

SUMMARY
Histone lysine acylations play an important role in regulation of gene transcription in chromatin. Unlike histone acetyl-lysine, molecular recognition of recently identified crotonyl-lysine mark is much less understood. Here, we report that the YEATS domain of AF9 preferentially binds crotonyl-lysine over acetyl-lysine in histone H3. NMR structural analysis reveals that crotonyl-lysine of histone H3 lysine 18 is engulfed deep into an aromatic cage of the YEATS domain where carbonyl oxygen of crotonyl-lysine forms a hydrogen bond to backbone amide of protein residue Tyr78. The crotonyl-lysine through its unique electron-rich double bond side chain engages π–π aromatic stacking and extended hydrophobic/aromatic interactions with the YEATS domain as compared to acetyl-lysine. Our mutational analysis confirmed key protein residues Phe59 and Tyr78 for crotonyl-lysine recognition. Importantly, our findings present a new structural mechanism of protein-protein interactions mediated by histone lysine crotonylation, and show how the cells interpret acyl-lysine marks in different biological contexts.
eTOC Blurb
Zhang et al. report AF9 YEATS domain preferential recognition of crotonyl-lysine over acetyl-lysine in histone H3 via a unique π–π–π stacking structural mechanism. This study presents a new molecular mechanism underlying the role of histone lysine crotonylation in regulation of gene transcription in chromatin.
INTRODUCTION
As the major component of chromatin, histones function to maintain genome integrity and regulate gene transcription. Histone functions are tightly controlled by post-translational modifications (PTMs) such as acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, and ADP ribosylation (Kouzarides, 2007; Ruthenburg et al., 2007), and recently reported but much less understood lysine acylations including crotonylation, butyrylation, propionylation, butyrylation, formylation, and citrullination (Tan et al., 2011). It is thought that histone lysine acylations contribute to chromatin-templated cellular processes via two major mechanisms. First, histone lysine acylations can directly modulate the packaging of chromatin by altering the net charge of histone molecules or inter-nucleosomal interactions, thereby changing chromatin structure and DNA access by transcription factors. Second, they regulate chromatin structure and function by recruiting PTM-specific binding proteins, which recognize specific histone PTM marks by functionally specialized structural folds such as bromo-, chromo-, PHD and YEATS domains (Andrews et al., 2016a; Margueron et al., 2005; Zeng and Zhou, 2002).
Of the newly reported histone lysine acylations, the “writer” and “eraser” enzymes for lysine crotonylation have been identified. It was shown that co-activator p300 can catalyze histone lysine crotonylation and stimulate gene transcription to a greater degree than lysine acetylation, and Sirt3 can specifically remove crontonylation of histone H3 at lysine 4 (Bao et al., 2014; Sabari et al., 2015). Chromatin immunoprecipitation sequencing (ChIP-seq) analyses have mapped histone lysine crotonylation to regulatory elements of actively transcribed regions of the genome, generally coincident with the localization of lysine acetylation (Dai et al., 2014; Tan et al., 2011). Further, histone lysine crotonylation confers the resistance to transcription repressor and could be a dominant element in maintaining X/Y-linked gene activation in globally repressive environment of haploid cell sex chromosomes. These studies indicate that histones at active regulatory elements are modified with chemically distinct acylation marks, suggesting a role for these modifications in gene transcriptional regulation. The functional readout of crotonyl-lysine, however, has remained elusive, due to in part a lack of understanding of molecular recognition of this new histone PTM. Recently, a subset of bromodomain proteins including TAF1, Brd9 and CERCR2 have been reported to bind to crotonyl-lysine and butyryl-lysine, but with much weaker affinity than to acetyl-lysine (Flynn et al., 2015). Further, their biological significance has not been established.
The YEATS (Yaf9, ENL, AF9, Taf14, and Sas5) domain is an evolutionarily conserved module present in 4 proteins (ENL, AF9, GAS41, and YEATS2) in humans and 3 proteins (Sas5, Taf14, and Yaf9) in yeast. The YEATS domain proteins are found in major chromatin-remodeling and histone acetyl-transferase (HAT) complexes and implicated in regulation of chromatin structure, histone acetylation and deposition, gene transcription and DNA damage response (Schulze et al., 2009). The structural studies of AF9 (Li et al., 2014) and Taf14 YEATS domains (Shanle et al., 2015) show that the YEATS domains utilizes a serine/threonine-lined aromatic cage for acetyl-lysine recognition, distinct from the well-known bromodomains (Dhalluin et al., 1999; Smith and Zhou, 2016) or the tandem PHD finger module of human DPF3b (Zeng et al., 2010). In this study, we report that the AF9 YEATS domain is capable of binding to crotonyl-lysine with higher affinity than that to acetyl-lysine, and represent a detailed structural mechanism for this new acyl-lysine mark recognition by the AF9 YEATS domain.
