

Abstract
Background and Objectives
Obesity is a global epidemic which increases the risk of the metabolic syndrome. Cathelicidin (LL-37 and mCRAMP) is an antimicrobial peptide with an unknown role in obesity. We hypothesize that cathelicidin expression correlates with obesity and modulates fat mass and hepatic steatosis.
Materials and Methods
Male C57BL/6J mice were fed a high-fat diet. Streptozotocin was injected into mice to induce diabetes. Experimental groups were injected with cathelicidin and CD36 overexpressing lentiviruses. Human mesenteric fat adipocytes, mouse 3T3-L1 differentiated adipocytes, and human HepG2 hepatocytes were used in the in vitro experiments. Cathelicidin levels in non-diabetic, prediabetic, and Type II diabetic patients were measured by ELISA.
Results
Lentiviral cathelicidin overexpression reduced hepatic steatosis and decreased the fat mass of high-fat diet-treated diabetic mice. Cathelicidin overexpression reduced mesenteric fat and hepatic fatty acid translocase (CD36) expression that was reversed by lentiviral CD36 overexpression. Exposure of adipocytes and hepatocytes to cathelicidin significantly inhibited CD36 expression and reduced lipid accumulation. Serum cathelicidin protein levels were significantly increased in non-diabetic and prediabetic patients with obesity, compared to non-diabetic patients with normal body mass index (BMI) values. Prediabetic patients had lower serum cathelicidin protein levels than non-diabetic subjects.
Conclusions
Cathelicidin inhibits the CD36 fat receptor and lipid accumulation in adipocytes and hepatocytes, leading to a reduction of fat mass and hepatic steatosis in vivo. Circulating cathelicidin levels are associated with increased BMI. Our results demonstrate that cathelicidin modulates the development of obesity.
Introduction
Obesity is a national health epidemic in the United States. The Centers for Disease Control and Prevention (CDC) reported that more than one-third of US adults and 17% of US children and adolescents are obese1. Obesity is associated with heart disease, stroke, Type II diabetes, and cancer 2. In the presence of obesity, many organs such as adipose tissues and liver produce inflammatory mediators 3. These inflammatory mediators are implicated in the development of type II diabetes and other diseases 4, 5.
Cathelicidin belongs to a family of endogenous peptides (LL-37 in humans and mCRAMP in mice) with potent antimicrobial and anti-inflammatory effects6. However, the involvement of cathelicidin in obesity has not been studied extensively. Previous studies reported that peripheral blood cathelicidin mRNA is reduced in patients with Type II diabetes, and biopsies of diabetic foot ulcers in patients show either very low or no LL-37 expression relative to healthy skin7,8. Similarly, in animal studies Otsuka Long-Evan Tokushima Fatty rats develop long-term hyperglycemia when cathelicidin gene expression is impaired9. Also, LL-37 expressing adenoviruses improved re-epithelialization of excisional wounds in ob/ob mice10.
Non-alcoholic fatty liver disease (NAFLD), also known as hepatic steatosis, is becoming a significant problem in the United States, affecting about 20% of the population11. In the early stages, hepatic steatosis is mostly asymptomatic. However, without treatment, hepatic steatosis can progress to nonalcoholic steatohepatitis (NASH), which is characterized by inflammation and liver damage12. In severe cases, untreated NASH can eventually lead to cirrhosis and permanent liver damage 13. Most end-stage severe fatty liver disease patients will require liver transplantation14. Since none of the subjects in our study had a diagnosis of NAFLD stated in their medical records, we are currently not able to determine whether serum LL-37 levels are associated with the fatty liver disease.
Based on these findings, we hypothesize that a link between cathelicidin expression and obesity may exist. We have determined the role of systemic cathelicidin expression in the modulation of fat mass in obese and streptozotocin-induced diabetic mice using lentiviral vectors. We also have identified a molecular target that appears to mediate the effect of cathelicidin in obesity. Also, we have found a correlation between serum LL-37 levels and the BMI of non-diabetic and prediabetic patients.
Materials and Methods
Cell culture of mouse 3T3-L1 preadipocytes and differentiation to adipocytes
Mouse 3T3-L1 preadipocytes (#CL-173) were puchased from American Type Culture Collection (ATCC, Manassas, VA) and immediately cultured in Dulbecco's modified Eagle's medium (DMEM) #11965-084 (Life Technologies, Grand Island, NY) with 10% fetal bovine serum (FBS) #10437-028 (Life Technologies) and 1% penicillin/streptomycin/glutamine (P/S/G) #10378-016 (Life Technologies) mixture upon arrival. To differentiate the preadipocytes, 3T3-L1 cells were grown to 60% confluence and changed to DMEM (with 1% P/S/G) media with 10% calf serum #16170-078 (Life Technologies) for 48 hours. Differentiation was initiated by incubation with induction media containing DMEM with FBS, P/S/G, bovine insulin (I-5500; 1 μg/mL, Sigma, St. Louis, MO), dexamethasone (Sigma D-4902; 1 μM) and isobutylmethylxanthine (IBMX; Sigma I-5500; 115 μg/mL) for 48 hours. The cells were then further incubated with insulin media containing DMEM with FBS, P/S/G and insulin (1 microg/mL) for another 48-72 hours. When the cell confluence reached 90%, 3T3-L1 cells were serum-starved overnight followed by treatment with cathelicidin and/or other reagents to study the role of cathelicidin in lipid accumulation.
Human mesenteric fat acquisition, isolation of human mesenteric fat preadipocytes and differentiation to adipocytes
Human mesenteric fat tissues were resected during gastrointestinal surgery for the management of gastrointestinal diseases from subjects who had given informed consent. The tissues were obtained from Cedars-Sinai Inflammatory Bowel and Immunobiology Research Institute ‘MIRIAD' Tissue Repository under Cedars-Sinai IRB #3358 and #23705 and UCLA IRB #11-001527. Subjects with malignancies were excluded to avoid contamination of the cultures by the malignant cells. Subjects ranged from 42–53 years old, and their mean body mass index was 49 ± 8.7 kg/m2. Preadipocyte isolation and culture was performed as previously described15. The tissue was homogenized, the homogenates were centrifuged (10 minutes at 1200 rpm), and the pellets were resuspended in erythrocyte lysis buffer and placed in a 37°C shaking water bath before re-centrifugation as above. Pellets were resuspended in plating medium and vortexed, and cells were then plated. After 24 h, the cells were trypsinized and centrifuged, the supernatants were aspirated, and the cells were resuspended in plating medium and counted before plating. The cells were incubated at 37°C until they reached confluence16. At full confluence, differentiation medium was added to the cells and replaced every 48 hours. After four weeks, the level of differentiation was determined as previously described17. The authenticity of adipocytes was verified by lipid deposition. Our laboratory's incubators were tested negative of mycoplasma contamination.
