

Abstract
Protein phosphorylation is one of the most important post-translational modifications in nature. However, the site-specific incorporation of O-phosphotyrosine into proteins in vivo has not yet been reported. Endogenous phosphatases present in cells can dephosphorylate phosphotyrosine as a free amino acid or as a protein residue. Therefore, we deleted the genes of five phosphatases from the genome of Escherichia coli with the aim of stabilizing phosphotyrosine. Together with an engineered aminoacyl-tRNA synthetase (derived from Methanocaldococcus jannaschii tyrosyl-tRNA synthetase) and an elongation factor Tu variant, we were able to co-translationally incorporate O-phosphotyrosine into the super-folder green fluorescent protein at a desired position in vivo. This system will facilitate future studies of tyrosine phosphorylation.
Keywords: phosphotyrosine, genetic code expansion, phosphatase, tyrosine phosphorylation, aminoacyl-tRNA synthetase, elongation factor
Phosphorylation of tyrosine residues in proteins is a crucial post-translational modification that plays key roles in regulating many prokaryotic and eukaryotic processes including growth control, cell cycle control, differentiation, cell shape and movement, gene transcription, synaptic transmission, and bacterial virulence [1–4]. The human genome contains at least 90 protein tyrosine kinases (PTKs) which phosphorylate tyrosine residues in proteins and 107 protein tyrosine phosphatases (PTPs) that can remove the phosphate from O-phospho-tyrosine (pTyr) in proteins, opposing the functions of PTKs [5,6]. The balanced actions of PTKs and PTPs regulate the phosphorylation of tyrosine residues, and abnormalities result in the pathogenesis of numerous inherited or acquired human diseases from cancer to immune deficiencies [1].
Due to the importance of tyrosine phosphorylation, many strategies have been adopted to synthesize peptides and proteins containing purely phosphorylated tyrosine residues. Using the method of in vitro translation which involves read-through of an amber (UAG) stop codon by a chemically misacylated suppressor tRNA, pTyr was incorporated into proteins at controlled positions [7]. Later, caged pTyr was chemically synthesized to overcome the low incorporation efficiency caused by the poor binding of the elongation factor Tu (EF-Tu) with charged phosphate group of pTyr-tRNA; subsequently the caging group could be removed by light [8]. Recently, several pTyr analogs were genetically incorporated into proteins in vivo [9,10]. Sulfotyrosine was incorporated into proteins for the study of tyrosine sulfation [9]. Another in vivo incorporated pTyr mimic is p carboxylmethy-phenylalanine which was previously used as a replacement for pTyr in SH2 domain binding [10,11]. Moreover, p (phosphonoamino)-phenylalanine was incorporated into proteins by chemically modifying a genetically installed p azidophenyalanine residue in vitro [12]. However, the direct incorporation of pTyr into proteins in vivo has not been reported yet. Here, we described an approach with integrated strategies to genetically incorporate pTyr into proteins in E. coli cells.
Materials and methods
General molecular biology and plasmid constructions
Oligonucleotide synthesis, DNA sequencing and mass spectrometry were performed by the Keck Foundation Biotechnology Resource Laboratory at Yale University. The chemicals in this study were purchased from Sigma-Aldrich or ChemImpex. E. coli TOP10 cells (Life Technologies) were used for general cloning. Plasmids: The genes of M. jannaschii tyrosyl-tRNA synthetase (mjTyrRS), tRNATyr, and EF-Tu were cloned by PCR from laboratory inventory and inserted into the pTech plasmid. The synthetase and EF-Tu genes were placed under the constitutive lpp promoter and the tRNA gene was placed under constitutive proK promoter. The gene of codon optimized super-folder green fluorescent protein (sfGFP) with C terminal His6 tags was cloned into the pBAD plasmid and placed under the control of the inducible arabinose promoter. All the cloning experiments were performed by using the Gibson Assembly kit (New England Biolabs). The mutations in protein genes were made by the QuikChange II mutagenesis kit (Agilent Life Sciences).