RESULTS AND DISCUSSION
AF9 YEATS domain binds histone crotonyl-lysine better than histone acetyl-lysine
While the evolutionarily conserved bromodomains have long been established as histone acetyl-lysine reader domains (Dhalluin et al., 1999), recently studies have shown that some tandem PHD fingers (Zeng et al., 2010) as well as the YEATS domains (Li et al., 2014; Shanle et al., 2015) can also function to recognize histone acetyl-lysine. Notably, our close inspection of the AF9 YEATS domain/H3K9ac complex structure (PDB code: 4TMP) revealed that un-occupied space in the acetyl-lysine binding pocket may accommodate larger acylation modifications such as crotonyl-lysine and butyryl-lysine (Figure 1A). To corroborate this hypothesis, we performed NMR analysis of the AF9 YEATS domain interactions with histone H3 peptides containing crotonyl-, acetyl- and butyryl-lysine using 2D 1H-15N heteronuclear single quantum coherence (HSQC) spectra (Figure 1B). Indeed, in addition to acetyl-lysine, the AF9 YEATS domain is also capable of binding to crotonyl-lysine or butyryl-lysine containing H3 peptides, as shown by substantial changes in protein backbone amide resonances globally. An unmodified H3 peptide did not cause any protein resonance changes, confirming that the YEATS domain and the H3 peptide interact in a modification-sensitive manner (Figure 1B). Moreover, the pattern of protein NMR resonance perturbations is similar upon addition of the corresponding crotonyl-, acetyl-, or butyryl-lysine containing H3K9 or H3K18 peptide, indicating that the different acy-lysine moieties likely bind to the same region in the AF9 YEATS domain.
Figure 1. AF9 YEATS domain binds crotonyl-lysine better than acetyl-lysine.

(A) Chemical structures of acetyl-lysine (Kac), crotonyl-lysine (Kcr), and butyryl-lysine (Kbu).
(B) 2D 1H-15N-HSQC NMR spectra of the AF9 YEATS domain depicting changes of the protein backbone amide resonances upon the addition of histone H3 peptides containing acetylation (blue), crotonylation (red) or butyrylation (green) at lysine 9 or lysine 18 (residues 3–15, TKQTAR-Kcr, Kac or Kbu-STGGKA; residues 12–24, GGKAPR-Kcr or Kac-QLATKA). The unmodified histone H3 (residues 3–15, TKQTAR-K-SSGGKA) shows no binding to the protein (left panel).
(C) Isothermal titration calorimetry (ITC) measurements of the AF9 YEATS domain binding to histone H3 peptides containing acetyl-lysine, crotonyl-lysine, or butyryl-lysine at H3K9, H3K18 or H3K27. The H3K9 and H3K18 peptides are same as in B, and H3K27 peptides consist of ATKAAR-Kcr or Kac-SAPATG (residues 21–33).
To determine relative binding preference of the AF9 YEATS domain for crotonyl-, acetyl- and butyryl-lysine, we performed quantitative isothermal titration calorimetry (ITC) binding study (Figure 1C). The ITC data showed that the YEATS domain consistently prefers binding to crotonyl-lysine over acetyl-lysine at all three major histone H3 lysine acylation sites of H3K9, H3K18 and H3K27. Overall, the binding affinity of AF9 YEATS to crotonyl-lysine is 2.5 to 3.5-fold higher than that of acetyl-lysine. Moreover, the binding affinity to butyryl-lysine is also stronger than acetyl-lysine at H3K9 with the order of H3K9cr > H3K9bu > H3K9ac (i.e. Kd of 4.6 μM vs. 10.0 μM vs. 16.6 μM, respectively). Collectively, our biochemical study clearly demonstrated that the AF9 YEATS domain prefers binding lysine crotonylation over lysine acetylation mark in histone H3.
