and SNC 80, a β-arrestin-2-biased agonist in primary sensory neurons (Rowan et al., 2014), equally inhibited KCl-evoked Ca2+ influx following pretreatment by BK (Figure 1D), which suggests that β-arrestin-2 neither mediates constitutive DOR incompetence nor BK-induced functional DOR competence. Furthermore, genetic ablation of β-arrestin-2 has been reported to have no effect on DOR-mediated inhibition of VGCCs or analgesia in the absence or presence of inflammation (Pradhan et al., 2013). These data suggest that the hierarchical nature of DOR phosphorylation by GRK2 does not necessarily regulate DOR activity in sensory neurons.
GRK2 regulation of GPCR coupling independent of receptor phosphorylation has been reported for Gαq/11- (Dicker et al., 1999), Gαs- (Reiter et al. 2001), and Gαi/o-coupled receptors (Lembo et al., 1999; Namkung et al., 2009). In our overexpression studies, we found that GRK2 suppression of DOR signaling in DRG is not attributable to GRK2 kinase activity because the ability of a kinase-deficient mutant to attenuate DOR activity is indistinguishable from that of GRK2 (Figure 3B-3C). Furthermore, GRK2 overexpression also attenuates optimal DOR-G protein coupling (Figure 7D). These data implicate that GRK2 protein-protein interaction with DOR, rather than kinase activity and receptor phosphorylation, governs DOR responsiveness to agonist stimulation in peripheral sensory neurons. In immortalized cells, GRK2 constitutive association with D2R attenuates receptor signaling and G protein coupling independent of receptor phosphorylation by GRK2 (Namkung et al., 2009). Whether GRK2 chronically downregulates GPCRs other than DOR at the PM in sensory neurons remains to be elucidated.
An orchestration of signaling events are likely necessary to induce peripheral functional DOR competence in physiologically relevant systems. BK receptors are fundamental to peripheral opioid analgesia following inflammatory insult or after chronic constriction injury (Cayla et al., 2012). In about half of small-sized TG and DRG that overexpress DOR-GFP, a mild increase in DOR trafficking to the PM is induced by BK (200 nM) within 5-10 min (Pettinger et al., 2013). We did not observe any notable differences in native DOR trafficking to the PM following BK exposure (Figures 2A, 7A-D); however, this effect could be diluted in our cultures for biochemistry that included neurons with sizes ranging from small to large along with support cells (glial, etc.). In addition to an increase in DOR targeting to the PM in small neurons, Pettinger et al. also observed a doubling of CAP-sensitive TG neurons that respond to a DOR agonist following BK (200 nM; 15 min) treatment. We also observed this phenomenon in DRG; the total population of neurons that responded to DPDPE rose from 22.9-25.0% to 48.3-52.9% following BK (200 nM; 5 min) treatment (Figure S2). Interestingly, our population data also revealed that BK increased the total population of DRG that responded to CAP (1 μM) from 50-59.5% to 70.3-79.4%. These data are consistent with another report that found that approximately 70% of DRG are sensitive to this dose of CAP (Wang et al., 2008). It has been demonstrated that BK lowers the threshold for heat-activation of TRPV1 in DRG (Sugiura et al. 2002), and our data suggests that BK also enhances CAP-sensivity in DRG.
Comprehensively, results presented herein contribute to a collection of findings that characterize mechanisms driving DOR responsivity in multiple cellular models. For instance, allosteric modulation of DOR by sodium ions (Fenalti et al., 2014) could also allosterically affect constitutive GRK2 association with the receptor. Additionally, GRK2 association with DOR, which contributes to receptor internalization, likely facilitates reduced DOR responsiveness following biased ligand administration (Pradhan et al., 2009). However, it is difficult to determine whether STAT5 signalosome formation with DOR is affected by chronic GRK2 association with the receptor, since both utilize the same C-terminal amino acids (Georganta et al., 2010). Importantly, many of these studies utilize non-physiologic model cell systems, including transfected immortalized cells, which can overexpress receptor proteins relative to other endogenous regulatory proteins. Results presented here employ more physiologically-relevant sensory neurons, providing analysis of endogenously expressed receptors and regulatory proteins that correlate more with behavioral measures, and hence, clinical relevance.