Phosphatase activity assay
The phosphatase activity was determined by the Tyrosine Phosphatase Assay System (Promega) measuring the absorbance of a molybdate:malachite green:phosphate complex following the manufacturer’s instructions. The reactions were read in the BioTeK micro-plate reader.
Protein expression and purification
The expression strains were grown in 500 ml of LB media supplemented with 0.1 mg/ml ampicillin at 37°C to an absorbance of 0.6 0.8 at 600 nm, then protein expression was induced by the addition of 1 mM isopropyl β D-thiogalactopyranoside (IPTG) or 10 mM arabinose. Cells were incubated at 25°C for an additional 12 h and harvested by centrifugation at 5000 × g for 10 min at 4 °C. The cell paste was suspended in 15 ml of lysis buffer (50 mM Tris pH 8, 300 mM NaCl, 20 mM imidazole) and broken by sonication. The crude extract was centrifuged at 20,000 × g for 30 min at 4°C. The soluble fraction was loaded onto a column containing 1 ml of Ni-NTA resin (Qiagen) previously equilibrated with 20 ml of lysis buffer. The column was washed with 20 ml of lysis buffer, and then eluted with 2 ml of 50 mM Tris (pH 7.5), 300 mM NaCl, 200 mM imidazole. The purified protein was dialyzed with 50 mM HEPES-KOH (pH 7.5), 50 mM KCl, 1mM DTT and 50% glycerol, and stored at −80°C for further studies.
tRNA transcription, purification, and 32P labeling
Template plasmids containing tRNA genes were purified with the plasmid maxi kit (Qiagen), and 100 µg of plasmid was digested with BstNI (New England Biolabs). The BstNI digested template DNA was purified by phenol/chloroform extraction, followed by ethanol precipitation and dissolved in double distilled water. The transcription reaction (40 mM Tris pH 8; 4 mM each of UTP, CTP, GTP, and ATP at pH 7.0; 22 mM MgCl2; 2 mM spermidine; 10 mM DTT; 6 µg pyrophosphatase (Roche Applied Science); 60 µg/mL BstNI digested DNA template, approximately 0.2 mg/ml T7 RNA polymerase) was performed in 10 ml reaction volumes for overnight at 37°C. The tRNA was purified on a 12% denaturing polyacrylamide gel containing 8 M urea and TBE buffer (90 mM Tris, 90 mM boric acid, 2 mM EDTA). UV shadowing identified the pure tRNA band, which was excised and extracted three times with 1M sodium acetate pH 5.3 at 4°C. The tRNA extractions were then ethanol precipitated, dissolved in RNase-free distilled water, pooled, and finally desalted using a Biospin 30 column (BioRad). The tRNA was refolded by heating to 100°C for 5 min and slow cooling to room temperature. At 65°C, MgCl2 was added to a final concentration of 10 mM to aid folding. 16 µM folded tRNA in 50 mM Tris (pH 8.0), 20 mM MgCl2, 5 mM DTT and 50 µM NaPPi was incubated at room temperature for 1 hr with approximately 0.2 mg/ml CCA-adding enzyme and 1.6 µCi/µl of [α-32P]-labeled ATP (PerkinElmer). The sample was phenol/chloroform extracted and then passed over a Bio-spin 30 column (Bio-Rad) to remove excess ATP.