High fat diet treatment and streptozotocin-induced diabetes
Eight-week-old male C57BL/6J mice (stock #000664) were purchased from Jackson Laboratories and maintained at the University of California at Los Angeles animal facility under standard environmental conditions with a 12-hour light period and a 12-hour dark period per day; 25°C room temperature. Camp-/- mice in C57BL/6J strain were obtained from Dr. Richard Gallo at the University of California, San Diego and were bred and maintained at the University of California, Los Angeles (UCLA) animal facility under standard environmental conditions. They were housed in disposable plastic cages with HEPA filtered air circulation, autoclaved bedding, animal chow, and sterile water ad libitum. Genotypes of mice were identified by Transnetyx genotyping service. Mice were fed with either regular chow provided by UCLA animal facility (6% fat, #7013 Harlan Laboratories) or high-fat diet (45% Kcal from fat; #D12451) purchased from, Research Diets, Inc., New Brunswick, NJ ad libitum. After eight weeks, some mice were injected with streptozotocin in citrate buffer (200 mg/kg) intraperitoneally to induce diabetes. The control group received intraperitoneal citrate buffer (200 microliters) alone. After three weeks (11 weeks after experiment initiation), blood samples were collected via tail veins while mesenteric fat and liver tissues of mice were collected after carbon dioxide euthanasia. The Institutional Animal Care and Use Committee of the David Geffen School of Medicine at the University of California at Los Angeles approved all procedures (#2007-116). n=6 mice per group. Additional experimental details are described in Figure 1A and 3D.
Figure 1. Lentiviral cathelicidin over-expression reduced fat mass and increased lean mass in high-fat diet-treated diabetic mice.

(A) Experimental plan of animal experiments. C57BL/6 mice were fed with high-fat diet for a total of 11 weeks. Streptozotocin was injected intraperitoneally to induce diabetes on week 8. Cathelicidin expressing lentivirus particles were also injected at the same time via tail veins. Details are described in Materials and Methods section. (B) Cathelicidin (Camp) mRNA expression of mouse mesenteric fat was determined by real-time RT-PCR. High-fat diet-treated diabetic mice had significantly reduced Camp mRNA expression (p=0.0001), compared to regular diet-treated non-diabetic mice. The mesenteric fat Camp mRNA expression of cathelicidin expressing group was significantly higher than those of control lentivirus expressing group (p=0.0001), showing the high expression efficacy of the lentivirus. (C) Body weight of the mice at week 11. High-fat diet-treated non-diabetic mice had significantly higher body weight (p=0.0002 and p=0.0007) than those treated with regular diet. Body weights of high-fat diet-treated diabetic mice were similar to regular diet-treated normal mice. Lentiviral cathelicidin overexpression did not affect their body weights. (D) Fat mass of the live mice at week 11 was measured by EchoMRI machine. High-fat diet treatment increased the fat mass of both non-diabetic and diabetic mice. Lentiviral cathelicidin overexpression significantly decreased the fat mass of the high-fat diet-treated diabetic mice but not high-fat diet-treated non-diabetic mice. (E) Lean mass of the live mice at week 11 was measured by EchoMRI machine. High-fat diet treatment decreased the lean mass of both non-diabetic and diabetic mice. Lentiviral cathelicidin overexpression significantly increased the fat mass of the high-fat diet-treated diabetic mice but not high-fat diet-treated non-diabetic mice. (F) Fasting blood glucose levels at week 10. High-fat diet treatment increased fasting blood glucose levels that were further exacerbated by streptozotocin- induced diabetes. Lentiviral cathelicidin overexpression did not affect the hyperglycemia. n=6 mice per group.
Figure 3. Cathelicidin reduced lipid accumulation via inhibition of CD36 expression.

(A) Serum-starved mouse differentiated 3T3-L1 adipocytes were transfected with mouse CD36 expressing construct or empty control vector overnight, followed by exposure to TFA (vehicle control) or mCRAMP (1-5 μM) for 24 hours. Mouse Cd36 mRNA expression was significantly increased in Cd36 transfection group to demonstrate transfection efficiency. (B) Cathelicidin (1-5 μM) significantly reduced lipid accumulation in adipocytes that was partially reversed by Cd36 overexpression. (C) Mesenteric fat was collected on week 11 (the end of the experimental period). Cd36 mRNA expression was measured by real-time RT-PCR. Mesenteric fat Cd36 mRNA expression was significantly increased in high-fat diet-treated diabetic and non-diabetic groups. Lentiviral cathelicidin overexpression led to significant reduction of mesenteric fat Cd36 mRNA expression in the high-fat diet-treated diabetic group but not in the non-diabetic group. (D) Experimental plan of animal experiments. Mice were fed with high-fat diet for a total of 11 weeks. Streptozotocin was injected to induce diabetes on the 8th week. Cathelicidin expressing lentivirus particles and Cd36 expressing lentivirus particles were also injected at the same time via tail veins on the same day. Details are described in Materials and Methods section. (E) Mesenteric fat Cd36 mRNA expression of the Cd36 lentivirus-infected group was significantly higher than that of the control lentivirus-infected group. (F) High-fat diet treatment increased fat mass of high-fat diet-treated diabetic mice. Lentiviral cathelicidin overexpression significantly decreased the fat mass of the high-fat diet-treated diabetic mice that was reversed by Cd36 overexpression. n=6 mice per group.
Power analysis
We performed power analysis once we began the experiments. For animal studies, we included 6 mice per group to achieve statistically significant difference of fat mass between high-fat diet-treated diabetic mice and regular diet-treated normal mice (29% vs. 14%) with SD = 8%, alpha = 0.05, and power = 0.8. For human serum experiments, we included 11 patients per group to achieve statistically significant difference of circulating LL-37 between non-diabetic normal BMI group and non-diabetic obese BMI group (28 vs. 39 ng/ml) with SD = 9%, alpha = 0.05, and power = 0.8. We did not perform power analysis for cell culture experiments, but it is common practice to perform in vitro experiments for 2-3 times independently. Quantitative ELISA and real-time RT-PCR experiments were performed in triplicate. Details of n-number per group are shown in respective tables and figure legends.
Statistical analysis
All mice were randomized and allocated to each cage (3-4 mice per cage) by animal facility personnel before experiments started. Investigators, except Hon Wai Koon, were blinded to the group allocation. For cell culture experiments, such as Western blot and Oil Red O staining, we did not make adjustments for multiple experiments. Instead, we present the data from a representative experiment.
