

Abstract
Indole alkaloids are a diverse class of natural products known for their wide range of biological activities and complex chemical structures. Rarely observed in this class are indolic nitrones, such as avrainvillamide and waikialoid, which possess potent bioactivities. Herein the oxa gene cluster from the marine-derived fungus Penicillium oxalicum F30 is described along with the characterization of OxaD, a flavin-dependent oxidase that generates roquefortine L, a nitrone-bearing intermediate in the biosynthesis of oxaline. Nitrone functionality in roquefortine L was confirmed by spectroscopic methods and 1,3-dipolar cycloaddition with methyl acrylate. OxaD is a versatile biocatalyst that converts an array of semisynthetic roquefortine C derivatives bearing indoline systems to their respective nitrones. This work describes the first implementation of a nitrone synthase as a biocatalyst and establishes a novel platform for late-stage diversification of a range of complex natural products.
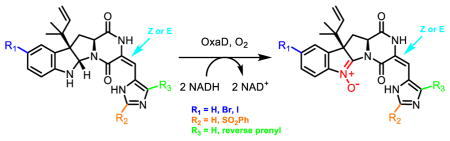
Graphical Abstract
INTRODUCTION
Prenylated indole fungal alkaloids constitute a structurally complex class of natural products, for which potent antibacterial, anticancer, antiparasitic, and insecticidal activities have been reported.1,2 These alkaloids are typically derived from a tryptophan-proline diketopiperazine skeleton, with a prenyl group frequently attached in the reverse orientation to the indole moiety. The biosynthesis of fungal prenylated indole alkaloids involves a multi-step cascade of reactions beginning with a non-ribosomal peptide synthetase (NRPS) catalyzed assembly of the diketopiperazine core. Subsequent sets of enzymes catalyze various oxidative/reductive reactions, C-C coupling, rearrangements, and in some cases, Diels-Alder reactions. 1,2
Roquefortines, glandicolines, meleagrin and oxaline comprise a select group of biosynthetically related prenylated indole alkaloids (Fig. 1). The archetypic roquefortine C (1) was first isolated from Penicillium roqueforti and has been detected in over 30 different Penicillium species.3–5 Oxaline (2) and meleagrin (3) have been less frequently isolated from fungal cultures.6,7 Roquefortine C (1) is found in low levels in industrially produced blue cheese,8 grain,9–12 and animal feedstocks,13 and while cytotoxic effects of varying degrees have been observed in mice14,15 and cockerels,16 roquefortine C has shown little to no cytotoxicity in a number of human cell lines.12,17,18 Roquefortine C is known to bind to iron, inactivating hepatic cytochrome P450s19 and inhibiting growth of gram-positive bacteria containing heme proteins.20,21 Roquefortine C may also be significant in indoor air contamination, as it has been isolated from numerous mold-contaminated indoor environments. Inflammatory responses have been reported for mouse lungs intratracheally instilled with roquefortine C.14,23–25 Despite their threat to human and animal health, as well as the economic loss due to food contamination by these compounds, 26 1–3 have also attracted interest as potential therapeutics. Recent reports indicate that 3 is a promising lead for the treatment of breast cancer,27 while meleagrin B, a diterpene-substituted biosynthetic derivative of 3, was shown to induce HL-60 cell apoptosis.28 Additionally, 3 has demonstrated inhibition of bacterial type II fatty acid synthesis while 2 has been shown to inhibit tubulin polymerization in Jurkat cells.29,30
Figure 1.

Roquefortine C (1) and derived alkaloids oxaline (2), meleagrin (3), glandicoline A (4), and glandicoline B (5). The 1,4-diazonane-2,5,6-trione 6 was proposed by Steyn and Vleggaar22 as an intermediate in the biosynthesis of 4.
Oxaline (2) and meleagrin (3) feature a triazaspirocyclic skeleton derived from an unusual rearrangement of the diketopiperazine core of 1.22 The first proposed mechanism for the biosynthesis of 2 from 1 included the formation of a 1,4-diazonane-2,5,6-trione intermediate 6 as the key step, prior to the transannular attack of the indole by the histidine-derived α-nitrogen, leading to the formation of the tri-nitrogen-substituted core in 2 and 3.22 Experiments performed with cultures of P. glandicola, P. atramentosum and P. farinosum provided additional evidence that glandicolines A (4) and B (5), meleagrin (3), and oxaline (2) are derived from roquefortine C (1).31
Whole genome sequencing, analysis, and substrate modeling with P. chrysogenum Wis54-1255 enabled the determination of the gene cluster that encodes the biosynthesis of 1.32 Open reading frames within the roquefortine-meleagrin gene cluster in the P. chrysogenum genome have been subjected to a series of RNA silencing experiments, which enabled a putative biochemical assignment for each step of the biosynthetic pathway.33 Transformation of roquefortine C (1) into glandicoline A (4) is promoted by a MAK1-like monooxygenase RoqM (encoded by Pc21g15460), as evidenced by loss of production of 3, 4, and 5 upon gene silencing. Subsequent gene deletion analysis of the P. chrysogenum strain DS54555 roquefortine C gene cluster34 confirmed the results obtained by gene silencing,33 leading to a mechanism identical to the one previously proposed by Steyn and Vleggaar.22 More recently, a rigorous structural elucidation of the metabolites of this pathway prompted a revision of the biosynthetic mechanism for this particular rearrangement.35 The key revision to this newly proposed metabolic pathway is the generation of the nitrone-bearing intermediate roquefortine L (7) by RoqM from 1 (Fig. 2). Since roquefortine L (7) has a molecular weight identical to that of glandicoline A (4), it was proposed that 7 had been mistakenly characterized as 4 in the initial gene silencing and gene deletion studies. Cytochrome P450 monooxygenase RoqO (encoded by Pc21g15450) was proposed to catalyze the rearrangement of 7 to give the triazaspirocyclic core of glandicoline B (5) as deletion of this gene resulted in loss of production of 5 and 3.