Aminoacylation assay
The assay was modified from the original method [13]. A 20 µl reaction contained the following components: 50 mM HEPES-KOH (pH 7.2), 25 mM KCl, 10 mM MgCl2, 5 mM DTT, 10 mM ATP, 25 µg/ml pyrophosphatase (Roche Applied Science), 1 µM tRNA. All tRNA aminoacylation levels were determined at 37°C with synthetase, 10 nM 32P-labeled tRNA. Time points were taken at 5 min, 10 min and 30 min by removing 2 µl aliquots from the reaction and immediately quenching the reaction into an ice-cold 3 µl quench solution (0.66 µg/µl nuclease P1 (Sigma) in 100 mM sodium citrate (pH 5.0)). For each reaction, 2 µl of blank reaction mixture (containing no enzyme) was added to the quench solution as the start time point. The nuclease P1 mixture was then incubated a room temperature for 30 min and 1 µl aliquots were spotted on PEI-cellulose plates (Merck) and developed in running buffer containing 5% acetic acid and 100 mM ammonium acetate. Radioactive spots of AMP and AA-AMP (representing free tRNA and aminoacyl-tRNA, respectively) were separated and then visualized and quantified by phosphorimaging in a Molecular Dynamics Storm 860 phosphorimager (Amersham Biosciences). The ratio of amino-acyl-tRNA to total tRNA was determined to monitor reaction progress.
Mass spectrometry analyses
The purified proteins were trypsin digested by a standard in-gel digestion protocol, and analyzed by LC-MS/MS on an LTQ Orbitrap XL (Thermo Scientific) equipped with a nanoACQUITY UPLC system (Waters). A Symmetry C18 trap column (180 µm×20 mm; Waters) and a nanoACQUITY UPLC column (1.7 µm, 100 µm×250 mm, 35°C) were used for peptide separation. Trapping was done at 15 µL min-1, 99% buffer A (water with formic acid (0.1 %)) for 1 min. Peptide separation was performed at 300 nL min-1 with buffer A and buffer B (CH3CN containing 0.1% formic acid). The linear gradient (51 min) was from 5% buffer B to 50% B at 50 min, to 85% B at 51 min. MS data were acquired in the Orbitrap with one microscan, and a maximum inject time of 900 ms followed by data-dependent MS/MS acquisitions in the ion trap (through collision induced dissociation, CID). The Mascot search algorithm was used to search for the appropriate noncanonical substitution (Matrix Science, Boston, MA).
Results
Deleting phosphatases for phosphotyrosine from E. coli
One challenge for in vivo incorporation of pTyr into proteins is the stability of pTyr in living cells which have many endogenous phosphatases. These enzymes may dephosphorylate pTyr either as free amino acid, or as a constituent of proteins. In E. coli there are about 90 phosphatases according to gene ontology [14], some of them may be specific for pTyr. Therefore, we wanted to decrease the phosphatase activity for pTyr in E. coli by deleting the genes of relevant phosphatases. As detailed biochemical knowledge on pTyr phosphatases does not exist, we selected 14 possible ones including SerB, SurE, PgpA, PgpB, PgpC, PphA, PphB, NudJ, HisB, YedJ, YedP, AphA, PhoA, and YnbD. We utilized the ASKA strain collections which overexpress individual gene of E. coli [15], and measured the phosphatase activities in the crude extracts of the corresponding strains for selected genes. The results showed that five phosphatases had obviously higher activities for pTyr than other phosphatase candidates; these are SerB, the phosphoserine phosphatase; PgpA, a phosphatidyl-glycerophosphatase in membranes; PphB, a phosphoprotein phosphatase involved in the misfolded protein stress; PhoA, a nonspecific alkaline phosphatase; and AphA, a nonspecific acidic phosphatase (Fig. 1). Then we knocked out the genes of these five phosphatases from the genome of E. coli TOP10 by recombination [16] to form the Δ5P strain, which grew at with a doubling time of 28 ± 4 min (12% slower than the wild-type parent) in LB medium. The phosphatase activity for pTyr in the crude extract of Δ5P cells was only about 30% of that in wild-type TOP10 cells.
Fig. 1.