B2R couples to multiple classes of G proteins, including Gαq and Gαs, and stimulates differential signaling cascades (Lliebmann & Böhmer, 2000). B2R activation of Gαq primarily activates the PLC-PKC pathway, whereas Gαs initiates cAMP-PKA signaling. Additionally, studies in immortalized cell lines have demonstrated that GRK2 can be directly phosphorylated by either PKC (Chuang et al., 1995; Pronin & Benovic, 1997) or PKA (Cong et al., 2001) to affect GPCR desensitization. We found that PLC and PKC, but not PKA, were involved in BK-induced GRK2 dissociation from PM DOR and BK priming of DOR in sensory neurons (Figure S3). Indeed, work in neuroblastoma cells demonstrated PKC and GRK2 mediate DOR desensitization (Marie et al., 2008). Although the concentration of BK used in our study (200 nM) activates B2R's primary PLC-PKC pathway, it may not sufficiently activate cAMP in DRG (Wang et al., 2008). Thus at higher concentrations of BK it may be possible that PKA contributes to functional DOR competence. Although PKA was not investigated, Patwardhan et al. demonstrated that PKC inhibition blocks peripheral DOR competence induced by a more potent dose of BK (10 μM) in sensory neurons (Patwardhan et al., 2006).
The importance of BK priming on functional DOR competence observed in vitro, by this study and others (Patwardhan et al., 2005; Patwardhan et al., 2006; Sullivan et al., 2015), was recapitulated in vivo. A peripherally restrictive dose of DPDPE was unable to elicit an anti-allodynic response unless primed by BK in MM-ODN-treated rats. This finding is similar to observations in rats in the absence of i.t. ODN treatments prior to behavioral experimentation (Rowan et al., 2009; Sullivan et al., 2015). This experiment identified that downregulation of GRK2 eliminates the need for BK priming of DOR-mediated anti-allodynia. Levine and colleges found that 3 days of i.t. GRK2 AS-ODN administration produces a 39% decrease in GRK2 protein expression in the saphenous nerve (Ferrari et al., 2012), yet we observed near-ablation of GRK2 protein at the level of the DRG. The difference in knockdown suggests that the support cells adjacent to distal portions of the afferent nerve were unaffected by i.t. GRK2 AS-ODN administration. Residual GRK2 that remains in fibroblasts, microglia, and other cells that run adjacent from the saphenous nerve to the peripheral terminals in the hindpaw may account for the enhanced onset of DPDPE anti-allodynia following BK priming in GRK2 knockdown animals without comparable changes in magnitude or duration of BK-induced mechanical and thermal allodynia. Because BK priming of DPDPE inhibition of PGE2-induced allodynia is mediated by PKC (Rowan et al., 2009), we theorize that BK may have been needed to induce the PKC-dependent sequestration of remaining GRK2 in order to behaviorally observe the earlier thermal anti-allodynic effects of DPDPE.
Our behavioral data also suggest that GRK2 chronically downregulates peripheral DOR anti-nociception. However, we observed a transient, enhanced thermal allodynia 5 min following DPDPE/PGE2 co-injection in GRK2 AS-ODN-treated rats. Similarly, PGE2-induced thermal allodynia was increased in two mouse models with reduced GRK2 expression: sensory neuron specific heterozygous GRK2 mice, and tamoxifen-treated inducible whole body heterozygous GRK2 mice (Eijkelkamp et al., 2010). Another study in GRK2 AS-ODN-treated rats found that reduced GRK2 expression enhances PGE2-induced mechanical allodynia (Ferrari et al., 2012). However, our results demonstrate that PGE2-induced mechanical allodynia is fully inhibited by DPDPE in GRK2 AS-ODN-treated rats (Figure 4C and 4G). Together with our findings, these data suggest that, although it can enhance the response to certain inflammatory mediators (including PGE2, not BK), targeting GRK2 can also be beneficial by enhancing peripheral opioid analgesia.
In conclusion, experimental results demonstrate that GRK2 chronically downregulates DOR functional competence at the PM in peripheral sensory neurons, as well as peripheral DOR anti-nociception in vivo. Prior to this study, there was no identified mechanism for DOR priming by BK beyond PKC-dependence. The phenomenon of peripheral DOR incompetence in the absence of an inflammatory priming stimulus would be expected to limit the effectiveness of locally administered DOR agonists to individuals with severe inflammatory pain. Since chronic GRK2 association with DOR contributes to receptor incompetence in the absence of inflammation, we propose that peripherally targeting GRK2 in combination with DOR may improve analgesic efficacy in non-inflammatory pain conditions.