Figure 2. Known indolic nitrones.

Roquefortine L (7) is a labile intermediate in the biosynthesis of 2. Indolic nitrone bearing natural products versicamide E (8), alstoyunine D (9), perakine N1,N4-dioxide (10), avrainvillamide (11), waikialoid A (12), and stephacidin B (13).
Fungal production of roquefortine L (7) is particularly relevant, since very few indoline nitrones are known as natural products.36–40 With the exception of the recently isolated versicamide E (8), alstoyunine D (9), and perakine N1,N4-dioxide (10), the remaining fungal indolic nitrones share a common avrainvillamide (11) core (Fig. 2). Nitrone incorporation into these compounds can promote several biologically relevant events. For example, the nitrone functional group on avrainvillamide is required for dimerization to stephacidin B (13).41 The nitrone function of avrainvillamide also plays a vital role in its anti-proliferative activity, with the proposed mechanism of anti-proliferation involving nucleophilic attack of the nitrone functionality by cysteine residues in cellular proteins.40,42–44 Such properties generate great interest in the chemical synthesis of these molecules.45–47
Chemical conversion of indolines into their respective nitrone has been reported in only a few instances. It requires either mild oxidative conditions to give very moderate yields,46,48 or a multi-step procedure affording good yield.49 Discovery of a biocatalyst that could efficiently perform such a reaction in a single high-yield step would be of major interest for the synthesis of nitrone functionalized indolines. Previous accounts of enzymatic nitrone formation have focused on metabolism of xenobiotic secondary amines and hydroxylamines by hepatic microsomal enzymes,50–57 however, in the majority of these instances, nitrone formation was accompanied by a number of additional oxidative side reactions. Therefore the complex and biologically active indole alkaloids remain important targets for this type of functionalization.
We herein report the isolation, characterization, and enzymatic activity of OxaD, which can be produced in significant yields by heterologous expression. OxaD is a flavoprotein that has been harnessed for the biocatalytic conversion of roquefortine C and roquefortine C semisynthetic derivatives to afford the resulting indoline nitrones in very good yield and in a single step. To the best of our knowledge, this work represents the first biocatalytic synthesis of indoline nitrones.
RESULTS
Oxaline Biosynthetic Gene Cluster
In order to investigate the biogenesis of oxaline in mechanistic detail, we sequenced the genome of P. oxalicum F30 and performed gene cluster analysis using antiSMASH followed by a deep annotation of the oxaline biosynthetic pathway (Gen- Bank: KX601657)(Scheme 1A, Table S1). The composition of the oxaline (oxa) gene cluster is highly similar to that of the roquefortine/meleagrin (roq) cluster from P. chrysogenum33 (Table S2), with an average 78.4% similarity between homologous ORFs in the respective systems. One exception is a second methyltransferase (OxaC), which is absent in P. chrysogenum and suggests that this enzyme is responsible for the enol O-methylation that distinguishes 2 from 3. By homology, we reason that the initial biosynthetic steps toward 2 match those previously reported for 3 (Scheme 1B)34,58,59. Briefly, the first biosynthetic intermediate His-Trp diketopiperazine (HTD, 16) is generated by an NRPS-mediated condensation of the corresponding L-amino acids. HTD undergoes reverse prenylation at C-3 catalyzed by OxaF and dehydrogenation catalyzed by OxaB to give 1. The flavin oxidase, OxaD, and the P450 monooxygenase, OxaH, are homologous to P. chrysogenum RoqM and RoqO, respectively. RoqM and RoqO have been proposed to generate the core intermediate 5 from 1 by proceeding through intermediate 7, as identified by Ries et al.35 The conversion of 7 to 5 is proposed to arise from a P450-dependent hydroxylation catalyzed by OxaH, although this transformation has not been observed in vitro. Two O-methylations by OxaG and OxaC generate the final product 2 (manuscript in preparation).
Scheme 1. Oxaline biosynthetic gene cluster.

(A) Schematic representation of the ORFs in the oxaline (oxa) gene cluster. (B) Proposed biosynthetic pathway in P. oxalicum F30. The homologous enzymes from the roquefortine C/meleagrin (roq) gene cluster in P. chrysogenum are shown. A second methyltransferase, OxaC, lacks a homolog in P. chrysogenum.