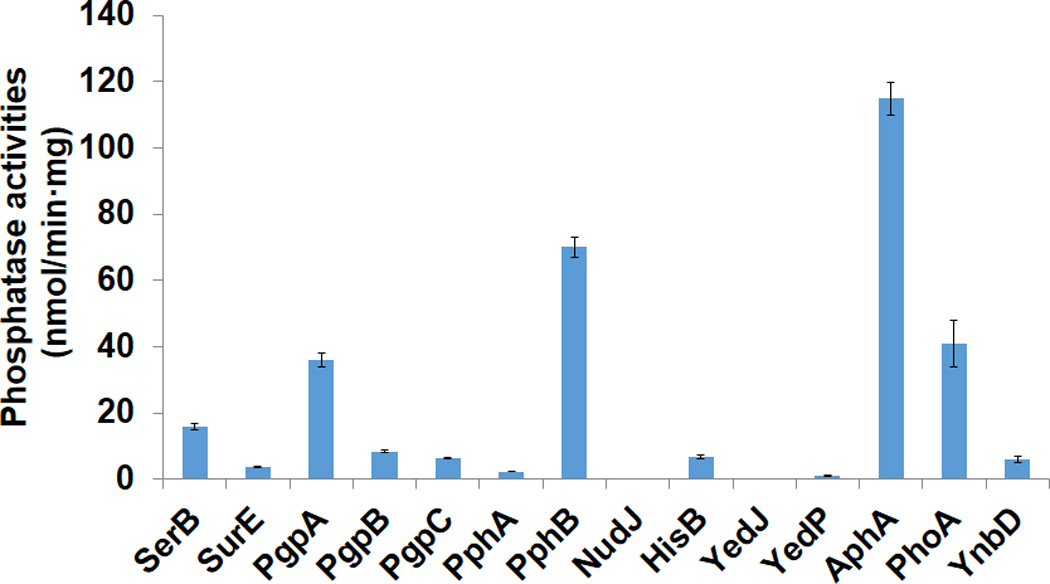
Activities of selected phosphatases for phosphotyrosine. The assays were performed according to the manufacturer’s instructions. 10 µL crude extracts were added to the reaction mixtures for measurement. The background value for wild-type TOP10 cells was set as zero, and the mean values and standard errors were calculated from three replicates.
Screening tyrosyl-tRNA synthetase (TyrRS) variants for pTyr-tRNA formation
Due to the poor cellular uptake of pTyr, the instability of pTyr in cells, as well as the need of evolving multiple translational components (TyrRS and EF-Tu) for pTyr incorporation, traditional strategies in genetic code expansion cannot be used to select TyrRS variants specific for pTyr efficiently [17]. To address this issue, we performed an in vitro screening approach based on the TyrRS variant activities of charging the tRNA with pTyr. Previously, an orthogonal pair of M. jannaschii TyrRS (mjTyrRS) and tRNATyrCUA was described [9] to incorporate sulfotyrosine into proteins, an amino acid structurally similar to pTyr. Thus, we started with this variant (Y32L, L65P, D158G, I159C, L162K) for further engineering. Based on the crystal structures of mjTyrRS and its variants [18–20], we chose four residues (positions of 32, 65, 108, and 109) which possibly contact for the additional negatively charged oxygen atom (Fig. 2A), to be replaced with all other 19 amino acids at each position. We also included the amino acid substitution of D286R in each variant which was shown to increase tRNATyrCUA anticodon recognition by the mjTyrRS [18]. In total 76 variants were made. Each variant was individually overexpressed, purified, and tested for aminoacylation activity with pTyr. Finally, we obtained two variants with the highest activities: pYRS1 (Y32L, L65R, D158G, I159C, L162K, D286R) and pYRS2 (Y32L, L65K, D158G, I159C, L162K, D286R) (Fig. 2B). The positively charged arginine and lysine residues at position 65 may facilitate the binding of negatively charged pTyr.
Fig. 2.

(A) The crystal structure of tyrosine binding site of the wild-type mjYRS (based on Protein Data Bank structure 1J1U). (B) The aminoacylation assay [13] of wild-type mjYRS for tyrosine and its variants for pTyr. 10 µM wild-type mjTyrRS or 100 µM mjTyrRS variants and 1 mM tyrosine or 10 mM pTyr were used in the assay. The mean values and standard errors were calculated from three replicates.