Results were expressed as mean±standard error of the mean and analyzed by using the Prism professional statistics software program (GraphPad, San Diego, CA). Unpaired Student's t-tests were used for intergroup comparisons. The data follow a normal distribution, and the variances of the two compared groups are similar. Error bars represent standard error of mean. P values of statistical significance are shown in each figure.
Results
Exogenous cathelicidin modulates fat mass of high-fat diet-treated and streptozotocin-induced diabetic mice
To understand whether relative increases of cathelicidin can reduce obesity, we overexpressed cathelicidin in diabetic and non-diabetic mice systemically using lentiviral vectors (Figure 1A). A group of high-fat diet-treated mice was injected with streptozotocin to induce diabetes (Figure 1A). Consequentially, the mesenteric fat of the high-fat diet-treated diabetic mice showed a significant reduction in cathelicidin mRNA expression when compared to the regular diet-treated normal group (Figure 1B). The cathelicidin overexpressing diabetic and non-diabetic groups also had significantly higher cathelicidin mRNA expression in mesenteric fat than their respective groups without cathelicidin overexpression (Figure 1B).
The lentiviral cathelicidin overexpression did not significantly alter the overall body weight of the high-fat diet-treated diabetic and non-diabetic mice (Figure 1C), but significantly reduced the percentage of fat mass and significantly increased the percentage of lean mass in high-fat diet-treated diabetic mice (Figure 1D and 1E). Also, lentiviral cathelicidin overexpression had no effect on the fat and lean mass of high-fat diet-treated non-diabetic mice (Figure 1D and 1E).
Lentiviral cathelicidin overexpression did not affect blood glucose levels in fasting and oral glucose tolerance test settings (Figure 1F and Supplementary Figure 2A). It also did not affect the hydration ratio, total blood cholesterol levels, serum free fatty acid levels, and food consumption (Supplementary Figure 2B, 2C, 2D, and 2E). Thus, cathelicidin reduced fat mass without affecting glucose metabolism in high-fat diet-treated diabetic mice.
Cathelicidin inhibits lipid accumulation in adipocytes
To determine the role of cathelicidin in fat metabolism in vitro, Oil Red O staining was used to measure intracellular lipid accumulation in differentiated adipocytes. Exposure to LL-37 and mCRAMP (1 to 5 μM) in differentiated human adipocytes, which were isolated from mesenteric fat tissues and mouse 3T3-L1 adipocytes, led to significantly reduced levels of lipid accumulation, as reflected by the Oil Red O Staining (Figure 2A and 2B). LL-37 and mCRAMP at 10 μM caused an increase in non-specific Oil Red O staining, which was possibly caused by observed apoptotic cell death in human differentiated adipocytes and mouse 3T3-L1 adipocytes respectively (data not shown). Taken together, our data indicates that exposure to cathelicidin inhibits lipid accumulation in adipocytes.
Figure 2. Cathelicidin inhibited lipid accumulation and CD36 expression in adipocytes.

(A) Serum-starved human differentiated adipocytes were exposed to TFA or LL-37 (1-5 μM) for 16 hours. The lipid accumulation was determined by Oil red O staining at 500nm. LL-37 (1-5 μM) significantly reduced lipid accumulation in human adipocytes. (B) Serum-starved mouse differentiated 3T3-L1 adipocytes were exposed to TFA or mCRAMP (1-5 μM) for 16 hours. The lipid accumulation was determined by Oil red O staining at 500 nm. mCRAMP (1-5 μM) significantly reduced lipid accumulation in mouse adipocytes. (C) Real-time RT-PCR of human differentiated adipocytes. Human FFAR2 and FFAR3 mRNA expression were not affected by LL-37 exposure. (D) Real-time RT-PCR of mouse differentiated 3T3-L1 adipocytes. Mouse Ffar3 and Ffar3 mRNA expression was not affected by mCRAMP exposure. (E) Human CD36 mRNA expression in adipocytes was significantly reduced by LL-37 (1-5 μM) (F) Mouse Cd36 mRNA expression in adipocytes was significantly reduced by mCRAMP (1-5 μM). Results are representative of three independent experiments.
Cathelicidin inhibits fat accumulation by suppressing CD36 mRNA expression in differentiated adipocytes
Several fat receptors, such as free fatty acid receptor 2 (FFAR2), free fatty acid receptor 3 (FFAR3), and CD36, were reported to mediate lipid accumulation in adipocytes18. We measured FFAR2/Ffar2, FFAR3/Ffar3, and CD36/Cd36 mRNA expression in human and mouse adipocytes to assess their roles in cathelicidin associated lipid accumulation. Exposure to LL-37 and mCRAMP led to reduced mRNA expression of CD36/Cd36, but not FFAR2/Ffar2, and FFAR3/Ffar3, in both human and mouse adipocytes (Figure 2C-F).
To determine if cathelicidin plays a role in inhibiting lipid accumulation via inhibition of CD36 expression, Cd36 over-expressing constructs were transfected into differentiated mouse adipocytes. Cd36 mRNA expression in the transfected group was increased 18 fold when compared to the control group, as validated by real-time RT-PCR (Figure 3A). The Cd36 over-expression was shown to reverse the inhibitory effects of cathelicidin on lipid accumulation, as shown by the Oil Red O Staining (Figure 3B).
Cathelicidin inhibits CD36 mRNA expression in mesenteric fat
An increase in mesenteric fat Cd36 mRNA expression was observed in high-fat diet-treated non-diabetic and diabetic mice (Figure 3C). Lentiviral cathelicidin overexpression significantly reduced mesenteric fat Cd36 mRNA expression in high-fat diet-treated diabetic but not non-diabetic mice (Figure 3C). This trend is consistent with the cathelicidin dependent reduction of fat mass in high-fat diet-treated diabetic mice as shown in Figure 1D.
Cathelicidin reduces fat mass in high-fat diet-treated diabetic mice via CD36 inhibition
To understand whether cathelicidin reduces fat mass via inhibition of CD36 expression in vivo, high-fat diet-treated diabetic mice were infected with Camp- and/or Cd36-overexpressing lentiviruses (Figure 3D). Mesenteric fat Cd36 mRNA expression in the CD36-overexpressing lentivirus-infected group was increased 17 fold when compared to the control group (Figure 3E).
Cd36 overexpression reversed the cathelicidin-mediated reduction of fat mass and augmentation of lean mass in high-fat diet-treated diabetic mice (Figure 3F and Supplementary Figure 3B). This data suggests that cathelicidin inhibits obesity via inhibition of CD36 expression. However, Cd36 overexpression did not affect body weight, hydration ratio, fasting/OGTT glucose levels, blood total cholesterol levels, free fatty acid levels, and food consumption regardless of cathelicidin overexpression (Supplementary Figure 3A, 3C, 3D, 3E, 4A, 4B, and 4C). Cd36 overexpression alone did not alter fat mass and lean mass in high-fat diet-treated diabetic and non-diabetic mice (Figure 3F and Supplementary Figure 3B).