OxaD is a flavin-dependent nitrone synthase
OxaD was cloned from a P. oxalicum F30 cDNA library and heterologously expressed in Escherichia coli. The purified enzyme has an intense yellow color. HPLC-MS analysis of the supernatant of boiled OxaD shows near quantitative incorporation of flavin adenine dinucleotide (FAD) (Fig. S1). In order to obtain sufficient quantities of the starting substrate 1, a fermentation-based isolation using P. crustosum was developed.60 Gram quantities of pure 1 were obtained for enzymatic testing and chemical derivatization. Both NADH and NADPH were shown to be effective in the oxidation of 1 by OxaD. Incubation of OxaD with 1 and reduced nicotinamide cofactor resulted in the formation of several products (Fig. S2). After 4 hours incubation of 1 with OxaD, HPLC-MS analysis (50:50 MeOH:H2O) showed peaks with molecular weights corresponding to the nitrone 7, the hydrated product 14, and a presumed MeOH adduct. Low levels of the hydroxylamine 15 (Fig. 3) were also detected. Preparative scale enzymatic reactions were conducted using 1 (20 mg) in order to characterize the major reaction product. Since reverse-phase C18 chromatography led exclusively to the hydrolysis product 14, subsequent reactions were designed to minimize the exposure of 7 to aqueous conditions, acidic or basic pH, and light. Biocatalytic reactions were performed for 1 h prior to extraction with EtOAc and normal phase chromatography purification on deactivated silica gel in the absence of small alcohols. These conditions led to 7 in a single step in good yield. The presence of the nitrone functionality in 7 was confirmed by analysis of 1H NMR, 13C NMR, and IR spectra. Additionally, P. crustosum was cultured on 15N-enriched medium to give 15N-labeled 1 (86% incorporation, Table S4). This material was subjected to the same reaction conditions as 1 to provide 15N-labeled 7, which was characterized by 15N-NMR. Comparison with the spectrum for labeled 1 clearly demonstrated the presence of a nitrone at N-1 in roquefortine L (7) (Fig. S17). A final confirmation of the nitrone functionality in roquefortine L (7) resulted from its 1,3-dipolar cycloaddition with methyl acrylate (Scheme 2, and vide infra).
Figure 3.

Reactions with OxaD and 1 led to the detection of a hydroxylamine intermediate 15 and the hydrolysis product 14.
Scheme 2.

Roquefortine L (7) undergoes 1,3-dipolar cycloaddition with methyl acrylate to generate 18.
Investigating the mechanism of oxidation
The observation of hydroxylamine 15 (Fig. 3) led us to investigate the mechanism of roquefortine C (1) oxidation by OxaD. The full catalytic cycle encompasses a four-electron oxidation, in which two equivalents of reduced nicotinamide cofactor (NADH) are consumed en route to the nitrone product (Fig. S3). Therefore, a single turnover experiment was performed, in which only one equivalent of NADH was provided to equimolar concentrations of OxaD and roquefortine C. Analysis of the reaction products showed an approximate 1:2 ratio of singly-oxidized hydroxylamine 15 species relatively to the doubly-oxidized nitrone product (Fig. S3), conclusively demonstrating that the reaction proceeds through a hydroxylamine intermediate. The abundance of 15 with respect to 7 under single turnover conditions also suggested the possibility that oxidation by OxaD is catalyzed in an iterative rather than a processive manner. To further support this proposal, single-turnover reactions were performed using a 10-fold stoichiometric excess of roquefortine C with respect to NADH and OxaD in an effort to preclude re-binding of the dissociated hydroxylamine species. Under these conditions, 15 was the dominant reaction product (Fig. S3), further supporting an iterative mechanism for OxaD. This iterative N-oxidation mechanism has been previously reported with human FMO3 using various substrates.61,62 The steady state kinetic constants of OxaD with roquefortine C were determined using an HPLC-based assay. These data show a kcat of 0.017 s−1 with a KM of 71 nM resulting in a catalytic efficiency (kcat/KM) of 2.3 × 105 M−1×s−1 (Fig. S4).
Roquefortine C nitrone undergoes 1,3-dipolar cycloaddition
As a means to further validate the enzymatic production of 7, and in order to explore indoline nitrones as a handle for further indole alkaloid diversification, 7 was screened with known dipolarophiles for 1,3-dipolar cycloaddition reactions. Methyl acrylate showed the best reactivity toward 7. Optimized reaction conditions generated cycloadduct 18 in a single step with 84% conversion, 24% yield (Scheme 2). The observed regiochemistry of this reaction is consistent with predictions based on frontier molecular orbital interactions.63
Roquefortine C derivatives are converted to nitrones by OxaD
In order to explore the substrate scope of OxaD, the roquefortine C derivatives 19 – 23 were assayed under the same conditions as described above for the native substrate 1. Compounds 19 – 23 were generated by semisynthesis from 1 (Fig. 4). Isoroquefortine C (19) was obtained by photochemical reaction of 1 using broad spectrum UV light.64 Electrophilic aromatic substitution of 1 using N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS) yielded 11-bromoroquefortine C (20)65 and 11-iodoroquefortine C (21), respectively. Phenylsulfonylroquefortine C (22) was prepared using N-fluorobenzenesulfonimide (NFSI) as a phenylsulfonyl group transfer reagent.66 Roquefortine E (23) was purchased from Enzo (Ann Arbor, MI). Processing 19 – 23 to their corresponding nitrone products (24–28) demonstrated that OxaD can accommodate variation at several positions in the roquefortine C skeleton.
Figure 4.

Semisynthetic roquefortine C derivatives. Modifications to the roquefortine C scaffold highlighted in red.
