

Abstract
Objective
The Epi4K consortium recently identified four de novo mutations in the γ-aminobutyric acid type A (GABAA) receptor β3 subunit gene GABRB3 and one in the β1 subunit gene GABRB1 in children with epileptic encephalopathies (EEs) Lennox-Gastaut syndrome (LGS) or infantile spasms (IS). Since the etiology of EEs is often unknown, we determined the impact of GABRB mutations on GABAA receptor function and biogenesis.
Methods
GABAA receptor α1 and γ2L subunits were co-expressed with wild-type and/or mutant β3 or β1 subunits in HEK 293T cells. Currents were measured using whole cell and single channel patch clamp techniques. Surface and total expression levels were measured using flow cytometry. Potential structural perturbations in mutant GABAA receptors were explored using structural modeling.
Results
LGS-associated GABRB3(D120N, E180G, Y302C) mutations located at β+ subunit interfaces reduced whole cell currents by decreasing single channel open probability without loss of surface receptors. In contrast, IS-associated GABRB3(N110D) and GABRB1(F246S) mutations at β-subunit interfaces produced minor changes in whole cell current peak amplitude but altered current deactivation by decreasing or increasing single channel burst duration, respectively. GABRB3(E180G) and GABRB1(F246S) mutations also produced spontaneous channel openings.
Interpretation
All five de novo GABRB mutations impaired GABAA receptor function by rearranging conserved structural domains, supporting their role in EEs. The primary effect of LGS-associated mutations was reduced GABA-evoked peak current amplitudes while the major impact of IS-associated mutations was on current kinetic properties. Despite lack of association with epilepsy syndromes, our results suggest GABRB1 as a candidate human epilepsy gene.
Introduction
Epileptic encephalopathies (EEs) are a diverse group of severe childhood epilepsy syndromes with intractable seizures, neurodevelopmental delay and regression, resistance to treatment and poor clinical outcomes. According to the International League Against Epilepsy’s revised terminology of seizures and epilepsies, “EEs embodies the notion that the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone (e.g., cortical malformation), and that these can worsen over time.” 1 Often the etiologies of EEs are unknown, and patients have limited or no family history of epilepsy. Due to advances in sequencing technologies, several de novo single nucleotide mutations have been discovered in EE patients and are emerging as genetic risk factors for EEs.
A recent study by the Epi4K Consortium and Epilepsy Phenome/Genome project (EPGP) identified four novel de novo mutations in the GABAA receptor β3 subunit gene (GABRB3(N110D, D120N, E180G, Y302C)) in patients with two rare, but severe, EEs, the Lennox-Gastaut syndrome (LGS) and infantile spasms (IS) 2. Additionally one IS patient had a mutation in the GABAA receptor β1 subunit gene (GABRB1(F246S)). Sequence alignment analysis among GABR genes (Figure 1A) and structural modeling of the GABAA receptor (Figure 1B) revealed that these mutations are located in conserved structural domains that are important for function of the receptor 3, 4.
Figure 1. Location of the de novo GABAA receptor β3 and β1 subunit mutations found in LGS and IS patients.

(A) Sequence alignments of human β1-3, α1-6 and γ1-3 GABAA receptor subunits show the conserved residues altered by the de novo mutations (shown in red). The residues highlighted in grey are conserved across all of the subunits. Secondary structures are represented above the alignments as α-helices (black bar) or β-sheets (arrows). (B) 3D structural model of the GABAA receptor with the β subunits in blue, α subunits in gray and γ subunit in yellow. GABRB de novo mutations are mapped onto the structure and represented respectively in orange and green for LGS- and IS-associated mutations.
GABAA receptors are heteropentameric GABA-gated chloride ion channels formed by the assembly of 2 α, 2 β, and 1 γ subunits, which mediate the majority of fast inhibitory neurotransmission in the brain. Several mutations in GABRs that impair GABAA receptor function by gating or trafficking deficiencies have been identified in patients that exhibit a broad spectrum of epilepsy syndromes 5. Previously three GABRB3 mutations, P11S, S15F and G32R, have been associated with childhood absence epilepsy 6, 7, and heterozygous Gabrb3+/− mice exhibit absence-like seizures 8, 9. Moreover, β3 subunits are abundantly expressed in the developing brain and are critically involved in early stages of development 10, 11. However, characterization of the contribution of GABRB3 and GABRB1 to catastrophic childhood epilepsies is missing, and GABRB1 has not been associated with epilepsy syndromes. While not directly demonstrated, the strong genetic evidence and the important role of β3 subunits in neurodevelopment suggest that the de novo GABRB3 mutations identified by the Epi4K consortium in EEs are likely to be pathogenic.