Structural Basis of Crotonyl-Lysine Recognition by the AF9 YEATS Domain
It was recently reported that co-activator p300 catalyzes histone H3K18 crotonylation, which results in stimulation of gene transcription to a greater degree than lysine acetylation (Sabari et al., 2015). Given that the bromodomain of p300/CBP does not bind to H3K18cr containing H3 peptide (data not shown), whereas the AF9 YEATS domain shows at least 3-fold higher affinity for H3K18cr over H3K18ac (Kd of 19.4 μM vs. 59.5 μM) (Figure 1C), we postulate that the YEATS domain may function as reader for H3K18cr mark.
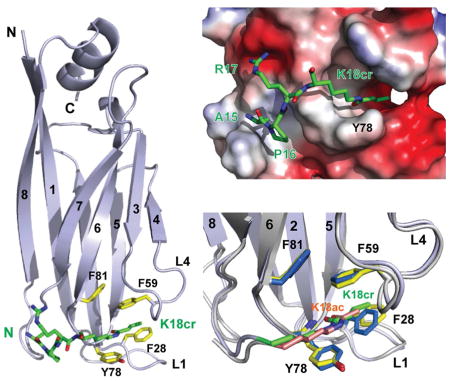
To understand the detailed molecular basis of histone crotonyl-lysine recognition, we determined 3D solution structure of the AF9 YEATS domain bound to an H3K18cr peptide (residues 12–24) using heteronuclear NMR spectroscopy (Figure S1A and Table 1). The overall AF9 YEATS domain structure in solution is similar to its crystal structure determined in complex with an H3K9ac peptide (Li et al., 2014), which consists of the conserved immunoglobin fold made of a two-layer β sandwich with eight antiparallel β strands (Figures 2A and Figure S1B). While two α helices (α1, αC) cap the β sandwich at one end, the H3K18cr peptide is bound into a surface-exposed cavity formed by inter-β strand loops of L1 and L4 (Figure 2B). We also determined the solution of the AF9 YEATS domain bound to an H3K18ac peptide in order to appreciate the detailed molecular basis of the preferred recognition of H3K18cr (Figure S1A and Table 1). As compared to acetyl-lysine of H3K18ac, notably, crotonyl-lysine of H3K18cr extends more fully into the aromatic cage and is sandwiched between Phe59 and Tyr78 through π-π stacking. The latter for crotonyl-lysine recognition is likely further enhanced as compared to acetyl-lysine due to the electron-rich double bond moiety of crotonyl-lysine. In addition, owing to its extended side chain, crotonyl-lysine also seems to establish additional hydrophobic/aromatic interactions with the aromatic side chain of Phe28 than acetyl-lysine. Our new detailed structural insights can explain the binding affinity order of the AF9 YEATS domain for crotonyl-lysine over butyryl-lysine over acetyl-lysine as shown with a set of H3K9 peptides (Figure 1C) in that as compared to acetyl-lysine, crotonyl-lysine has the advantages of both having the π-π stacking interactions with Phe59 and Tyr78, and the extended side-chain interactions with Phe28, whereas butyryl-lysine has only the extended side-chain interaction.
Table 1.
Summary of Statistics of NMR Structures of AF9 YEATS Domain in Complex with H3-K18cr and H3-K18ac Peptides
| H3K18cr | K3K18ac | |
|---|---|---|
| Protein NMR distance and dihedral constraints | ||
| Distance constraints | ||
| Total NOE | 2534 | 2402 |
| Intra-residue | 1019 | 1017 |
| Inter-residue | 1515 | 1385 |
| Sequential (|i − j| = 1) | 461 | 449 |
| Medium-range (1 < |i − j| ≤ 5) | 252 | 214 |
| Long-range (|i − j| > 5) | 802 | 722 |
| Inter-molecular constraints | 65 | 62 |
| Hydrogen bonds | 56 | 56 |
| Total dihedral angle restraints | ||
| Phi angle | 130 | 130 |
| Psi angle | 130 | 130 |
| Ramachandran Map Analysis a (%) | ||
| Most favored regions | 89.7 | 89.7 |
| Additional allowed regions | 10.3 | 10.3 |
| Generally allowed regions | 0.0 | 0.0 |
| Disallowed regions | 0.0 | 0.0 |
| Structure statistics | ||
| Violations (mean ± s.d.) | ||
| Distance constraints (Å) | 0.050 ± 0.0039 | 0.050 ± 0.011 |
| Dihedral angle constraints (°) | 0.92 ± 0.094 | 0.64 ± 0.10 |
| Max. dihedral angle violation (°) | 1.10 | 0.81 |
| Max. distance constraint violation (Å) | 0.061 | 0.081 |
| Deviations from idealized geometry | ||
| Bond lengths (Å) | 0.0052 ± 0.00016 | 0.0050 ± 0.00013 |
| Bond angles (°) | 0.62 ± 0.012 | 0.62 ± 0.061 |
| Impropers (°) | 1.8 ± 0.069 | 1.7 ± 0.065 |
| Average pairwise r.m.s. Deviation b (Å) | ||
| Heavy | 0.63 ± 0.066 | 0.65 ± 0.058 |
| Backbone | 0.30 ± 0.033 | 0.29 ± 0.045 |
Procheck residue numbers are 8–24, 28–42, 51–56, 65–70, 72–80, 82–92, 100–110, 115–128, and 130–141.