Total turnover numbers (TTN) were measured in order to quantify the effect of these variations on the OxaD biocatalyst (Fig. 5). Modifications to the histidine moiety, including both alkene isomerization (24) and reverse prenylation (28), were well tolerated. Only the phenylsulfonyl derivative (22) showed a substantial decrease in TTN. In order to test whether the diminished turnover of 22 was observed due to the poor solubility of this substrate, TTNs were measured using both 10% and 20% DMSO cosolvent. While elevated cosolvent increased the solubilization of 22, only a modest increase in TTN was observed, likely indicating that the benefit was offset by attenuated enzymatic activity. Modifications to the indole ring via halogenation were also processed efficiently by OxaD.
Figure 5.

Modifications to the native substrate 1 at several positions were processed by OxaD. TTN was calculated as moles product/moles enzyme.
OxaD catalyzes indole 2,3-oxidation of notoamide S
Based on the broad reactivity that OxaD displayed with roquefortine C derivatives, additional indole alkaloids were subjected to the enzyme to examine whether this substrate flexibility extended beyond the roquefortine C scaffold. These include brevianamide F (29), 6-OH-deoxybrevianamide E (30), notoamide S (31), notoamide E (32), (+)-stephacidin A (33), notoamide T (34) and preparaherquamide (35) (Scheme 3A). Of particular interest was to assay 33, a presumed biosynthetic precursor to the nitrone bearing avrainvillamide (11);67 however no reaction was observed for substrates 29, 30, and 32–35. Enzyme-dependent generation of a new product with m/z = +16 Da was uniquely observed by LC-MS with 31. The total conversion (20%) was lower than that of the roquefortine C alkaloids. Biocatalytic reaction of 31 with OxaD yielded 36 (Scheme 3B), rather than the expected indole N-hydroxylamine. The product 36 is presumed to arise via a pinacol-like rearrangement, following 2,3-epoxidation in an analogous fashion to the NotB catalyzed formation of notoamides C (37) and D (38) (Scheme 4).68
Scheme 3. OxaD reactivity with notoamides.

(A) Synthetic notoamides tested with OxaD. Only 31 was converted by the enzyme. (B) Pinacol-like rearrangement of the OxaD catalyzed 2,3-indole epoxidation on 31 gives rise to the novel product 36.
Scheme 4.

NotB-catalyzed formation of notoamides C (37) and D (38) from notoamide E (32).
DISCUSSION
While the biosynthesis of the roquefortine C derived alkaloids 1–5 has been the subject of investigations employing genetic approaches,33–35 enzymatic characterization relating to assembly and processing steps is completely lacking for these indole alkaloid pathways. By sequencing the P. oxalicum F30 genome, we have identified the oxaline biosynthetic gene cluster, which is homologous to the meleagrin system.33 Large scale heterologous expression of the indoline nitrone synthase OxaD enabled its complete enzymatic characterization. OxaD catalyzes the key oxidation steps leading to the conversion of 1 to 5. It is a robust biocatalyst for nitrone installation on the wild type substrate roquefortine C as well as a range of structurally related alkaloids. Large quantities of 1, obtained by fermentation of P. crustosum, enabled semisynthetic preparation of roquefortine C analogues and preparative scale biocatalytic reactions with these substrates. P. crustosum produced 1 for a fraction of the cost of total synthesis69 and promotes green chemistry practices by avoiding the use of large quantities of organic solvents and unnecessary derivatizations.
Identification of nitrones has long been a challenge with regard to their differentiation from other products of N-oxidation, such as imines, amine oxides, or oxaziridines. Since nitrones undergo 1,3 dipolar cycloadditions with dipolarophiles such as alkenes, alkynes, and nitriles, roquefortine L (7) was subjected to 1,3-dipolar cycloaddition with methyl acrylate to give 18, in agreement with the frontier molecular orbital interactions described by Sustmann and Trill.63 While this cycloaddition is highly indicative of the presence of a nitrone functionality in 7, oxaziridines are also known to thermally rearrange to nitrones before undergoing 1,3-dipolar cycloadditions.70,71
Preparative scale (>20 mg) reactions of 1, 15N-enriched 1, 19, 20, and 22 with OxaD were conducted to provide sufficient material for spectroscopic analysis of nitrones 7, 24, 25, and 27. Analysis of the 1H NMR spectrum of 7 confirms the double oxidation event at the indoline nitrogen, demonstrated conclusively by the disappearance of the H-6 signal at δ 6.35 ppm. Moreover, chemical shifts of H-9, H-10, H-11 and H-12 were observed further downfield. Analysis of the 13C NMR spectrum of 7 was further indicative of the nitrone formation, as the chemical shift of the C-6 peak shifted from δ 78 ppm to δ 150 ppm. Additionally, previous infrared analysis of N-phenyl nitrones shows a very strong band between 1000–1100 cm−1 corresponding to the N-O stretch.72 This band disappears upon isomerization of N-phenyl nitrones to the corresponding oxaziridines. Following the oxidation of roquefortine C by OxaD, an intense band was observed at 1014 cm−1 in the IR spectrum of 7, as additional support for the presence of a nitrone functionality instead of an oxaziridine. The presence of the nitrone group was also confirmed by analysis of 1H-15N HMBC spectrum of 15N-enriched 7. Nitrogen atoms in N-aryl nitrones display a characteristic peak between δ 270–300 ppm, while the corresponding amine nitrogens are observed between δ 0–100 ppm. In the 1H-15N HMBC spectrum of 15N-enriched 7 the signal at δ 83 ppm disappeared, and a new peak was observed at δ 281 ppm, which correlated to H-9 and H-10.