Since the pathological impact of these mutations remains unknown, we sought to determine the effects of the de novo GABRB3 and GABRB1 mutations on GABAA receptor function and biogenesis in vitro. Using the HEK293T cell expression system, we found that the mutations disrupted whole cell and single channel GABA currents without reducing the surface expression of GABAA receptors. Furthermore, structural modeling predicted mutation-induced rearrangements of inter- and intra-subunit secondary structures and side chains that may underlie both assembly and channel kinetic defects of GABAA receptors, thus causing disinhibition and EEs.
Subjects/Materials and Methods
Complementary DNA (cDNA) constructs
The cDNAs encoding human GABAA receptor subunits α1 (NM_000806.5), β1 (NM_000812.3), β3 (NM_021912.4 variant 2), γ2L (NM_198904.2) and EGFP (LC008490.1) were cloned into the pcDNA3.1(+) vector. Point mutations in the cDNAs encoding β1, β3 subunits and hemagglutinin (HA) epitope tag (YPYDVPDYA) between amino acids 4 and 5 of the mature β1, β1(F246S), and γ2L subunit were introduced and sequenced prior to use as previously described 7. Amino acids were numbered according to the immature peptide sequence that includes the signal peptide.
Expression of recombinant GABAA receptors
Whole cell recordings were obtained from HEK293T cells (HEK 293T/17, ATCC® CRL-11268™) that were cultured as monolayers in 60 mm dishes (Corning) as previously described 12. For the wild-type (wt) or homozygous (hom) condition, 0.6 μg cDNA of each α1, β (β3, β1, β3(mut) or β1(mut)) and γ2L subunit, and 0.1 μg cDNA of EGFP (to identify transfected cells) were transfected using X-tremeGENE9 DNA Transfection Reagent (Roche Diagnostics, 1.15 μl per μg cDNA). For the heterozygous (het) condition, 0.6 μg α1 and γ2L and 0.3 μg of wt β3 or β1, 0.3 μg of mutant β3 or β1 subunit, and 0.1 μg of EGFP cDNAs were used. For convenience the terms wt, het and hom were used for different expression conditions of GABAA receptor subunits and did not refer to any genetic conditions. Single channel recordings were obtained from HEK293T cells plated onto 12 mm cover glasses at a density of 4x104 in 35 mm culture dishes (Corning), and transfected after 24 hours with 0.3 μg cDNA of each α1, β3, β1 and γ2L subunit, and 0.05 μg of EGFP. Recordings were obtained 48 hours after transfection. For flow cytometry experiments, cells were plated at a density of 4–6x105 in 60 mm culture dish (Corning), and transfected 24 hours after plating using polyethyleneimine (MW 40,000 KD, 24765, Polysciences Inc.). For mock or single subunit expression, empty pcDNA3.1 vector was added to make a final cDNA transfection amount to 1.8 μg.
Electrophysiology
Whole cell recordings from lifted HEK293T cells and cell attached single channel recordings were obtained at room temperature as previously described 12. For whole cell recordings the external solution was composed of (in mM): 142 NaCl, 8 KCl, 10 D(+)-glucose, 10 HEPES, 6 MgCl2.6H2O, and 1 CaCl2 (pH 7.4, ~326 mOsm). The internal solution consisted of (in mM): 153 KCl, 10 HEPES, 5 EGTA 2 Mg-ATP, and 1 MgCl2.6H2O (pH 7.3, ~300 mOsm). The Cl− reversal potential was near 0 mV, and cells were voltage clamped at −20 mV. One or 10 mM GABA was applied for 4 s. Drugs were gravity-fed to four-barrel square glass connected to a SF-77B Perfusion Fast-Step system (Warner Instruments Corporations). The 10–90% rise times of open-tip liquid junction currents were 200–800 μs.
Single-channel currents were recorded in an external solution containing (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH 7.4). The internal solution consisted of (in mM): 120 NaCl, 5 KCl, 10 MgCl2, 0.1 CaCl2, 10 glucose, 10 HEPES (pH 7.4), and 1 mM of GABA. The micropipette potential was +80 mV. GABAA receptor spontaneous activity was recorded in absence of GABA, and blocked by adding 100 μM picrotoxin and 100 μM Zinc in the external solution.