Experimental Procedures
All procedures utilizing animals were approved by the Institutional Animal Care and Use Committee of University of Texas Health Science Center at San Antonio, and were conducted in accordance with the policies for the ethical treatment of animals established by the National Institutes of Health (NIH). Every effort was made to limit animal discomfort and the number of animals used.
Animals
Adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 200-250 g (biochemistry, Ca2+ Imaging) or 350-400 g (oligodeoxynucleotide-treated) were used in this study. Animals were housed in clean cages with a 12 h light/dark cycle for one week with food and water ad libitum before use.
Neuronal Cultures
For biochemistry, trigeminal ganglia (TG) were dissected bilaterally from male rats. TG were dissociated by 30 min collagenase (Worthington, Lakewood, NJ) treatment followed by 30 min trypsin (Sigma Aldrich, St. Louis, MO) treatment, with gentle rocking every 10 min. Cells were then resuspended in complete media (Dulbecco's modified Eagle's medium, Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen), 100 ng/mL nerve growth factor (NGF; Harlan Laboratories Indianapolis, IN), mitotic inhibitors (Sigma), 1% penicillin/streptomycin (Invitrogen), and 1% glutamine (Sigma)) and plated on poly-d-lysine-coated plates (Corning, Glendale, AZ). Cultures were maintained at 37°C and 5% CO2 and grown for 5-6 days with media changed the following day and every two days thereafter. TG were utilized for biochemical experiments to satisfy NIH requirements to reduce animal use in research.
For Ca2+ imaging, TG or dorsal root ganglia (DRG) dissected bilaterally at L4-L6 were dissociated by 40 min co-treatment with collagenase (Worthington) and dispase (Sigma) with gentle rocking every 10 min. Next, cells were resuspended in complete media and plated on poly-d-lysine/laminin-coated coverslips (BD Biosciences). Media was changed the following day and experiments were conducted within 24-48 h of initial culture.
Knockdown and overexpression strategies described in the Supplemental Experimental Procedures.
Crude Membrane Preparation
Primary TG cultures were pretreated as indicated. Cells were harvested and homogenized in homogenization buffer (25 mM HEPES, 25 mM sucrose, 1.5 mM MgCl2, 50 mM NaCl (pH 7.4), 1 mM sodium pyrophosphate, 1 mM sodium orthovanadate (Sigma), 1 μg/mL pepstatin (Sigma), 1 μg/mL leupeptin (Sigma), 1 μg/mL aprotinin (Sigma), and 100 nm phenylmethylsulphonyl fluoride (PMSF, Sigma)) with 20 strokes using a Potter-Elvehjem pestle and glass homogenizer tube. Homogenates were placed on ice for 15 min incubation and then centrifuged at 1000 × g for 1 min to remove nuclei and unlysed cells from the homogenate. Resulting supernatant was centrifuged at 16000 × g for 30 min at 4°C to separate cell membrane proteins from cytosolic proteins. Cytosolic supernatant was separated from the pellet (crude membrane fraction), which was re-suspended in 250 μL homogenization buffer containing 1% Triton X-100 (Fisher Scientific). Total protein was quantified using Bradford assay (Sigma) prior to co-immunoprecipitation (Co-IP). For Co-IP protocol and Western blot (WB) analysis details, please see Supplemental Experimental Procedures.
Ca2+ Imaging
Fura-2 AM was used to image individual neurons within a population of cultured ganglionic cells. Following 2 h serum-starvation, cells were loaded with fura-2 AM (1 μM; Molecular Probes, Carlsbad, CA) in the presence of pluronic F-127 (0.04%; Molecular Probes) for 1 h at 37°C in the dark, in standard extracellular solution (SES) containing (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 d-(+)-glucose, pH 7.40. Cells were viewed on an inverted Nikon Eclipse Ti-U microscope fitted with a 40×/1.35 numerical aperture Fluor objective and imaged using MetaFluor System for Ratio Fluorescence (MetaMorph, Downingtown, PA). Fluorescent images were taken alternately every 3 s with 340 and 380 nm excitation wavelengths in combination with a 510 nm emission filter with 200 ms exposure. Ratio of ΔF340/F380 was plotted for each cell versus time. Intracellular Ca2+ levels were analyzed as ΔF340/F380 ratios background corrected and normalized to initial value, R0. Corresponding filters were used to select FITC-siRNA- or GFP-positive DRG.