Hepatic cathelicidin overexpression reduces lipid accumulation in the liver in high-fat diet-treated diabetic mice via CD36 inhibition
Obesity can also lead to fatty liver disease19. Through Masson Trichrome lipid staining, livers from high-fat diet-treated diabetic and non-diabetic mice showed extensive lipid accumulation, indicating hepatic steatosis (Figure 4A). Cathelicidin over-expression reduced lipid accumulation in liver tissues in both high-fat diet-treated diabetic and non-diabetic mice (Figure 4A). Lentiviral Cd36 overexpression reversed the inhibitory effects of cathelicidin on hepatic steatosis in high-fat diet-treated diabetic mice (Figure 4A).
Figure 4. Lentiviral cathelicidin overexpression inhibited hepatic steatosis via CD36.
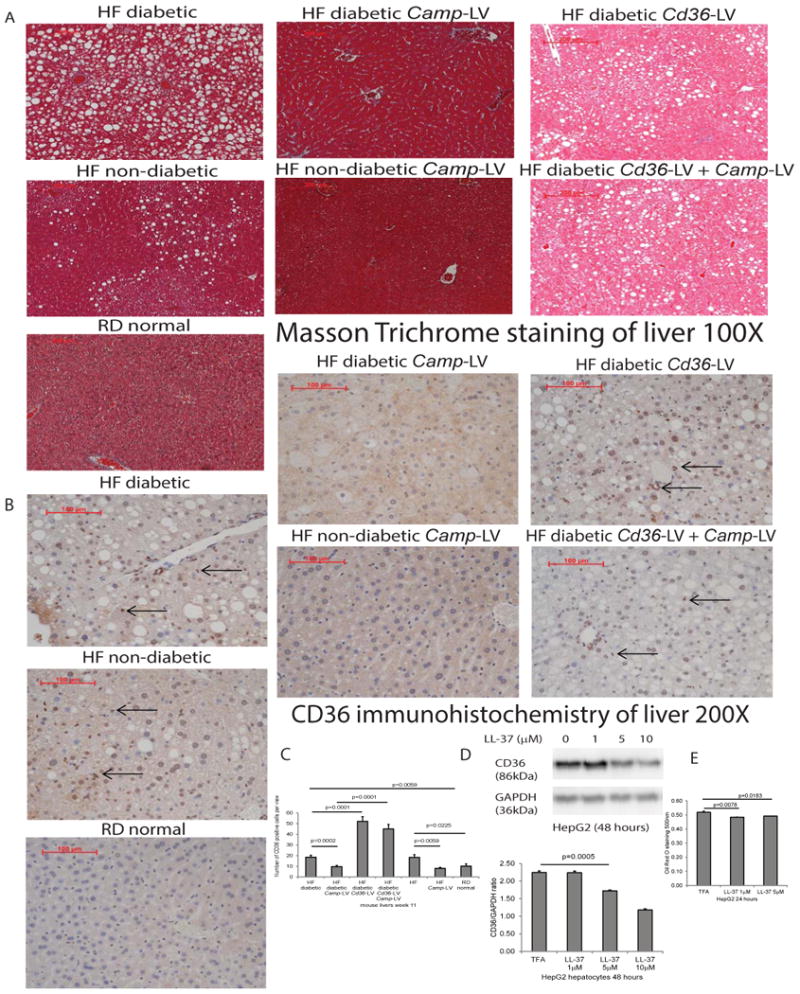
(A) Masson Trichrome staining of liver tissues of mice dissected on week 11. The background was stained in red color. Lipid accumulation was presented as white dots. High-fat diet-treated diabetic and non-diabetic mice all developed steatosis, compared with normal mice with regular diet. Steatosis was ameliorated in Camp-expressing groups that were reversed by Cd36 overexpression, regardless of diabetic status. (B) Immunohistochemistry of CD36 protein in liver tissues of mice dissected on week 11. (C) Quantification of CD36 protein in liver tissues. CD36 protein expression, as indicated by brown color, was significantly increased in high-fat diet-treated diabetic and non-diabetic mice, compared with normal mice with regular diet. Hepatic CD36 protein expression was significantly reduced in Camp-expressing groups, compared with control counterparts. CD36 protein expression was significantly increased in Cd36-expressing groups, indicating successful lentiviral transfection. n=6 mice per group. (D) Serum-starved HepG2 cells were exposed to TFA (vehicle control) or LL-37 (1-10 μM) for 24 hours. Western blot of HepG2 cells with quantification. LL-37 (1-10 μM) significantly reduced CD36 protein expression in human hepatocytes. (E) Oil red O staining was used to determine lipid accumulation. LL-37 (1-5 μM) significantly reduced lipid accumulation in hepatocytes. Experiments are representative of 2 independent experiments.
Through immunohistochemistry and image quantification, CD36 protein expression was significantly increased in the liver of high-fat diet-treated diabetic and non-diabetic mice (Figure 4B and 4C). Cathelicidin significantly reduced hepatic CD36 expression of high-fat diet-treated diabetic and non-diabetic mice (Figure 4B and 4C). Again, the inhibitory effect of cathelicidin in the hepatic steatosis of high-fat diet-treated diabetic mice was reversed by lentiviral Cd36 overexpression (Figure 4B and 4C). Reduced CD36 protein expression was also observed in cathelicidin-treated HepG2 hepatocytes (Figure 4D).
There were low basal levels of cathelicidin protein expression in the livers of regular diet-treated non-diabetic mice (Supplementary Figure 5). Hepatic cathelicidin expression of high-fat diet-treated non-diabetic mice was significantly higher than that of regular diet-treated non-diabetic mice while hepatic cathelicidin expression of high-fat diet-treated diabetic mice was significantly lower than that of regular diet-treated non-diabetic mice. The lentiviral cathelicidin overexpressing group had significantly stronger hepatic cathelicidin expression than the non-expressing group (Supplementary Figure 5).
To demonstrate the cathelicidin-mediated inhibition of lipid accumulation in hepatocytes in vitro, we measured lipid accumulation in well-established human hepatocyte HepG2 cell lines using Oil Red O staining. Cathelicidin (1-5 μM) significantly reduced lipid accumulation in HepG2 cells (Figure 4E).