The enzymatic characterization of OxaD, along with the results of gene disruptions,34 indicates that formation of roquefortine L (7) is a key biosynthetic intermediate toward formation of the glandicoline core. OxaD conversion of 1 into 7 generates the electrophilic center required for the attack by the amide nitrogen in the proposed 1,4-diazonane-2,5,6-trione intermediate (6) (Scheme 5).35 Hydroxylation at C-16 of 1 leading to 39 appears insufficient to induce rearrangement, since this metabolite has been previously isolated and identified;73,74 therefore, both hydroxylation at C-16 and nitrone generation to give 41 are likely required for the core rearrangement to occur. Roquefortine L (7) involvement is supported by the fact that glandicoline A (5) has not been observed in P. chrysogenum, and that disruption of roqM (81% homology with oxaD) abolished the production of glandicoline B (6).35 The first report of 5 in P. glandicola is sufficiently supported by spectroscopic data;75 however, subsequent reports of 5 were based on mass spectrometry analysis, chromatographic retention time (Rf) and/or UV signature, but not by NMR analysis.31,33,34 As substantial structural characterization of metabolites is imperative in differentiating glandicoline A from 7, we have unequivocally assigned the nitrone group in 7 by NMR, IR, and chemical transformation.
Scheme 5. Proposed biogenesis of glandicoline A (4) and B (5).

The nitrone intermediate 7 described by Ries et al. and in this work is likely a key priming step for the rearrangement to 6. Compound 5 is not observed in P. chrysogenum or P. oxalicum F30 but is a known natural product. One possible route to 5 is through dehydration of the hydroxylamine intermediate 15 followed by hydroxylation and rearrangement. Enzymatic processing of 5 to 4 must also be considered.
At present, the mechanism of formation of 5 in P. glandicola is unclear. It could require a yet unknown route to the 1,2-imine in 4 proposed by Steyn and Vleggaar33 or arise by modification of 6 (Scheme 5). Investigation of the biosynthetic gene clusters in strains that produce 4 will be necessary to identify candidate enzymes that could give rise to 4, either through a different oxidation cascade involving 1 or enzymatic processing of 5. Understanding the role of the oxidative enzymes involved in the rearrangement of 1 to 5 will be critical for in vitro reconstitution and for future mutasynthetic efforts based on derivatives of 1. There is ample precedence for plasticity in these pathways, as mutasynthesis with roqA gene disruption mutant strains has been used to generate novel roquefortine D derivatives.76 Characterization of OxaD reported in this study also reveals the ability of the roquefortine C nitrone synthase to accept an array of modified substrates that enables the generation of novel indoline nitrones that would be synthetically appealing for the preparation of further elaborated alkaloid scaffolds.
Indole alkaloids 29 – 35 were tested primarily as a means to define the substrate scope of OxaD and also to explore the possibility that a flexible OxaD enzyme could reveal a functional link between nitrone biogenesis in roquefortine L (7) and avrainvillamide (11). While no reaction was observed with OxaD and (+)-stephacidin A (33), OxaD was shown to catalyze oxidation of notoamide S (31) to the rearranged product 36. Surprisingly, in this case the enzyme catalyzed a 2,3-indole epoxidation rather than the expected N-hydroxylation. Compound 36 is likely derived from a pinacol-type rearrangement, as reported for the homologous NotB, which shows 53% similarity and 37% identity with OxaD (Scheme 3B).68 Compound 36 was observed as a single reaction product between 31 and OxaD, as opposed to the reaction between notoamide E (32) and NotB, which generated notoamides C (37) and D (38) (Scheme 4). OxaD appears to play a role in directing reaction selectivity, although low enzyme activity with notoamide S (31) could limit the detection of minor reaction products. This outcome shows that OxaD presents a remarkable functional plasticity in addition to its broad substrate scope. Indeed, bioinformatic analysis of OxaD reveals that biochemically characterized homologs promote a variety of oxidative reactions (Table S3), indicating that subtle structural differences likely enable these enzymes to differentially catalyze oxidative processes. Further structural investigation of OxaD or its structural and functional homologs would be required to determine the mechanistic basis of this branch in reaction specificity.
The discovery of OxaD as an indolic nitrone synthase enables a unique strategy in late stage chemical derivatization of nitrone functionalized indolines through 1,3-dipolar cycloadditions. There are limited reports of the occurrence of 1,3-dipolar cycloadditions using natural products or their biosynthetic intermediates as substrates. 77 Roquefortine L (7) undergoes effective 1,3-dipolar cycloaddition with methyl acrylate to give 18 (Scheme 2), a derivatization outcome that may be extended to an array of other dipolarophiles with electron-withdrawing substituents.78 By coupling this derivatization strategy with the broad substrate scope of OxaD, we have established a platform in which novel indole alkaloid frameworks can be paired with an array of dipolarophiles in a cycloaddition cascade. Recent reports describing the therapeutic potential of 229 and 327,30 provide strong motivation to develop these tools. Future efforts in our laboratories will also seek to extend the substrate scope of OxaD beyond alkaloids in the roquefortine family.