To quantify DOR activity in sensory neurons, Ca2+ imaging was used to assess DOR agonist inhibition of KCl-evoked Ca2+ influx (Figure S1). Although time in culture varied by protocol, BK pretreatment significantly enhanced DPDPE inhibition of KCl-evoked Ca2+ influx at all timepoints utilized in this study (Figure S2). For details on primary afferent neuron selection, opioid inhibition of KCl-evoked Ca2+ influx protocol, equations, and drug stock concentrations, refer to Supplemental Experimental Procedures.
Electrophysiology
Whole-cell patch-clamp recordings were used to measure DOR-mediated inhibition of VGCCs in cultured rat DRG neurons (20-35 pF). Following 2 h incubation in 2% serum at 37°C, whole-cell patch-clamp configuration was performed at room temperature on neurons viewed on an upright Nikon Eclipse E600FN microscope fitted with a 40×/0.80W numerical aperture objective. Borosilicate glass patch clamp capillaries (Sutter, Novato, CA, USA) were polished to resistances of 2-4 MΩ and filled with internal solution containing (in mM): 140 CsCl, 10 EGTA, 1 CaCl2, 1 MgCl2, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, pH 7.20. Whole-cell configuration was established in extracellular solution containing (in mM): 140 TEA-Cl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 D–glucose, pH 7.30. Axopatch 200B amplifier and pCLAMP9.6 software (Molecular Devices, Axon) were used to acquire and analyze data. From a holding potential of -60 mV, VGCC currents were activated by single pulse from −60 to 0 mV (50 ms duration). Waveform was applied repetitively every 10s. DPDPE (1 s a was applied via local application. Coverslips were incubated at room temperature for 10 min either with vehicle or with BK (200 nM). All recorded cells sized 20-35 pF were included in analysis.
Behavioral Test for BK Priming of DOR Functional Competence
This study utilized custom GRK2 antisense (AS)- and mismatch (MM)- oligodeoxynucleotide (ODN) sequences synthesized by Invitrogen first described by Levine and colleagues (Ferrari et al., 2012). ODNs were intrathecally (i.t.) administered to rats anesthetized with 2.5% isoflurane once daily for 3 days prior to behavioral testing. On day 4, BK-induced DPDPE inhibition of prostaglandin (PGE2)-induced thermal and mechanical allodynia was assessed in ODN-treated rats to determine the role of GRK2 in BK priming of peripheral DOR antinociception. All measurements were conducted by blinded observers. For further explanation, see Supplemental Experimental Procedures.
Statistics
GraphPad Prism 5.0 was used for statistical analyses (GraphPad Software, Inc., La Holla, CA). Quantitative data expressed as mean ± S.E.M. Statistical significance was determined by student's unpaired t-test, one-way ANOVA, or two-way ANOVA with Bonferroni post-hoc analyses as needed. p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We acknowledge Dr. Jeffrey L. Benovic (Thomas Jefferson University, USA) for gifting the GRK2-K220R cDNA and Dr. Kristina Lorenz (University of Wuerzburg, Germany) for gifting RKIPWT and RKIPS153A cDNAs. We would like to thank Dr. Elaine Por for critical discussions on RKIP. NINDS/RO1 NS082746 (N.A.J.), NIDCR/T32 DE14318 (A.D.B.), and NIDCR/F31 DE025551 (A.D.B.) from NIH funded this work.