Screening EF-Tu variants for pTyr-tRNA binding
It is known that the EF-Tu binds tRNAs bearing negatively charged amino acids poorly [21]. Previously, we engineered EF-Tu to efficiently facilitate incorporation of another phosphoamino acid, phosphoserine, into proteins [22–24]. Phosphotyrosine also has two negative charges, so we rationally designed EF-Tu variants for better binding with pTyr to increase its incorporation into proteins. Based on the co-crystal structure of EF-Tu with Phe-tRNAPhe (Fig. 3A), we introduced the amino acid substitutions of E216V and D217G from the EF-Tu variant which could facilitate phosphoserine incorporation [24]. These two substitutions of negatively charged residues (Asp and Glu) facilitate the binding of EF-Tu with negatively charged amino acids. As for pTyr, it needs more binding space than phosphoserine due to its rigid aromatic ring, thus we replaced the residue at position 219, which may contact with the phosphate group in pTyr, with the other 19 amino acids and utilized the well-established super-folder green fluorescent protein (sfGFP) readthrough assay [25] to test the residue substitution effects on pTyr incorporation. The Δ5P strain constructed above was used as the host, and the orthogonal pair of engineered pYRS1 and Nap1 tRNATyr which was shown to comprehensively increase non-canonical amino acid incorporation by the mjYRS system [26] was co-expressed with each EF-Tu variant, individually. By using the sfGFP bearing an amber stop codon at a permissive position 143 as the reporter [27], we quantitated the readthrough of the amber stop codon from fluorescence readings. The results showed that the replacement of position 219 with amino acids with smaller side chains (Ala, Gly, and Ser) could increase the pTyr incorporation (Fig. 3B). Here, we selected the best variant (E216V, D217G, F219G) as EF-pY in later experiments.
Fig. 3.

(A) The model of the amino acid binding pocket of E. coli EF-Tu with phenylalanine charged tRNA (based on Protein Data Bank structure 1OB2). (B) The sfGFP readthrough assay for EF-Tu variants. The background fluorescence for wild-type TOP10 cells was set as zero. The mean values and standard errors were calculated from three replicates.
Incorporation of pTyr into protein in vivo
First we used the sfGFP readthrough assay to evaluate the essentiality of each component mentioned above in pTyr incorporation (Fig. 4A). The results showed that the phosphatase knockout strain, the engineered TyrRS and EF-Tu variants were equally important. Without any one of the Δ5P strain, the pYRS variant, or the EF-pY variant, the fluorescence reading dropped to the background level (no pTyr in the media). Then we purified the sfGFP bearing an amber stop codon at position 143 which was expressed with all these components (Fig. 4B). The yield of pTyr-containing sfGFP was about 20 mg/L in LB media. The mass spectrometric analysis confirmed pTyr incorporation at the designed position 143 (Fig. 5).
Fig. 4.

(A) The effects of components in pTyr incorporation. The background fluorescence for wild-type TOP10 cells was set as zero. 10 mM pTyr was used in the growth media. The mean values and standard errors were calculated from three replicates. (B) The purification of pTyr-containing and wild-type sfGFP. Lane 1, 2, and 3 are the crude extract, the soluble fraction, and the elution from the cells expressing the wild-type sfGFP, individually. Lane 4, 5, and 6 are the crude extract, the soluble fraction, and the elution from the cells expressing pTyr-containing sfGFP, separately. The same volumes of fractions were loaded on the SDS-PAGE gel.
Fig. 5.

The tandem mass spectrum of the peptide (residues 141–156) LEYPHNFNSHNVYITADK (ion score 57) from purified sfGFP with one amber codon at position 143. YPH denotes pTyr incorporation. The partial sequences of the peptides containing the pTyr can be read from the annotated b or y ion series.