Whole cell and single channel currents were amplified and low-pass filtered at 2 kHz using an Axopatch 200B amplifier, digitized at 10 kHz (whole cell recordings) or 20 kHz (single channel recordings) using Digidata 1322A, and saved using pCLAMP 9 (Axon Instruments). Data were analyzed offline using Clampfit 10.3 (Axon Instruments), TAC 4.2 and TACFit 4.2 (Bruxton Corporation) software 12, 13. Whole cell peak currents, 10–90% rise time, desensitization, deactivation, % Zn inhibition were calculated using Clampfit 10.3 as previously described 12, 14. Holding current was calculated as the average baseline current before GABA application. The outward Zn2+ current was calculated by subtracting the 6 s average baseline current before 10 μM Zn2+ application from the average current during the last 6 s (of 10s) of Zn2+ application.
Single-channel open and closed events were analyzed using the 50% threshold detection method and visually inspected before accepting the events. Single-channel openings occurred as bursts and clusters of bursts. Bursts were defined as one or more consecutive openings separated by closed times shorter than 5 ms. Clusters were defined as a series of bursts separated by closed intervals longer than 10 ms. Single channel Po and opening frequency were determined within clusters to eliminate very long closed times.
Flow cytometry
Flow cytometry protocols have been previously described in detail 7, 15. Cell surface expression levels of α1, β3, β1HA or γ2LHA subunits were determined using primary antibodies against human α1 subunits (N-terminal, clone BD24, Millipore; 2.5 g/ml), human β3 subunits (N-terminal, monoclonal, β2/3-PE, clone 62-3G1, Millipore; 2.5 g/ml), and the HA epitope tag (clone 16B12, Covance; 2.5 g/ml), respectively. Following antibody incubation, cells were washed four times with FACS buffer and incubated with Alexa647-conjugated anti-mouse IgG1 secondary antibody (Invitrogen) before washing and fixation with 2% w/v paraformaldehyde. For total cellular protein detection, cells were permeabilized using Cytofix/Cytoperm (BD Biosciences) and washed twice in Perm/Wash (BD Biosciences) prior to staining. Following antibody incubations, cells were washed four times in Perm/Wash and twice in FACS buffer before fixation.
Fluorescence intensity (FI) levels of cells were determined using a BD LSR II 3/5-laser flow cytometer (BD Biosciences) and analyzed offline using FlowJo 7.5.5 (Tree Star). For each condition, 10,000 cells in the final gate were acquired. Mean FI for each condition was calculated after subtracting the mean FI of the cells transfected with blank pcDNA3.1(+) vector. The relative FI for each condition was obtained by normalizing to the mean FI of the wt condition.
Structural modeling and simulation
GABAA receptor subunit raw sequences in FASTA format were individually loaded into Swiss-PdbViewer 4.10 for template search against the ExPDB database. The structure of the Caenorhabditis elegans glutamate-gated chloride channel (GluCl; PDB: 3RHW) was identified as the best template with 33% and 36% sequence identity for γ2 and α1 subunits respectively. For β3 subunits, the human GABAA receptor β3 homopentamer (PDB: 4COF) crystal structure was used per se with no further modifications. The long cytoplasmic regions of the γ2 and α1 subunits were excluded from modeling as they were absent in the solved GluCl structure, and separate alignments were generated for the TM4 domains. Full-length multiple alignments were submitted for automated comparative protein modeling incorporated in SWISS-MODEL program suite. The resulting subunit models were energy-optimized using GROMOS96 of the Swiss-PdbViewer. Then, pentameric GABAA receptor homology models were generated by combining α1, β3 and γ2 structural models in 2β:2α:1γ stoichiometry and subunit arrangement of β-α-β-α-γ as viewed from the synaptic cleft. Structural conformational changes induced by a single mutated amino acid at the β+ and β− interface of the human β3 subunit were simulated using Rosetta 3.1 of the Rosetta Backrub program suite. Since Rosetta 3.1 does not allow Cysteine substitutions, Cysteine mutations were exchanged with Alanine. Up to twenty of the best-scoring structures were generated for each mutation by choosing parameters recommended by the application as follows: β3(D120N), β3(E180G), and β3(Y302A) at the β3+/α1− interface, and β3(N110D) and β3(F246S) at the α1+/β3− and γ2+/β3− interfaces. All single point mutations were incorporated in the β3 subunit since the full-length alignment between the β3 and β1 displayed high sequence similarity (91.2%). RMS (Root mean square deviation) was calculated between the initial (wt) structures and superimposed simulated (mutated) structures. For each mutation the RMS average over ten lowest energy structures was computed. Chimera 1.7 was used to display conformational changes among neighborhood structural domains.