Footnotes
Author Contributions: Conceptualization, A.D.B. and N.A.J.; Methodology, A.D.B. and N.A.J.; Validation, N.A.J.; Investigation, A.D.B., N.A.J., A.K.A., and M.A.H.; Formal Analysis, A.D.B. and A.K.A.; Resources, A.D.B., N.A.J., R.G., A.K.A., and M.A.H.; Writing – Original Draft, A.D.B. and N.A.J.; Writing – Review and Editing, A.D.B., N.A.J., and A.K.A.; Visualization, A.D.B. and N.A.J.; Project Administration, A.D.B. and N.A.J.; Supervision, N.A.J.; Funding Acquisition, A.D.B. and N.A.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvez ID, Ciano KA, Boguslavski V, Varga E, Salamon Z, Yamamura HI, Hruby VJ, Tollin G. Selectivity, cooperativity, and reciprocity in the interactions between the delta-opioid receptor, its ligands, and G-proteins. J Biol Chem. 2004;279:44673–82. doi: 10.1074/jbc.M404713200. [DOI] [PubMed] [Google Scholar]
- Bradbury FA, Zelnik JC, Traynor JR. G protein independent phosphorylation and internalization of the delta-opioid receptor. J Neurochem. 2009;109:1526–35. doi: 10.1111/j.1471-4159.2009.06082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon SN, Rothman RB, Porreca F, Flippen-Anderson JL, McNutt RW, Xu H, Smith LE, Bilsky EJ, Davis P, Rise KC. Probes for narcotic receptor mediated phenomena. 19. Synthesis of (+)-4-[(alpha R)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC 80): a highly selective, nonpeptide delta opioid receptor agonist. J Med Chem. 1994;37:2125–8. doi: 10.1021/jm00040a002. [DOI] [PubMed] [Google Scholar]
- Cayla C, Labuz D, Machelska H, Bader M, Schäfer M, Stein C. Impaired nociception and peripheral opioid antinociception in mice lacking both kinin B1 and B2 receptors. Anesthesiology. 2012;116:448–57. doi: 10.1097/ALN.0b013e318242b2ea. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–24. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chuang TT, LeVine H, 3rd, DeBlasi A. Phosphorylation and activation of beta-adrenergic receptor kinase by protein kinase C. J Biol Chem. 1995;270:18660–5. doi: 10.1074/jbc.270.31.18660. [DOI] [PubMed] [Google Scholar]
- Cong M, Perry SJ, Lin FT, Fraser ID, Hu LA, Chen W, Pitcher JA, Scott JD, Lefkowitz RJ. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J Biol Chem. 2001;276:15192–9. doi: 10.1074/jbc.M009130200. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trrakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory potein. J Biol Chem. 2003;278:13061–8. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- Deiss K, Kisker C, Lohse MJ, Lorenz K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein-coupled receptor kinase (GRK) 2. J Biol Chem. 2012;287:23407–17. doi: 10.1074/jbc.M112.363812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron. 2002;34:209–20. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Dicker F, Quitterer U, Winstel R, Honold K, Lohse MJ. Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc Natl Acad Sci U S A. 1999;96:5476–81. doi: 10.1073/pnas.96.10.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Wang H, Garza-Carbajal A, Willemen HL, Zwartkruis FJ, Wood JBB, Dantzer R, Kelley KW, Heijnen CJ, Kavelaars A. Low nociceptor GRK2 prolongs prostaglandin E2 hyperalgesia via biased cAMP signaling to Epac/Rap1, protein kinase Cepsilon, and MEK/ERK. J Neurosci. 2010;30:12806–15. doi: 10.1523/JNEUROSCI.3142-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of δ-opioid receptor signaling. Nature. 2014;506:191–6. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang CS, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Molecular basis for interactions of G protein betagamma subunits with effectors. Science. 1998;280:1271–4. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- Fu T, Okano Y, Hagiwara M, Hidaka H, Nozawa Y. Bradykinin-induced translocation of protein kinases C in neuroblastoma NCB-20 cell: dependence on 1,2-diacylglycerol content and free calcium. Biochem Biophys Res Commun. 1989;162:1279–86. doi: 10.1016/0006-291x(89)90812-7. [DOI] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci. 2008;27:2558–67. doi: 10.1111/j.1460-9568.2008.06223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georganta EM, Agalou A, Georgoussi Z. Multi-component signaling complexes of the delta-opioid receptor with STAT5B and G proteins. Neuropharmaoclogy. 2010;25:139–48. doi: 10.1016/j.neuropharm.