CONCLUSION
The results herein illustrate that the combination of biological and chemical approaches for the generation of new and diverse natural products can lead to a number of applications in chemistry and chemical biology. Understanding the genetic and metabolic information of biosynthetic systems can provide access to complex chiral molecular scaffolds, which can be further synthetically elaborated into novel molecules. These natural product-based modified frameworks are of considerable interest in chemistry, biochemistry, and chemical biology as bioactive chemical entities or as unique biochemical tools. Heterologous production and enzymatic characterization of OxaD constitutes a proof of concept that unique biocatalysts can generate versatile and powerful chemical and biochemical tools for structural diversification.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the NIH (CA070375), NSF CHE 1220121, the FAPESP (grants 2012/50026-3, 2013/50228-8 and 2014/05670-7), a post-doctoral fellowship awarded to SR (CAPES, BEX 4498/14-3), and pre-doctoral training grant fellowships awarded to CMG (NIH, 5T32GM071339-09, 5T32GM071339-10, 2T32GM071339-11).
The authors would like to thank Dr. George Furst, Dr. Jun Gu, and Dr. Wendy Feng for their support obtaining NMR spectra.
Footnotes
The authors declare no competing financial interest.
Supporting Information. Full experimental details and NMR spectra are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li SM. Nat Prod Rep. 2010;27:57. doi: 10.1039/b909987p. [DOI] [PubMed] [Google Scholar]
- 2.Finefield JM, Frisvad JC, Sherman DH, Williams RM. J Nat Prod. 2012;75:812. doi: 10.1021/np200954v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohmomo S, Sato T, Utagawa T, Abe M. Agr Biol Chem Tokyo. 1975;39:1333. [Google Scholar]
- 4.Ohmomo S, Utagawa T, Abe M. Agr Biol Chem Tokyo. 1977;41:2097. [Google Scholar]
- 5.Polonsky J, Merrien MA, Scott PM. Ann Nutr Aliment. 1977;31:963. [PubMed] [Google Scholar]
- 6.Qu P, Wu ZY, Zhu WM. Acta Crystallogr, Sect E: Struct Rep Online. 2012;68:o1626. doi: 10.1107/S1600536812019423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagel DW, Pachler KGR, Steyn PS, Wessels PL, Gafner G, Kruger GJ. J Chem Soc, Chem Commun. 1974:1021. doi: 10.1039/c39740001021. [DOI] [Google Scholar]
- 8.Hymery N, Vasseur V, Coton M, Mounier J, Jany JL, Barbier G, Coton E. Compr Rev Food Sci F. 2014;13:437. doi: 10.1111/1541-4337.12069. [DOI] [PubMed] [Google Scholar]
- 9.Driehuis F, Spanjer MC, Scholten JM, te Giffel MC. J Dairy Sci. 2008;91:4261. doi: 10.3168/jds.2008-1093. [DOI] [PubMed] [Google Scholar]
- 10.Mansfield MA, Jones AD, Kuldau GA. Phytopathology. 2008;98:330. doi: 10.1094/PHYTO-98-3-0330. [DOI] [PubMed] [Google Scholar]
- 11.Spanjer MC, Rensen PM, Scholten JM. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2008;25:472. doi: 10.1080/02652030701552964. [DOI] [PubMed] [Google Scholar]
- 12.Rasmussen RR, Rasmussen PH, Larsen TO, Bladt TT, Binderup ML. Food Chem Toxicol. 2011;49:31. doi: 10.1016/j.fct.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Monbaliu S, Van Poucke C, Detavernier C, Dumoulin F, Van De Velde M, Schoeters E, Van Dyck S, Averkieva O, Van Peteghem C, De Saeger S. J Agric Food Chem. 2010;58:66. doi: 10.1021/jf903859z. [DOI] [PubMed] [Google Scholar]
- 14.Rand TG, Giles S, Flemming J, Miller JD, Puniani E. Toxicol Sci. 2005;87:213. doi: 10.1093/toxsci/kfi223. [DOI] [PubMed] [Google Scholar]
- 15.Arnold DL, Scott PM, McGuire PF, Harwig J, Nera EA. Food Cosmet Toxicol. 1978;16:369. doi: 10.1016/s0015-6264(78)80009-1. [DOI] [PubMed] [Google Scholar]
- 16.Wagener RE, Davis ND, LDU Appl Environ Microb. 1980;39:882. doi: 10.1128/aem.39.4.882-887.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsen TO, Gareis M, Frisvad JC. J Agric Food Chem. 2002;50:6148. doi: 10.1021/jf020453i. [DOI] [PubMed] [Google Scholar]
- 18.Bunger J, Westphal G, Monnich A, Hinnendahl B, Hallier E, Muller M. Toxicology. 2004;202:199. doi: 10.1016/j.tox.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Aninat C, Andre F, Delaforge M. Food Addit Contam. 2005;22:361. doi: 10.1080/02652030500073287. [DOI] [PubMed] [Google Scholar]
- 20.Kopp-Holtwiesche BB. J Environ Pathol Toxicol Oncol. 1990;10:41. [PubMed] [Google Scholar]