Cathelicidin reduces CD36 expression in adipocytes and hepatocytes via extracellular signal-regulated kinase (ERK) inhibition
A previous report has shown that ERK mediates CD36 expression20. In our studies, exposure of differentiated human adipocytes to LL-37 (1-10 μM) reduced ERK phosphorylation, which was partially reversed by the ERK activator epidermal growth factor (EGF) (Figure 5A). Also, cathelicidin-mediated inhibition of CD36 expression in the adipocytes was partially reversed by EGF, suggesting that cathelicidin inhibits CD36 expression in adipocytes via ERK inhibition (Figure 5B). Co-incubation of adipocytes with EGF increased basal lipid accumulation and reversed the cathelicidin-mediated inhibition of lipid accumulation (Figure 5C). This finding suggests that cathelicidin inhibits lipid accumulation via an ERK-dependent mechanism.
Figure 5. Cathelicidin inhibited CD36 expression and lipid accumulation in human differentiated adipocytes and hepatocytes via an ERK-dependent pathway.
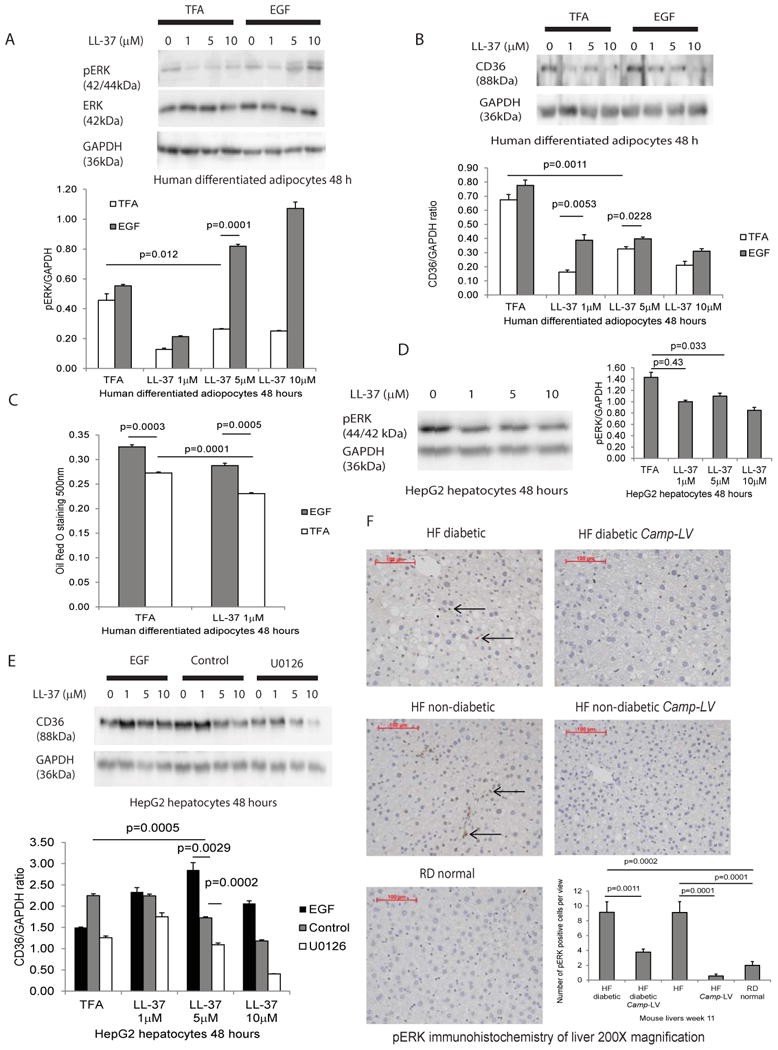
(A) Serum-starved human differentiated adipocytes were treated with 0.1% TFA or epidermal growth factor (EGF, 1 μg/ml) for 30 minutes followed by exposure to TFA or LL-37 (1-10 μM) for 48 hours. LL-37 inhibited ERK1/2 phosphorylation that was reversed by ERK activator EGF pretreatment. (B) LL-37 inhibited CD36 protein expression that was reversed by EGF pretreatment, suggesting cathelicidin mediated ERK-dependent CD36 inhibition in adipocytes. (C) Lipid accumulation in adipocytes was determined by Oil Red O staining. LL-37 inhibited lipid accumulation that was reversed by EGF pretreatment, suggesting the ERK-dependent mechanism. Results are representative of two independent experiments. (D) Serum-starved human HepG2 hepatocytes were exposed to 0.1% TFA or LL-37 (1-10 μM) for 48 hours. LL-37 inhibited ERK1/2 phosphorylation in hepatocytes. (E) Serum-starved human HepG2 hepatocytes were pretreated with ERK activator EGF (1 μg/ml) or ERK inhibitor U0126 for 30 minutes, followed by exposure to TFA or LL-37 (1-10 μM) for 48 hours. LL-37 inhibited CD36 expression in hepatocytes that was reversed by pretreatment of EGF. ERK inhibitor U0126 further reduced CD36 expression, suggesting an ERK-dependent mechanism. Results are representative of two independent experiments. (F) Immunohistochemistry and quantification of ERK phosphorylation in liver tissues of mice dissected on week 11. ERK phosphorylation, as indicated by brown color, was significantly increased in high-fat diet-treated diabetic and non-diabetic mice, compared with normal mice with regular diet. Hepatic ERK phosphorylation was significantly reduced in Camp-expressing groups, compared with control counterparts.
Similarly, LL-37 inhibited ERK phosphorylation in human HepG2 hepatocytes (Figure 5D). Co-incubation of HepG2 hepatocytes with EGF reversed the cathelicidin-mediated decrease of CD36 expression (Figure 5E). Pretreatment with ERK1/2 inhibitor U0126 reduced overall CD36 protein expression in hepatocytes (Figure 5E).
The hepatic ERK phosphorylation signal of high-fat diet-treated diabetic and non-diabetic mice was significantly stronger than those of regular diet-treated normal mice (Figure 5F). Lentiviral cathelicidin expression significantly diminished hepatic ERK phosphorylation of all high-fat diet-treated mice, regardless of diabetic status (Figure 5F).
Endogenous cathelicidin modulates obesity and hepatic steatosis in high-fat diet-treated non-diabetic mice
As the high-fat diet-treated non-diabetic mice had increased mesenteric fat cathelicidin mRNA expression (Figure 1B), we studied the role of endogenous cathelicidin in obesity development using high-fat diet-treated c57BL/6J wild-type and cathelicidin deficient Camp-/- mice (Supplementary Figure 6A). Although wild-type and Camp-/- mice showed no statistically significant difference in overall body weight after being treated with either a high-fat diet or a regular diet (Supplementary Figure 6B), high-fat diet-treated Camp-/- mice did show a significant increase in fat mass percentage, a significant reduction in lean mass, and severe hepatic steatosis (Supplementary Figure 6C-E). The endogenous cathelicidin deficiency did not appear to affect fasting blood glucose, OGTT blood glucose and total blood cholesterol levels in mice treated with either a high-fat diet or a regular diet (Supplementary Figure 6F-H). This data suggests that endogenous cathelicidin may modulate fat mass and hepatic steatosis in high-fat diet-treated non-diabetic mice.