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Graness A, Adomeit A, Ludwig B, Müller WD, Kaufmann R, Liebmann C. Novel bradykinin signalling events in PC-12 cells: stimulation of the cAMP pathway leads to cAMP-mediated translocation of protein kinase Cepsilon. Biochem J. 1997;327:147–54. doi: 10.1042/bj3270147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wu Y, Zhang W, Zhao J, Devi LA, Pei G, Ma L. Identification of G protein-coupled receptor kinase 2 phosphorylation sites responsible for agonist-stimulated delta-opioid receptor phosphorylation. Mol Pharmacol. 2000;58:1050–6. doi: 10.1124/mol.58.5.1050. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–62. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hong MH, Xu C, Wang YJ, Ji JL, Tao YM, Xu XJ, Chen J, Xie X, Chi ZQ, Liu JG. Role of Src in ligand-specific regulation of delta-opioid receptor desensitization and internalization. J Neurochem. 2009;108:102–14. doi: 10.1111/j.1471-4159.2008.05740.x. [DOI] [PubMed] [Google Scholar]
- Kahsabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–53. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsabova IA, Simone DA, Seybold VS. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediated-size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–25. doi: 10.1016/s0306-4522(02)00449-9. [DOI] [PubMed] [Google Scholar]
- Kong G, Penn R, Benovic JL. A beta-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the beta 2-adrenergic receptor. J Biol Chem. 1994;269:13084–7. [PubMed] [Google Scholar]
- Kouhen OM, Wang G, Solberg J, Erickson LJ, Law PY, Loh HH. Hierarchical phosphorylation of delta-opioid receptor regulates agonist-induced receptor desensitization and internalization. J Biol Chem. 2000;275:36659–64. doi: 10.1074/jbc.M006788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroslak T, Koch T, Kahl E, Höllt V. Human phosphatidylethanolamine-binding protein facilitates heterotrimeric G protein-dependent signaling. J Biol Chem. 2001;276:39772–8. doi: 10.1074/jbc.M106991200. [DOI] [PubMed] [Google Scholar]
- Law PY, Bergsbaken C. Properties of delta opioid receptor in neuroblastoma NS20Y: receptor activation and neuroblastoma proliferation. J Pharmacol Exp Ther. 1995;272:322–32. [PubMed] [Google Scholar]
- Lembo PM, Ghahremani MH, Albert PR. Receptor selectivity of the cloned opossum G protein-coupled receptor kinase 2 (GRK2) in intact opossum kidney cells: role in desensitization of endogenous alpha2C-adrenergic but not serotonin 1B receptors. Mol Endocrinol. 1999;13:138–47. doi: 10.1210/mend.13.1.0217. [DOI] [PubMed] [Google Scholar]
- Levy D, Zochodne DW. Increased mRNA expression of the B1 and B2 bradykinin receptors and antinociceptive effects of their antagonists in an animal model of neuropathic pain. Pain. 2000;86:265–71. doi: 10.1016/S0304-3959(00)00256-6. [DOI] [PubMed] [Google Scholar]
- Liebmann C, Böhmer FD. Signal transduction pathways of G protein-coupled receptors and their cross-talk with receptor tyrosine kinases: lessons form bradykinin signaling. Curr Med Chem. 2000;7:911–43. doi: 10.2174/0929867003374589. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–9. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- Marie N, Aguila B, Hasbi A, Davis A, Jauzac P, Allouche S. Different kinases desensitize the human delta-opioid receptor (hDOP-R) in the neuroblastoma cell line SK-N-BE upon peptidic and alkaloid agonists. Cell Signal. 2008;20:1209–20. doi: 10.1016/j.cellsig.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Dipace C, Urizar E, Javitch JA, Sibley DR. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J Biol Chem. 2009;284:34103–15. doi: 10.1074/jbc.M109.055707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopian AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci. 2005;25:8825–32. doi: 10.1523/JNEUROSCI.0160-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain. 2006;125:114–24. doi: 10.1016/j.pain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Agonist-dependent phosphorylation of the mouse delta-opioid receptor: involvement of G protein-coupled receptor kinases but not protein kinase C. Mol Pharmacol. 1995;48:173–7. [PubMed] [Google Scholar]