he residue number ranges used in protein/peptide complex root-mean-square deviation (rmsd) calculations are 8–40, 47–92, 100–110, and 116–140. Pairwise rmsd was calculated among 20 refined structures of total 200 structures.
Figure 2. 3D structure of the AF9 YEATS domain bound to an H3K18cr peptide.

(A) Left, ribbon diagram depicting the average minimized NMR structure of the YEATS domain bound to an H3K18cr peptide (green). Right, space-filled representation of the YEATS domain structure highlights the H3K18cr binding site.
(B) Electrostatic potential surface representation of the AF9 YEATS domain structure depicting its recognition of an H3K18cr peptide. Lower, depiction of the inter-molecular interactions between the protein and peptide.
(C) Comparison of H3K18cr (green) versus H3K18ac (salmon) recognition by the AF9 YEATS domain, showing key residues engaged in the inter-molecular protein/peptide interactions. The key residues involved in protein/peptide interactions are color-coded by atom type, with blue for H3K18cr and yellow for H3K18ac bound forms, respectively.
(D) Effects of mutations of key residues in the AF9 YEATS domain on histone lysine acylation binding, as assessed by NMR binding study using 1H-15N HSQC spectra.
Indeed, point mutation of Tyr78 to Ala markedly diminished YEATS domain binding to histone H3 lysine crotonylation and acetylation at H3K9, H3K18 and H3K27, and a double mutant of Y78A/F59A nearly completely abolished binding to lysine crotonylation, as demonstrated by NMR HSQC spectra (Figure 2D).
Distinct Modes of Crotonyl-Lysine Recognition by Histone Binding Domains
To better understand protein-protein interactions mediated by lysine crotonylation, we compared modes of crotonyl-lysine recognition by different histone binding domains reported thus far, including bromodomain proteins of TAF1, Brd9 and CERCR2 (Flynn et al., 2015). Structurally distinct from that of the YEATS domain (Figure 3A vs. 3B), bromodomains share a conserved left-handed four-helical bundle and recognize acetyl-lysine through a key hydrogen bond with a highly conserved Asn residue located in a well-defined hydrophobic/aromatic pocket at one end of the helix bundle. As shown in the crystal structure of the second bromodomain of TAF1 (TAF1-BD2) bound to an H4K5cr peptide (Figure 3B), the carbonyl oxygen of crotonyl-lysine is indeed hydrogen-bonded to the conserved Asn1583, which typically engages in acetyl-lysine recognition. Recognition of crotonyl-lysine is reinforced by hydrophobic/aromatic interactions with Tyr1540, Tyr1582 and Tyr1589, but does not involve π-π stacking interactions as seen in the YEATS domain. Notably, a set of stably bound water molecules that form a network of hydrogen-bond interactions with the bromodomain occupy major portion of the binding pocket, creating much less optimal environment for binding crotonyl-lysine than smaller acetyl-lysine. This explains that TAF1-BD2 binds weaker to crotonyl-lysine than to acetyl-lysine, e.g. Kd of 100 μM vs. 50 μM for H4K5cr/K8cr vs. H4K5ac/K8ac, respectively (Flynn et al., 2015).
Figure 3. Crotonyl-lysine recognition by distinct histone binding modules.

(A) H3K18cr recognition by the AF9 YEATS domain, as depicted in the overall protein structure (upper) and the detailed interactions at the crotonyl-lysine binding site (lower).
(B) H4K5cr recognition by the second bromodomain of TAF1 (TAF1-BD2), as shown in the crystal structure of the TAF1_BD2/H4K5cr complex (PDB: 4YYN). The side-chain carbonyl of the crotonyl-lysine H4K5cr is hydrogen-bonded to the conserved Asn1583, similar to what’s seen for acetyl-lysine recognition by the bromodomain.