Lentiviral cathelicidin reduces pro-inflammatory gene expression in the sciatic nerves of high-fat diet-treated diabetic mice
Inflammation is involved in peripheral diabetic neuropathy21. Lentiviral cathelicidin administration significantly decreased sciatic nerve aldose reductase and proinflammatory cytokine TNFα mRNA expression (Supplementary Figure 7A and 7B), suggesting that cathelicidin reduces pro-inflammatory gene expression associated with peripheral neuropathy.
Circulating serum cathelicidin protein levels are associated with BMI values of non-diabetic and prediabetic subjects
We also measured the serum LL-37 protein levels among non-diabetic, prediabetic, and Type 2 diabetic subjects. Baseline characteristics of non-diabetic, prediabetic, and Type II diabetic subjects are shown in Supplementary Table 1-3. We divided the entire test population into 3 groups according to the Centers for Disease Control and Prevention (CDC) definition of obesity, i.e. normal (BMI <24.9); overweight (BMI 25-29.9) and obese (BMI >30).
In non-diabetic subjects, the serum LL-37 levels of overweight and obese groups were significantly higher than those of normal groups (Figure 6A). Elevated serum LL-37 protein levels were associated with increasing BMI values of non-diabetic subjects. In prediabetic patients, serum LL-37 protein levels were also positively correlated with BMI values (Figure 6C). The difference in serum LL-37 protein levels between the overweight group (BMI 25-29.9) and the combined class II and class III obese group (BMI >35) was statistically significant (Figure 6B). Prediabetic subjects of class I obese group (BMI range 30-34.9) had significantly reduced serum LL-37 levels when compared to non-diabetic subjects in the same BMI range (Figure 6C). In the Type II diabetic group, we did not find a correlation between serum LL-37 levels and BMI values (Supplementary Figure 1).
Figure 6. Circulating cathelicidin LL-37 levels are positively correlated with BMI of non-diabetic subjects.

(A) The age-matched data pool of non-diabetic subjects is divided into 3 groups according to CDC BMI-obesity definitions (normal BMI<24.9 n=11; overweight BMI 25.0-29.9 n=12; obese BMI>30 n=11). The serum LL-37 levels of obese non-diabetic patients are significantly higher than those of normal non-diabetic subjects. (B) The data pool of prediabetic patients is divided into 3 groups according to CDC BMI-obesity definitions (overweight BMI<25-29.9 n=5; class I BMI 30-34.9 n=10; class II + III obese BMI>35 n=5). Class II obese and III obese prediabetic patients have significantly higher serum cathelicidin levels than overweight prediabetic patients. (C) The LL-37 levels of the non-diabetic and prediabetic groups. Circulating LL-37 levels of prediabetic groups with BMI range 30-34.9 are significantly lower than those of non-diabetic subjects with the same BMI range. LL-37 levels of prediabetic patients and non-diabetic subjects with BMI >35 are similar. Non-diabetic BMI 30-34.9 n=17; non-diabetic BMI >35 n=16; prediabetic BMI 30-34.9 n=10; prediabetic BMI >35 n=5. (D) Summary of cathelicidin-mediated effects.
Discussion
This is the first report in the literature showing the metabolic effects of cathelicidin in obesity and diabetes. We have discovered a novel cathelicidin dependent molecular target that modulates obesity and fatty liver disease in diabetic mice and its link to an ERK-CD36 dependent mechanism. We have also discovered a correlation between circulating cathelicidin levels and obesity among non-diabetic and prediabetic subjects.
The underlying cause for the difference in responses to cathelicidin overexpression among high-fat diet-treated non-diabetic and diabetic mice still requires further investigation. We speculate that cathelicidin overexpression was unable to change the fat mass of the high-fat diet-treated non-diabetic mice because the mesenteric fat already expressed high levels of endogenous cathelicidin, which possibly desensitized the cathelicidin-mediated signaling mechanism (Figure 1B). In contrast, high-fat diet-treated diabetic mice expressed relatively low levels of endogenous cathelicidin (Figure 1B); the mesenteric fat cells of these mice may have improved sensitivity to cathelicidin overexpression.
As shown by our in vitro studies, cathelicidin specifically inhibits CD36 and Cd36 mRNA expression in human and mouse adipocytes respectively (Figure 2E and 2F). CD36 is broadly expressed in a variety of tissues including livers and adipose tissues22, and it can bind to many lipid ligands23. Recently regarded as a lipid sensing mechanism24, CD36 expression is involved in a number of metabolic pathways related to obesity. Mesenteric fat Cd36 mRNA expression was increased in high-fat diet-treated diabetic and non-diabetic mice (Figure 3C), indicating that CD36 is involved in the gain of fat mass in vivo. Cd36 deficient mice have reduced adipose tissue compared to wild-type mice25. Similarly, lentiviral Cd36 overexpression reversed the effects of cathelicidin in decreasing the fat mass of high-fat diet-treated diabetic mice (Figure 3F). We also found that Cd36 overexpression in mouse 3T3-L1 adipocytes reversed the cathelicidin-mediated inhibition of lipid accumulation (Figure 3B). Thus, cathelicidin inhibits obesity in diabetic mice via modulating CD36 expression.
In another previous report, the cathelicidin peptide inhibited ERK phosphorylation in macrophages26. The ERK1 deficient mice were resistant to obesity in response to a high fat diet27. This finding indicated that the ERK pathway is necessary for lipid accumulation and obesity. Although we did not have enough fat tissue available for ERK immunohistochemistry, the ERK activator, EGF, reversed the cathelicidin-mediated inhibition of CD36 expression and lipid accumulation in adipocytes and hepatocytes (Figure 5). These findings demonstrate the ERK-dependent anti-obesity effect of cathelicidin.
A previous study reported that hepatic steatosis was linked to up-regulation of CD3628. In another study, CD36 deficient mice showed reduced hepatic steatosis29. Using immunohistochemistry, we found that hepatic CD36 protein expression is primarily up-regulated in high-fat diet-treated diabetic and non-diabetic mice, compared to regular diet-treated mice (Figure 4B). Lentiviral overexpression of cathelicidin in the high-fat diet-treated diabetic and non-diabetic mice ameliorated hepatic steatosis, which was reversed by Cd36 overexpression (Figure 4A and 4B). In a previous study, the liver of ob/ob and diet-induced obese mice showed increased ERK activity, while diet-induced obese ERK-deficient mice showed improvement in insulin sensitivity30. Similar to our finding in adipocytes, cathelicidin inhibited ERK phosphorylation and CD36 expression in cultured HepG2 hepatocytes (Figure 5D and 5E). We also observed elevated ERK phosphorylation in the liver of high-fat diet-treated mice, which was reduced by lentiviral cathelicidin treatment (Figure 5F). This finding indicates the role of ERK in the cathelicidin mediated regulation of CD36 expression in obesity and hepatic steatosis.