- Petcu M, Dias JP, Ongali B, Thibault G, Neugebauer W, Couture R. Role of kinin B1 and B2 receptors in a rat model of neuropathic pain. Int Immunopharmacol. 2008;8:188–96. doi: 10.1016/j.intimp.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Pettinger L, Gigout S, Linley JE, Gamper N. Bradykinin controls pool size of sensory neurons expressing functional δ-opioid receptors. J Neurosci. 2013;33:10762–71. doi: 10.1523/JNEUROSCI.0123-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan A, Smith M, McGuire B, Evans C, Walwyn W. Chronic inflammatory injury results in increased coupling of delta opioid receptors to voltage-gated Ca2+ channels. Mol Pain. 2013;9:8. doi: 10.1186/1744-8069-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Massotte D, Gavériaux-Ruff C, Kieffer BL. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS One. 2009;4:e5425. doi: 10.1371/journal.pone.0005425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Inglese J, Lefkowitz RJ. Proteein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 1995;9:175–82. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Benovic JL. Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J Biol Chem. 1997;272:3806–12. doi: 10.1074/jbc.272.6.3806. [DOI] [PubMed] [Google Scholar]
- Reiter E, Marion S, Robert F, Troispoux C, Boulay F, Guillou F, Crepieux P. Kinase-inactive G-protein-coupled receptor kinases are able to attenuate follicle-stimulating hormone-induced signaling. Biochem Biophys Res Commun. 2001;282:71–8. doi: 10.1006/bbrc.2001.4534. [DOI] [PubMed] [Google Scholar]
- Rodbell M, Birnbaumer L, Pohl SL, Krans HM. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem. 1971;246:1877–82. [PubMed] [Google Scholar]
- Rowan MP, Ruparel NB, Patwardhan AM, Berg KA, Clarke WP, Hargreaves KM. Peripheral delta opioid receptors require priming for functional competence in vivo. Eur J Pharmacol. 2009;602:283–7. doi: 10.1016/j.ejphar.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MP, Szteyn K, Doyle AP, Gomez R, Henry MA, Jeske NA. β-arrestin-2-biased agonism of delta opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) in primary sensory neurons. Mol Pain. 2014;10:50. doi: 10.1186/1744-8069-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–75. [PubMed] [Google Scholar]
- Stein C, Zöllner C. Opioids and sensory nerves. Handb Exp Pharmacol. 2009;194:495–518. doi: 10.1007/978-3-540-79090-7_14. [DOI] [PubMed] [Google Scholar]
- Steranka LR, Manning DC, DeHaas CJ, Ferkany JW, Borosky SA, Connor JR, Vavrek RJ, Steward JM, Snyder SH. Bradykinin as a pain mediator: receptors are localized to sensory neurons, and antagonists have analgesic actions. Proc Natl Acad Sci U S A. 1988;85:3245–9. doi: 10.1073/pnas.85.9.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol. 2002;88:544–8. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- Sullivan LC, Berg KA, Clarke WP. Dual regulation of δ-opioid receptor function by arachidonic acid metabolites in rat peripheral sensory neurons. J Pharmacol Exp Ther. 2015;353:44–51. doi: 10.1124/jpet.114.221366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippmer S, Quitterer U, Kolm V, Faussner A, Roscher A, Mosthaf L, Müller-Esterl W, Häring H. Bradykinin induces translocation of the protein kinase C isoforms alpha, epsilon, and zeta. Eur J Biochem. 1994;225:297–304. doi: 10.1111/j.1432-1033.1994.00297.x. [DOI] [PubMed] [Google Scholar]
- Vanderah TW. Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain. 2010;26(10):S10–5. doi: 10.1097/AJP.0b013e3181c49e3a. [DOI] [PubMed] [Google Scholar]
- Wang H, Heijnen CJ, Eijkelkamp N, Garza Carbajal A, Schedlowski M, Kelley KW, Dantzer R, Kavelaars A. GRK2 in sensory neurons regulates epinephrine-induced signalling and duration of mechanical hyperalgesia. Pain. 2011;152:1649–58. doi: 10.1016/j.pain.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Wang S, Dai Y, Fukuoka T, Yamanaka H, Kobayashi K, Obata K, Cui X, Tominaga M, Noguchi K. Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain. 2008;131:1241–51. doi: 10.1093/brain/awn060. [DOI] [PubMed] [Google Scholar]
- Xu C, Hong MH, Zhang LS, Hou YY, Wang YH, Wang FF, Chen YJ, Xu XJ, Chen J, Xie X, et al. Serine 363 of the {delta}-opioid receptor is crucial for adopting distinct pathways to activate ERK1/2 in response to stimulation with different ligands. J Cell Sci. 2010;123:4259–70. doi: 10.1242/jcs.073742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.