(C) H3K4cr recognition by Sirt3, as shown in the crystal structure of human Sirt3/H3K4cr complex (PDB: 4V1C). The side-chain amide of crotonyl-lysine is hydrogen-bonded to the backbone carbonyl of Val292 of the protein.
Human Sirt3 recognition of H3K4cr is achieved through a hydrogen bond formed between backbone carbonyl of Val292 and side-chain amide nitrogen of the crotonyl-lysine (Figure 3C). Binding of crotonyl-lysine involves hydrophobic/aromatic interactions with His248, Phe294 and Val324, but no π-π stacking interactions. The binding pocket for crotonyl-lysine is shallow, leaving the crotonyl-lysine side chain half nearly exposed to the solvent. This explains relatively modest affinity of Kd of 25 μM for the H3K4cr peptide, as determined by ITC measurement (Bao et al., 2014).
Conclusions
In summary, our study reported here provides a new structural mechanism of the AF9 YEATS domain as a reader module for histone lysine acylations. Particularly, our detailed structural and biochemical analyses show that the AF9 YEATS domain preferentially binds crotonyl-lysine over acetyl-lysine through the π-π stacking interactions and the extended hydrophobic/aromatic interactions of electron-rich double bond side-chain of crotonyl-lysine. Our structural analysis further indicates that the AF9 YEATS domain is likely a much better crotonyl-lysine reader domain than some bromodomains including TAF1-BD2 as well as Sirt3. Indeed, our findings in this study are fully supported by three independent studies of the YEATS domain recognition of crotonyl-lysine that were recently published while our study was under review (Andrews et al., 2016b; Li et al., 2016; Zhao et al., 2016). Given the growing evidence for the functional importance of histone lysine crotonylation, this new structural mechanism of YEATS/Kcr recognition is expected to guide future functional characterization of the fundamental molecular mechanism of histone lysine crotonylation mediated protein-protein interactions in regulation of gene transcription in the context of chromatin in human biology of health and disease.
EXPERIMENTAL PROCEDURES
Preparation of Protein and Peptides
AF9 YEATS domain (residues 1–138) fused with an N-terminal 6x His tag was expressed in E. Coli BL21(DE3) codon plus RIL strain cells induced by the addition of isopropyl-β-D-thiogalactopyranoside (0.3 mM) at 25°C. The His-tagged AF9 YEATS domain was purified with HiTrap IMAC FF column (GE Healthcare) followed by the removal of His-Tag via thrombin cleavage. The AF9 YEATS domain was further applied to a Superdex 75 column and eluted with PBS buffer of pH 7.4 containing 2.0 mM EDTA, 2.0 mM DTT and 500 mM NaCl. Uniformly 15N- and 15N/13C-labeled proteins were prepared from cells grown in the minimal medium containing 15NH4Cl with or without 13C6-glucose in H2O. The mutants of the AF9 YEATS domain were generated using QuikChange site-directed mutagenesis kit (Agilent Technologies), and the presence of appropriate mutations was confirmed by DNA sequencing. The synthetic histone peptides were synthesized by Mimotopes and confirmed by liquid chromatography-mass spectrometry analysis.
Protein Structure Determination by NMR
NMR samples of the AF9 YEATS domain (0.5 mM) in complex with H3K18cr or H3K18ac (residues 12–24) peptide of 0.5 mM were prepared in PBS buffer of pH 7.4 containing 2.0 mM perdeuterated DTT and 2.0 mM EDTA in H2O/2H2O (9/1) or 2H2O. All NMR spectra were collected at 30°C on NMR spectrometers of 800, 600, or 500 MHz. The 1H, 13C, and 15N resonances of a protein of the complex were assigned by triple-resonance NMR spectra collected with a 13C/15N-labeled and 75% deuterated protein bound to an unlabeled peptide (Clore and Gronenborn, 1994). The distance restraints were obtained in three-dimensional 13C- or 15N-NOESY spectra. Slowly exchanging amides, identified in two-dimensional 15N-HSQC spectra recorded after a H2O buffer was changed to a 2H2O buffer, were used with structures calculated with only NOE distance restraints to generate hydrogen-bond restraints for final structure calculations. The inter-molecular NOEs were detected in 13C-edited (F1), 13C/15N-filtered (F3), three-dimensional NOESY spectrum.