- 21.Kopp B, Rehm HJ. Eur J Appl Microbiol Biotechnol. 1979;6:397. [Google Scholar]
- 22.Steyn PS, Vleggaar R. J Chem Soc, Chem Commun. 1983;10:560. [Google Scholar]
- 23.Vishwanath V, Sulyok M, Labuda R, Bicker W, Krska R. Anal Bioanal Chem. 2009;395:1355. doi: 10.1007/s00216-009-2995-2. [DOI] [PubMed] [Google Scholar]
- 24.Taubel M, Sulyok M, Vishwanath V, Bloom E, Turunen M, Jarvi K, Kauhanen E, Krska R, Hyvarinen A, Larsson L, Nevalainen A. Indoor Air. 2011;21:368. doi: 10.1111/j.1600-0668.2011.00721.x. [DOI] [PubMed] [Google Scholar]
- 25.Vishwanath V, Sulyok M, Weingart G, Kluger B, Taubel M, Mayer S, Schuhmacher R, Krska R. Talanta. 2011;85:2027. doi: 10.1016/j.talanta.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 26.Marroquin-Cardona AG, Johnson NM, Phillips TD, Hayes AW. Food Chem Toxicol. 2014;69:220. doi: 10.1016/j.fct.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 27.Mady MS, Mohyeldin MM, Ebrahim HY, Elsayed HE, Houssen WE, Haggag EG, Soliman RF, El Sayed KA. Bioorg Med Chem. 2016;24(2):113. doi: 10.1016/j.bmc.2015.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du L, Feng T, Zhao B, Li D, Cai S, Zhu T, Wang F, Xiao X, Gu Q. J Antibiot (Tokyo) 2010;63:165. doi: 10.1038/ja.2010.11. [DOI] [PubMed] [Google Scholar]
- 29.Koizumi Y, Arai M, Tomoda H, Ōmura S. Biochim Biophys Acta. 2004;1693:47. doi: 10.1016/j.bbamcr.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Zheng CJ, Sohn MJ, Lee S, Kim WG. PLoS ONE. 2013;8:e78922. doi: 10.1371/journal.pone.0078922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overy DP, Nielsen KF, Smedsgaard J. J Chem Ecol. 2005;31:2373. doi: 10.1007/s10886-005-7107-y. [DOI] [PubMed] [Google Scholar]
- 32.van den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WH, Joardar V, Kiel JA, Kovalchuk A, Martin JF, Nierman WC, Nijland JG, Pronk JT, Roubos JA, van der Klei IJ, van Peij NN, Veenhuis M, von Dōhren H, Wagner C, Wortman J, Bovenberg RA. Nat Biotechnol. 2008;26:1161. doi: 10.1038/nbt.1498. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Estrada C, Ullan RV, Albillos SM, Fernandez-Bodega MA, Durek P, von Dohren H, Martin JF. Chem Biol. 2011;18:1499. doi: 10.1016/j.chembiol.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Ali H, Ries MI, Nijland JG, Lankhorst PP, Hankemeier T, Bovenberg RA, Vreeken RJ, Driessen AJ. PLoS ONE. 2013;8:e65328. doi: 10.1371/journal.pone.0065328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ries MI, Ali H, Lankhorst PP, Hankemeier T, Bovenberg RA, Driessen AJ, Vreeken RJ. J Biol Chem. 2013;288:37289. doi: 10.1074/jbc.M113.512665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenical W, Jensen PR, Cheng XC. Avrainvillamide, a cytotoxic marine natural product, and derivatives thereof. 6,066,635. US Patent. 2000 May 23;
- 37.Wang XR, You JL, King JB, Powell DR, Cichewicz RH. J Nat Prod. 2012;75:707. doi: 10.1021/np2009994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng JX, Gao HQ, Li J, Ai J, Geng MY, Zhang GJ, Zhu TJ, Gu QQ, Li DH. J Org Chem. 2014;79:7895. doi: 10.1021/jo5010179. [DOI] [PubMed] [Google Scholar]
- 39.Sugie Y, Hirai H, Inagaki T, Ishiguro M, Kim YJ, Kojima Y, Sakakibara T, Sakemi S, Sugiura A, Suzuki Y, Brennan L, Duignan J, Huang LH, Sutcliffe J, Kojima N. J Antibiot (Tokyo) 2001;54:911. doi: 10.7164/antibiotics.54.911. [DOI] [PubMed] [Google Scholar]
- 40.Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 41.von Nussbaum F. Angew Chem Int Ed. 2003;42:3068. doi: 10.1002/anie.200301646. [DOI] [PubMed] [Google Scholar]
- 42.Wulff JE, Siegrist R, Myers AG. J Am Chem Soc. 2007;129:14444. doi: 10.1021/ja075327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wulff JE, Herzon SB, Siegrist R, Myers AG. J Am Chem Soc. 2007;129:4898. doi: 10.1021/ja0690971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mikkola R, Andersson MA, Hautaniemi M, Salkinoja-Salonen MS. Toxicon. 2015;99:58. doi: 10.1016/j.toxicon.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Herzon SB, Myers AG. J Am Chem Soc. 2005;127:5342. doi: 10.1021/ja0510616. [DOI] [PubMed] [Google Scholar]
- 46.Baran PS, Guerrero CA, Hafensteiner BD, Ambhaikar NB. Angew Chem Int Ed. 2005;44:3892. doi: 10.1002/anie.200500655. [DOI] [PubMed] [Google Scholar]