Results
De novo GABRB3 and GABRB1 mutations identified in patients with LGS and IS were located in conserved structural domains of GABAA receptor β subunits
We found that the de novo GABRB3 and GABRB1 mutations in patients with LGS and IS were located in conserved structural domains of GABAA receptor β subunit that have important functional roles. By analyzing the sequence alignment among the GABR genes (Figure 1A), we found that D120, Y302 and F246 are invariant residues across all GABAA receptor subunits. All five mutations are part of major structural domains (Figure 1B, LGS-associated mutations are shown in orange and IS-associated mutations in green), such as loop A (D120), loop B (β7 sheet, E180), M2–M3 loop (Y302), transmembrane domain 1 (TM1, F246), and the α2 helix (N110), that are involved in the ligand binding-channel gating coupling mechanism and proper assembly of pentameric αβγ GABAA receptors 3, 4, 16, 17.
De novo GABAA receptor β1,3 subunit mutations altered GABA-evoked currents
We determined the functional consequences of LGS- and IS-associated mutations by measuring macroscopic GABA-evoked α1β1,3γ2L currents. In the hom condition GABA-evoked peak current amplitudes were unaltered for cells expressing β3(N110D) subunits, while GABA-evoked currents were significantly reduced from cells expressing β3(D120N), β3(E180G), β3(Y302C) and β1(F246S) subunits (Figure 2A, B). The current densities from cells expressing β3(D120N), β3(E180G) and β3(Y302C) subunits were reduced to ~24%, 1%, and 5% of those containing wt β3 subunits (Figure 2B, Table 1). Under similar hom expression conditions, current densities from cells expressing β1(F246S) subunits had a minor (25%) reduction of peak current densities (Figure 2C, D; Table 1).
Figure 2. The de novo GABAA receptor β subunit mutations found in LGS patients produced substantial reduction of GABA-evoked currents.

(A) Representative GABA current traces obtained following rapid application of 1 mM GABA for 4s to lifted HEK293T cells voltage clamped at −20mV. The current traces from GABAA receptors containing mutant β3 and β1 subunits in hom conditions are compared to their respective wt current traces. (B) Bar graphs showing average peak current densities from cells expressing mutant β subunits in hom and het conditions. Values are expressed as mean ± SEM (See Table 1 for details). One-way ANOVA with Dunnett’s post-test was used to determine significance. * and # represents significant difference compared to the wt and het condition, respectively. */‡ = p < 0.05, **/## = p < 0.001, ***/### = p <0.0001.
Table 1.
Effects LGS- and IS-associated mutations on whole cell GABA-evoked currents and expression levels of GABAA receptor subunits.
| α1β3γ2L (n) | α1β1γ2L (n) | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| wt β3 (8–16) | β3(N110D) (8–11) | β3(D120N) (5–8) | β3(E180G) (5–21) | β3(Y302C) (8–10) | wt β1 (6–12) | β1(F246S) (5–11) | |
| Peak GABA current density (pA/pF), hom | 244 ± 21 | 248 ± 31 | 58.3 ± 8.8*** | 2.8 ± 0.7*** | 12.4 ± 3.7*** | 252 ± 17 | 189 ± 15* |
| Peak GABA current density (pA/pF), het | - | 247 ± 29 | 155 ± 36* | 149 ± 17**### | 108 ± 25***## | - | 233 ± 21 |
| 10–90% rise time (ms), hom | 2.6 ± 0.3 | 5.6 ± 0.7*** | 64.9 ± 17.8*** | 53.3 ± 11.7*** | 107 ± 15*** | 2.9 ± 0.3 | 2.1 ± 0.2 |
| 10–90% rise time (ms), het | - | 3.3 ± 0.2## | 4.4 ± 1.2### | 3.1 ± 0.4### | 5.9 ± 1.2### | - | 1.3 ± 0.2** |
| Desensitization τ (ms), hom | 1530 ± 128 | 1652 ± 104.3 | 1218 ± 262 | 3202 ± 452*** | 2640 ± 755.4 | 1361 ± 107.4 | 1328 ± 140.3 |
| Desensitization τ (ms), het | - | 1477 ± 115 | 1559 ± 320.1 | 2024 ± 285.1 | 1701 ± 424 | - | 1239 ± 264.1 |
| Deactivation τ (ms), hom | 165 ± 24 | 60.1 ± 5.7*** | 49.9 ± 6.5** | ND | 42.5 ± 8.1*** | 220 ± 23.3 | 783 ± 76.6*** |