Structure Calculations
Structures of the AF9 YEATS/H3K18cr or H3K18ac complex were calculated with a distance geometry-simulated annealing protocol using the X-PLOR program (Brünger AT, 1998) Manually assigned NOE-derived distance restraints were used to calculate initial structures. ARIA (Nilges and O’Donoghue, 1998) assigned distance restraints agree with structures calculated using only the manually determined NOE restraints. Ramachandran plot analysis of the final structures was performed using Procheck-NMR program (Laskowski et al., 1996).
Isothermal Titration Calorimetry
Experiments were carried out on a MicroCal ITC200 instrument at 20°C while stirring at 750 rpm in PBS buffer of pH 7.4 buffer containing, 2 mM EDTA, 2 mM β-mercaptoethanol and 500 mM NaCl. Peptide concentration was determined by weight and confirmed by NMR, and protein concentrations by A280 measurements. The protein sample (0.3 mM) was placed in the cell, whereas the micro-syringe was loaded with histone peptides (4.0 mM) in the same buffer as protein sample. The titrations were conducted using 17 successive injections of 2.0 μL (the first at 0.4 μL and the remaining 16 at 2.0 μL) with the duration of 4 sec per injection and a spacing of 180 sec between injections. The collected data was processed using the Origin 7.0 software program (OriginLab) supplied with the instrument according to the “one set of sites” fitting model.
Supplementary Material
3D structure of the AF9 YEATS domain in complex with an H3K18cr peptide.
The YEATS domain recognizes crotonyl-lysine through a π–π–π stacking mechanism.
The YEATS domain prefers crotonyl-lysine binding over acetyl- and butyryl-lysine.
The structural mechanism of YEATS/crotonyl-lysine binding is conserved.
Acknowledgments
We thank Dr. Haitao Li for kindly providing the pET28b-AF9 YEATS domain construct, and the New York Structural Biology Center as well as the State Key Laboratory of Supramolecular Structure and Materials at Jilin University for the use of their NMR facilities. This work was supported in part by the research fund from the First Hospital of Jilin University (Changchun, China), and the grants from the National Institutes of Health (M.-M. Z.).
Footnotes
ACCESSION NUMBER
Protein Data Bank: Coordinates for the NMR structures of the AF9 YEATS domain in complex with H3K18cr or H3K18ac peptides have been deposited at Protein Data Bank under PDB ID code 2NDG and 2NDF, and the NMR spectral data are deposited at BioMagResBank (BMRB) under BMRB accession numbers 26060 and 26059, respectively.
Supplementary information includes one figure can be found with this article online at https://.
AUTHOR CONTRIBUTIONS
Q.Z. and M.-M.Z conceived the project and designed the experiments. Q.Z., C.C.Z., Y.J., L.Z., and T.K. performed protein preparation, mutagenesis, NMR binding, and ITC measurements. L.Z. determined the protein/peptide structures by NMR. All authors contributed to the writing of the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews FH, Shanle EK, Strahl BD, Kutateladze TG. The essential role of acetyllysine binding by the YEATS domain in transcriptional regulation. Transcription. 2016a;7:14–20. doi: 10.1080/21541264.2015.1125987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, Krajewski K, Shi X, Strahl BD, Kutateladze TG. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016b doi: 10.1038/nchembio.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, Li XD. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. Elife. 2014;3 doi: 10.7554/eLife.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger ATAP, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods in enzymology. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Flynn EM, Huang OW, Poy F, Oppikofer M, Bellon SF, Tang Y, Cochran AG. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure. 2015;23:1801–1814. doi: 10.1016/j.str.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y, et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol Cell. 2016;62:181–193. doi: 10.1016/j.molcel.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014;159:558–571. doi: 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Nilges M, O’Donoghue S. Ambiguous NOEs and automated NOE assignment. Prog NMR Spectroscopy. 1998;32:107–139. [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze JM, Wang AY, Kobor MS. YEATS domain proteins: a diverse family with many links to chromatin modification and transcription. Biochem Cell Biol. 2009;87:65–75. doi: 10.1139/O08-111. [DOI] [PubMed] [Google Scholar]
- Shanle EK, Andrews FH, Meriesh H, McDaniel SL, Dronamraju R, DiFiore JV, Jha D, Wozniak GG, Bridgers JB, Kerschner JL, et al. Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes Dev. 2015;29:1795–1800. doi: 10.1101/gad.269977.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SG, Zhou MM. The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem Biol. 2016;11:598–608. doi: 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Letters. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- Zhao D, Guan H, Zhao S, Mi W, Wen H, Li Y, Zhao Y, Allis CD, Shi X, Li H. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016;26:629–632. doi: 10.1038/cr.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.