- 47.Artman GD, Grubbs AW, Williams RM. J Am Chem Soc. 2007;129:6336. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ideguchi T, Yamada T, Shirahata T, Hirose T, Sugawara A, Kobayashi Y, Omura S, Sunazuka T. J Am Chem Soc. 2013;135:12568. doi: 10.1021/ja406657v. [DOI] [PubMed] [Google Scholar]
- 49.Hafensteiner BD, Escribano M, Petricci E, Baran PS. Bioorg Med Chem Lett. 2009;19:3808. doi: 10.1016/j.bmcl.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beckett AH, Sheikh AH. J Pharm Pharmacol. 1973;25(Suppl):171P. [PubMed] [Google Scholar]
- 51.Kadlubar FF, McKee EM, Ziegler DM. Arch Biochem Biophys. 1973;156:46. doi: 10.1016/0003-9861(73)90339-1. [DOI] [PubMed] [Google Scholar]
- 52.Bondon A, Macdonald TL, Harris TM, Guengerich FP. J Biol Chem. 1989;264:1988. [PubMed] [Google Scholar]
- 53.Cashman JR, Yang ZC, Hogberg T. Chem Res Toxicol. 1990;3:428. doi: 10.1021/tx00017a007. [DOI] [PubMed] [Google Scholar]
- 54.Clement B, Lustig KL, Ziegler DM. Drug Metab Dispos. 1993;21:24. [PubMed] [Google Scholar]
- 55.Rodriguez RJ, Proteau PJ, Marquez BL, Hetherington CL, Buckholz CJ, O’Connell KL. Drug Metab Dispos. 1999;27:880. [PubMed] [Google Scholar]
- 56.Sun H, Ehlhardt WJ, Kulanthaivel P, Lanza DL, Reilly CA, Yost GS. J Pharmacol Exp Ther. 2007;322:843. doi: 10.1124/jpet.107.121723. [DOI] [PubMed] [Google Scholar]
- 57.Barbara JE, Kazmi F, Parkinson A, Buckley DB. Drug Metab Dispos. 2013;41:1012. doi: 10.1124/dmd.113.051151. [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Estrada C, Ullan RV, Albillos SM, Fernandez-Bodega MA, Durek P, von Dohren H, Martin JF. Chem Biol. 2011;18:1499. doi: 10.1016/j.chembiol.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 59.Ries MI, Ali H, Lankhorst PP, Hankemeier T, Bovenberg RA, Driessen AJ, Vreeken RJ. J Biol Chem. 2013;288:37289. doi: 10.1074/jbc.M113.512665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gober C, Joullie MM. Athens Journal of Sciences. 2015;3:257. [PMC free article] [PubMed] [Google Scholar]
- 61.Sun H, Ehlhardt WJ, Kulanthaivel P, Lanza DL, Reilly CA, Yost GS. J Pharmacol Exp Ther. 2007;322:843. doi: 10.1124/jpet.107.121723. [DOI] [PubMed] [Google Scholar]
- 62.Cashman JR, Xiong YN, Xu L, Janowsky A. J Pharmacol Exp Ther. 1999;288:1251. [PubMed] [Google Scholar]
- 63.Sustmann R, Trill H. Angew Chem Int Ed. 1972;11:838. [Google Scholar]
- 64.Scott PM, Polonsky J, Merrien MA. J Agric Food Chem. 1979;27:201. [Google Scholar]
- 65.Silva JVd, Fill TP, Lotufo LV, Pessoa CdO, Rodrigues-Filho E. Helv Chim Act. 2014;97:1345. [Google Scholar]
- 66.Roy A, Schneller SW. Org Lett. 2005;7:3889. doi: 10.1021/ol051297e. [DOI] [PubMed] [Google Scholar]
- 67.Nising CF. Chem Soc Rev. 2010;39:591. doi: 10.1039/b900407f. [DOI] [PubMed] [Google Scholar]
- 68.Li S, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH. J Am Chem Soc. 2012;134:788. doi: 10.1021/ja2093212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shangguan N, Hehre WJ, Ohlinger WS, Beavers MP, Joullie MM. J Am Chem Soc. 2008;130:6281. doi: 10.1021/ja800067q. [DOI] [PubMed] [Google Scholar]
- 70.Williamson KS, Michaelis DJ, Yoon TP. Chem Rev. 2014;114:8016. doi: 10.1021/cr400611n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Padwa A, Koehler KF. Heterocycles. 1986;24:611. [Google Scholar]
- 72.Shindo H, Umezawa B. Chem Pharm Bull (Tokyo) 1962;10:492. doi: 10.1248/cpb.10.492. [DOI] [PubMed] [Google Scholar]
- 73.Trimble LA, Sumarah MW, Blackwell BA, Wrona MD, Miller JD. Tetrahedron Lett. 2012;53:956. [Google Scholar]
- 74.Shan WG, Ying YM, Yu HN, Liu WH, Zhan ZJ. Helv Chim Act. 2010;93:772. [Google Scholar]
- 75.Kozlovsky AGV, NG, Reshetilova TA, Sakharovsky VG, Baskunov BP, Seleznyov SG. Prikl Biohim Mikrobiol. 1994;30:410. [Google Scholar]
- 76.Ouchaou K, Maire F, Salo O, Ali H, Hankemeier T, van der Marel GA, Filippov DV, Bovenberg RA, Vreeken RJ, Driessen AJ, Overkleeft HS. Chembiochem. 2015;16:915. doi: 10.1002/cbic.201402686. [DOI] [PubMed] [Google Scholar]
- 77.Payne KA, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SE, Scrutton NS, Hay S, Leys D. Nature. 2015;522:497. doi: 10.1038/nature14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang J. Synlett. 2012;16:2293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.