Discussion
Compared with the expression of wild-type sfGFP under the same growth condition (Fig. 4B), the yield of pTyr-containing sfGFP was about 5%. The major amino acid incorporated at position 143 is pTyr, because we showed that if we did not provide pTyr in the growth media, the readthrough of the stop codon in sfGFP is only 20% of that with pTyr in the media (Fig. 4A). We also detected the incorporation of glutamine and lysine at this position, which was probably from the near cognate suppression of the amber stop codon [28]. Although the yield of pTyr-containing protein was relatively low, our work forms a solid step toward highly efficient incorporation of pTyr into proteins in vivo. Below are some approaches that could increase pTyr incorporation.
In this work, we used a high concentration of pTyr in the growth media to overcome the poor cellular uptake of pTyr [29]. However, it is necessary to look for proper transporters for pTyr which may be from other organisms. A similar strategy was successful in making a semi-synthetic E. coli with an expanded genetic alphabet by introducing an algal nucleotide triphosphate transporter for unnatural nucleoside triphosphates uptake [30].
Although we removed five phosphatase genes from the E. coli genome, there remained 30% pTyr phosphatase activity in the cells. We did detect by mass spectrometry analysis Tyr incorporation at position 143 in sfGFP; this may be due to dephosphorylation by unknown protein phosphatases. Thus, inactivating additional phosphatases or adding phosphatase inhibitors may help to increase pTyr incorporation. For this purpose, advanced techniques such as multiplex automated genome engineering (MAGE) can be utilized to inactivate multiple phosphatases simultaneously [31]. However, deleting more phosphatases may affect cell growth due to their important roles in cell physiology. And indeed, our five-phosphatase knockout strain grew relatively poorly in M9 minimal media.
In this study, we used rational design to engineer TyrRS and EF-Tu variants for pTyr. The advantage of advanced protein evolution systems such as MAGE and phage-assisted continuous evolution (PACE) [32,33] in creating larger libraries of variants and selecting variants automatically will help us to find more efficient and specific AARS and EF-Tu variants for pTyr incorporation.
Acknowledgments
We thank Dr. Jesse Rinehart (Yale University) for providing valuable suggestions. We are also grateful to Ana Crnkovic, Li-Tao Guo, Sergey Melnikov, Corwin Miller, Takahito Mukai, Noah Reynolds, Anastasia Sevostiyanova, Tateki Suzuki, and Oscar Vargas-Rodriguez for enlightened discussions. This work was supported by grants from the National Institute of General Medical Sciences (GM22854) to D.S. and from the National Institute of Allergy and Infectious Diseases (AI119813) to C.F.
Footnotes
Author Contributions
CF and DS conceived and designed experiments, and wrote the paper. CF and KI carried out experiments. CF and DS discussed and analyzed data.