In addition to demonstrating that cathelicidin reduces the fat mass of high-fat diet-treated diabetic mice, we also observed an increase in lean mass, which consists of muscle and bone tissue (Figure 1E). Peripheral neuropathy in diabetes is often associated with muscle wasting31, which causes loss of lean mass. We speculate that one of the mechanisms by which cathelicidin can increase lean mass is by reducing muscle wasting in diabetic mice through modulation of aldose reductase and TNFα expression in peripheral nerves (Supplementary Figure 7A-B). High aldose reductase expression in the sciatic nerve is well known to mediate diabetic peripheral neuropathy of the lower limbs32. Aldose reductase inhibitor has been shown to prevent loss of nerve conduction velocity, skeletal muscle mass, and contractile capability in streptozotocin diabetic rats33-36. High TNFα expression can further damage nerve structure and impair its function37. We did not measure a quantitative change in bone and muscle mass, which is beyond the scope of this investigation. We plan to study the physiological effects of cathelicidin in nerves, bones and muscles under diabetic conditions and other metabolic conditions in the future.
In our study, we failed to find any correlation between serum cathelicidin levels and BMI values among Type II diabetic patients (Supplementary Figure 1). We speculate that there are many variables among diabetic patients, including medications and lifestyles, which may influence cathelicidin expression. Similar to the non-diabetic groups of another study38, the non-diabetic group and prediabetic group in our study showed a positive correlation between serum cathelicidin levels and BMI values (Figure 6B). We speculate that the non-diabetic and prediabetic patients have relatively simpler clinical profiles than Type II diabetic patients (for example, the former groups in our study took fewer medications).
This study investigated the direct effect of cathelicidin in controlling lipid accumulation in adipocytes. Since cathelicidin also possesses antimicrobial effect, it will be interesting to explore whether cathelicidin can alter gut microbiota that indirectly modulate intestinal nutrient absorption in the future.
In conclusion, this is the first report to show the novel metabolic effects of cathelicidin. Circulating cathelicidin levels correlate with BMI values in non-diabetic and prediabetic subjects. Cathelicidin, through an ERK- and CD36-dependent mechanism, inhibits lipid accumulation in adipocytes and hepatocytes (Figure 6D). Cathelicidin may be a novel drug target for treating obesity and hepatic steatosis. Orally active cathelicidin mimic should be developed for the convenient administration of cathelicidin to patients.
Supplementary Material
Acknowledgments
This work was supported by CCFA (#2691), NIH K01(DK084256), and NIH R03 (DK103964) grants to HWK, CCFA (#287244, #3831 & #324000) to Michelle Cheng, Samantha Ho, and Deanna Tran, and United States PHS grant DK046763 to D.Q.S.
Clinical data and specimens were provided by the MIRIAD Biobank. MIRIAD is currently supported by F Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, NIH grant P01DK046763, The European Union Grant 305479, NIDDK Grant DK062413, U54 DE023798, and the Leona M and Harry B Helmsley Charitable Trust.
Footnotes
Author contribution: Deanna Hoang-Yen Tran, Diana Hoang-Ngoc Tran, Hon Wai Koon---acquired data and drafted the manuscript
Tamer Sallam, Lori Robbins, Elaine Lee, Christina Ortiz , Samantha Ho, Elizabeth Fisseha, Christine Shieh, Jung Eun Lee, Aristea Sideri, Michelle Vu, Tressia C. Hing, Kyriaki Bakirtzi, Michelle Cheng, Bowei Su, Ivy Law, Iordanes Karagiannides---acquired data and revised the manuscript
S Anjani Mattai, Richard Gallo, Zhaoping Li, David Q Shih, Philip Fleshner, Dermot PB McGovern, Stephan R Targan, ---provision of materials, acquired data, and revised manuscript
Hon Wai Koon---approved the final version of manuscript and study supervision
Conflict of interests: All authors disclose no conflict of interest.
References
- 1.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. Jama. 2014;311(8):806–14. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health affairs. 2009;28(5):w822–31. doi: 10.1377/hlthaff.28.5.w822. [DOI] [PubMed] [Google Scholar]
- 3.Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nature reviews Endocrinology. 2016;12(1):15–28. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Chen R, Wang H, Liang F. Mechanisms Linking Inflammation to Insulin Resistance. Int J Endocrinol. 2015;2015:508409. doi: 10.1155/2015/508409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and beta-cell dysfunction. Transl Res. 2016;167(1):228–56. doi: 10.1016/j.trsl.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Current pharmaceutical design. 2013;19(1):40–7. doi: 10.2174/13816128130108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivas-Santiago B, Trujillo V, Montoya A, Gonzalez-Curiel I, Castaneda-Delgado J, Cardenas A, et al. Expression of antimicrobial peptides in diabetic foot ulcer. Journal of dermatological science. 2012;65(1):19–26. doi: 10.1016/j.jdermsci.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Curiel I, Castaneda-Delgado J, Lopez-Lopez N, Araujo Z, Hernandez-Pando R, Gandara-Jasso B, et al. Differential expression of antimicrobial peptides in active and latent tuberculosis and its relationship with diabetes mellitus. Human immunology. 2011;72(8):656–62. doi: 10.1016/j.humimm.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 9.Park HY, Kim JH, Jung M, Chung CH, Hasham R, Park CS, et al. A long-standing hyperglycaemic condition impairs skin barrier by accelerating skin ageing process. Experimental dermatology. 2011;20(12):969–74. doi: 10.1111/j.1600-0625.2011.01364.x. [DOI] [PubMed] [Google Scholar]
- 10.Carretero M, Escamez MJ, Garcia M, Duarte B, Holguin A, Retamosa L, et al. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. The Journal of investigative dermatology. 2008;128(1):223–36. doi: 10.1038/sj.jid.5701043. [DOI] [PubMed] [Google Scholar]
- 11.Masarone M, Federico A, Abenavoli L, Loguercio C, Persico M. Non Alcoholic Fatty Liver. Epidemiology and Natural history. Reviews on recent clinical trials. 2014 doi: 10.2174/1574887109666141216111143. [DOI] [PubMed] [Google Scholar]
- 12.Yoon HJ, Cha BS. Pathogenesis and therapeutic approaches for non-alcoholic fatty liver disease. World journal of hepatology. 2014;6(11):800–11. doi: 10.4254/wjh.v6.i11.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobyliak N, Abenavoli L. The Role Of Liver Biopsy To Assess Non-Alcoholic Fatty Liver Disease. Reviews on recent clinical trials. 2014 doi: 10.2174/1574887109666141216102231. [DOI] [PubMed] [Google Scholar]
- 14.Zezos P, Renner EL. Liver transplantation and non-alcoholic fatty liver disease. World journal of gastroenterology : WJG. 2014;20(42):15532–15538. doi: 10.3748/wjg.v20.i42.15532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karagiannides I, Kokkotou E, Tansky M, Tchkonia T, Giorgadze N, O'Brien M, et al. Induction of colitis causes inflammatory responses in fat depots: evidence for substance P pathways in human mesenteric preadipocytes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(13):5207–12. doi: 10.1073/pnas.0600821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karagiannides I, Bakirtzi K, Kokkotou E, Stavrakis D, Margolis KG, Thomou T, et al. Role of substance P in the regulation of glucose metabolism via insulin signaling-associated pathways. Endocrinology. 2011;152(12):4571–80. doi: 10.1210/en.2011-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karagiannides I, Thomou T, Tchkonia T, Pirtskhalava T, Kypreos KE, Cartwright A, et al. Increased CUG triplet repeat-binding protein-1 predisposes to impaired adipogenesis with aging. The Journal of biological chemistry. 2006;281(32):23025–33. doi: 10.1074/jbc.M513187200. [DOI] [PubMed] [Google Scholar]
- 18.Hara T, Kimura I, Inoue D, Ichimura A, Hirasawa A. Free fatty acid receptors and their role in regulation of energy metabolism. Reviews of physiology, biochemistry and pharmacology. 2013;164:77–116. doi: 10.1007/112_2013_13. [DOI] [PubMed] [Google Scholar]
- 19.Larter CZ, Yeh MM, Van Rooyen DM, Teoh NC, Brooling J, Hou JY, et al. Roles of adipose restriction and metabolic factors in progression of steatosis to steatohepatitis in obese, diabetic mice. Journal of gastroenterology and hepatology. 2009;24(10):1658–68. doi: 10.1111/j.1440-1746.2009.05996.x. [DOI] [PubMed] [Google Scholar]
- 20.Turcotte LP, Raney MA, Todd MK. ERK1/2 inhibition prevents contraction-induced increase in plasma membrane FAT/CD36 content and FA uptake in rodent muscle. Acta physiologica Scandinavica. 2005;184(2):131–9. doi: 10.1111/j.1365-201X.2005.01445.x. [DOI] [PubMed] [Google Scholar]
- 21.Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr Diab Rep. 2016;16(3):29. doi: 10.1007/s11892-016-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. The Journal of clinical investigation. 2001;108(6):785–91. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, et al. Regulation of fatty acid transport by fatty acid translocase/CD36. The Proceedings of the Nutrition Society. 2004;63(2):245–9. doi: 10.1079/PNS2004331. [DOI] [PubMed] [Google Scholar]
- 24.Martin C, Chevrot M, Poirier H, Passilly-Degrace P, Niot I, Besnard P. CD36 as a lipid sensor. Physiology & behavior. 2011;105(1):36–42. doi: 10.1016/j.physbeh.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Christiaens V, Van Hul M, Lijnen HR, Scroyen I. CD36 promotes adipocyte differentiation and adipogenesis. Biochimica et biophysica acta. 2012;1820(7):949–56. doi: 10.1016/j.bbagen.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Pinheiro da Silva F, Gallo RL, Nizet V. Differing effects of exogenous or endogenous cathelicidin on macrophage toll-like receptor signaling. Immunology and cell biology. 2009;87(6):496–500. doi: 10.1038/icb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54(2):402–11. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 28.Miquilena-Colina ME, Lima-Cabello E, Sanchez-Campos S, Garcia-Mediavilla MV, Fernandez-Bermejo M, Lozano-Rodriguez T, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60(10):1394–402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 29.Clugston RD, Yuen JJ, Hu Y, Abumrad NA, Berk PD, Goldberg IJ, et al. CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. Journal of lipid research. 2014;55(2):239–46. doi: 10.1194/jlr.M041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiao P, Feng B, Li Y, He Q, Xu H. Hepatic ERK activity plays a role in energy metabolism. Molecular and cellular endocrinology. 2013;375(1-2):157–66. doi: 10.1016/j.mce.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen H. Motor dysfunction in diabetes. Diabetes/metabolism research and reviews. 2012;28(Suppl 1):89–92. doi: 10.1002/dmrr.2257. [DOI] [PubMed] [Google Scholar]
- 32.Schemmel KE, Padiyara RS, D'Souza JJ. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. Journal of diabetes and its complications. 2010;24(5):354–60. doi: 10.1016/j.jdiacomp.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Krause MP, Riddell MC, Hawke TJ. Effects of type 1 diabetes mellitus on skeletal muscle: clinical observations and physiological mechanisms. Pediatric diabetes. 2011;12(4 Pt 1):345–64. doi: 10.1111/j.1399-5448.2010.00699.x. [DOI] [PubMed] [Google Scholar]
- 34.Cotter MA, Cameron NE, Robertson S, Ewing I. Polyol pathway-related skeletal muscle contractile and morphological abnormalities in diabetic rats. Experimental physiology. 1993;78(2):139–55. doi: 10.1113/expphysiol.1993.sp003675. [DOI] [PubMed] [Google Scholar]
- 35.Cameron NE, Cotter MA, Robertson S. Changes in skeletal muscle contractile properties in streptozocin-induced diabetic rats and role of polyol pathway and hypoinsulinemia. Diabetes. 1990;39(4):460–5. doi: 10.2337/diab.39.4.460. [DOI] [PubMed] [Google Scholar]
- 36.Cameron NE, Cotter MA, Basso M, Hohman TC. Comparison of the effects of inhibitors of aldose reductase and sorbitol dehydrogenase on neurovascular function, nerve conduction and tissue polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. 1997;40(3):271–81. doi: 10.1007/s001250050674. [DOI] [PubMed] [Google Scholar]
- 37.Sacerdote P, Franchi S, Trovato AE, Valsecchi AE, Panerai AE, Colleoni M. Transient early expression of TNF-alpha in sciatic nerve and dorsal root ganglia in a mouse model of painful peripheral neuropathy. Neuroscience letters. 2008;436(2):210–3. doi: 10.1016/j.neulet.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 38.Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, et al. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science. 2015;347(6217):67–71. doi: 10.1126/science.1260972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.