Reference
- 1.Hunter T. The Croonian Lecture, 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos Trans R Soc Lond B Biol Sci. 1998;353:583–605. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitmore SE, Lamont RJ. Tyrosine phosphorylation and bacterial virulence. Int J Oral Sci. 2012;4:1–6. doi: 10.1038/ijos.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter T. The genesis of tyrosine phosphorylation. Cold Spring Harb Perspect Biol. 2014;6:a020644. doi: 10.1101/cshperspect.a020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grangeasse C, Cozzone AJ, Deutscher J, Mijakovic I. Tyrosine phosphorylation: an emerging regulatory device of bacterial physiology. Trends Biochem Sci. 2007;32:86–94. doi: 10.1016/j.tibs.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Alonso A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 6.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 7.Arslan T, Mamaev SV, Mamaeva NV, Hecht SM. Structurally modified firefly luciferase, effects of amino acid substitution at position 286. J Am Chem Soc. 1997;119:10877–10887. [Google Scholar]
- 8.Rothman DM, Petersson EJ, Vazquez ME, Brandt GS, Dougherty DA, Imperiali B. Caged phosphoproteins. J Am Chem Soc. 2005;127:846–847. doi: 10.1021/ja043875c. [DOI] [PubMed] [Google Scholar]
- 9.Liu CC, Schultz PG. Recombinant expression of selectively sulfated proteins in Escherichia coli. Nat Biotechnol. 2006;24:1436–1440. doi: 10.1038/nbt1254. [DOI] [PubMed] [Google Scholar]
- 10.Xie J, Supekova L, Schultz PG. A genetically encoded metabolically stable analogue of phosphotyrosine in Escherichia coli. ACS Chem Biol. 2007;2:474–478. doi: 10.1021/cb700083w. [DOI] [PubMed] [Google Scholar]
- 11.Tong L, Warren TC, Lukas S, Schembri-King J, Betageri R, Proudfoot JR, Jakes S. Carboxymethyl-phenylalanine as a replacement for phosphotyrosine in SH2 domain binding. J Biol Chem. 1998;273:20238–20242. doi: 10.1074/jbc.273.32.20238. [DOI] [PubMed] [Google Scholar]
- 12.Serwa R, Wilkening I, Del Signore G, Muhlberg M, Claussnitzer I, Weise C, Gerrits M, Hackenberger CP. Chemoselective Staudinger-phosphite reaction of azides for the phosphorylation of proteins. Angew Chem Int Ed Engl. 2009;48:8234–8239. doi: 10.1002/anie.200902118. [DOI] [PubMed] [Google Scholar]
- 13.Wolfson AD, Pleiss JA, Uhlenbeck OC. A new assay for tRNA aminoacylation kinetics. RNA. 1998;4:1019–1023. doi: 10.1017/s1355838298980700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keseler IM, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 16.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat Struct Biol. 2003;10:425–432. doi: 10.1038/nsb934. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Wang L, Schultz PG, Wilson IA. Crystal structures of apo wild-type M. jannaschii tyrosyl-tRNA synthetase (TyrRS) and an engineered TyrRS specific for O-methyl-L-tyrosine. Protein Sci. 2005;14:1340–1349. doi: 10.1110/ps.041239305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner JM, Graziano J, Spraggon G, Schultz PG. Structural plasticity of an aminoacyl-tRNA synthetase active site. Proc Natl Acad Sci U S A. 2006;103:6483–6488. doi: 10.1073/pnas.0601756103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dale T, Sanderson LE, Uhlenbeck OC. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry. 2004;43:6159–6166. doi: 10.1021/bi036290o. [DOI] [PubMed] [Google Scholar]
- 22.Park HS, et al. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinemann IU, et al. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586:3716–3722. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S, Oh S, Yang A, Kim J, Söll D, Lee D, Park HS. A facile strategy for selective incorporation of phosphoserine into histones. Angew Chem Int Ed Engl. 2013;52:5771–5775. doi: 10.1002/anie.201300531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan C, Xiong H, Reynolds NM, Söll D. Rationally evolving tRNAPyl for efficient incorporation of noncanonical amino acids. Nucleic Acids Res. 2015;43:e156. doi: 10.1093/nar/gkv800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo J, Melancon CE, 3rd, Lee HS, Groff D, Schultz PG. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew Chem Int Ed Engl. 2009;48:9148–9151. doi: 10.1002/anie.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albayrak C, Swartz JR. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013;41:5949–5963. doi: 10.1093/nar/gkt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Donoghue P, Prat L, Heinemann IU, Ling J, Odoi K, Liu WR, Söll D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinfeld JB, Aerni HR, Rogulina S, Liu Y, Rinehart J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem Biol. 2014;9:1104–1112. doi: 10.1021/cb5000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR, Jr, Romesberg FE. A semi-synthetic organism with an expanded genetic alphabet. Nature. 2014;509:385–388. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amiram M, et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat Biotechnol. 2015;33:1272–1279. doi: 10.1038/nbt.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]