| Deactivation τ (ms), het | - | 98.7 ± 8.7* | 78.6 ± 11.9* | 93.2 ± 15.4 | 70.6 ± 9.5*** | - | 541 ± 49**# |
| Holding current (pA), hom | −85.8 ± 12 | −6.4 ± 2.1*** | −35.3 ± 17.3 | −466 ± 87*** | −10.8 ± 4.7*** | −66.4 ± 12.1 | −362 ± 75*** |
| Holding current (pA), het | - | −22.8 ± 5.4*** | −31.7 ± 10.4* | −209 ± 69 | −25.8 ± 8.2*** | - | −264.2 ± 16.1 |
| Outward Zn2+ current (pA), hom | +7.2 ± 2.3 | +0.6 ± 0.7 | +0.9 ± 0.6 | +47.5 ± 13.8** | −0.9 ± 1.3* | −3.9 ± 4.6 | +61.3 ± 25.0* |
| Outward Zn2+ current (pA), het | - | +7.92 ± 3.0 | +3.6 ± 1.6 | +15.8 ± 3.9# | +3.9 ± 1.3 | - | +32.3 ± 12.1 |
| % Zn2+ inhibition, hom | 13.2 ± 2.1 | 13.6 ± 2.7 | 13.6 ± 1.3 | 33.7 ± 8** | −9.9 ± 6.0*** | 16.4 ± 2.8 | 16.6 ± 2.8 |
| % Zn2+ inhibition, het | - | 9.9 ± 1.5 | 15.0 ± 1.4 | 12.2 ± 2.7## | 10.8 ± 2.0### | - | 5.8 ± 1.6* |
|
| |||||||
| (15/8) | (12/5) | (12/5) | (15/8) | (11/8) | |||
|
| |||||||
| Surface/Total α1, hom | - | 1.06 ± 0.12/1.26 ± 0.24 | 0.93 ± 0.04/1.00 ± 0.14 | 1.01 ± 0.13/1.10 ± 0.13 | 1.08 ± 0.09/1.30 ± 0.15 | - | 1.02 ± 0.11/1.01 ± 0.15 |
| Surface/Total β1HA/3, hom | - | 1.42 ± 0.08**/1.70 ± 0.17* | 0.98 ± 0.05/1.10 ± 0.11 | 1.25 ± 0.08/1.26 ± 0.17 | 1.059 ± 0.06/1.73 ± 0.21** | - | 0.80 ± 0.07**/1.03 ± 0.34 |
| Surface/Total γ2LHA, hom | - | 0.92 ± 0.06/1.07 ± 0.11 | 0.98 ± 0.07/0.97 ± 0.17 | 1.01± 0.16/1.33 ± 0.45 | 1.04 ± 0.07/1.15 ± 0.14 | - | 1.07 ± 0.11/1.10 ± 0.30 |
Hom and het denote homozygous and heterozygous expression conditions. Values represent mean ± S.E.M.
indicate p < 0.05, p < 0.01, and p<0.001 statistically different from wt, and
indicate p < 0.05, p < 0.01, and p<0.001 for statistical differences between homozygous and heterozygous conditions (one-way ANOVA with Dunnett’s multiple comparisons test). ND, non-determinate. Surface and total expression levels were normalized against the wt condition.
Since the LGS and IS patients were heterozygous for the de novo GABRB3 and GABRB1 mutations, we also studied α1β1,3γ2L receptors containing these mutations in the in vitro het expression condition. In the het condition, current densities from cells expressing β3(N110D) subunits did not change; however, current densities from cells expressing β3(D120N), β3(E180G) and β3(Y302C) subunits were significantly reduced to 44 – 63% of wt condition (het β3(D120N) = 64%, het β3(E180G) = 61%, het β3(Y302C) = 44% of wt current densities, respectively; Figure 2A, B; Table 1). The β1(F246S) subunit mutation also had no effects on current density in the het condition (Figure 2A, B; Table 1). Despite our results demonstrating minor or no change in current densities from cells expressing β3(N110D) and β1(F246S) subunits, the severity of the IS phenotype in patients carrying the de novo GABRB3(N110D) and GABRB1(F246S) mutations suggests alternate mechanisms might be responsible for impairing receptor function.
De novo GABAA receptor β subunit mutations altered the kinetic properties of GABAA receptor currents
To gain insights into whether the β subunit mutations altered other properties of macroscopic currents, we examined their activation, desensitization and deactivation rates, properties that shape inhibitory postsynaptic currents (IPSCs). We found that all β3 subunit mutations significantly slowed current activation (longer 10–90% rise time) in the hom condition, but not in the het condition (Figure 3A, left panels; Table 1). In contrast, currents from cells expressing the β1(F246S) subunit had reduced rise time in the het condition but not in the hom condition (Figure 3A, right panels; Table 1). Similar effects on rise times were seen with brief (10 ms) application of GABA for currents from β3(N110D) subunit-containing receptors, while the current rise times were slower for β1(F246S) subunit-containing receptors (Figure 3B).
Figure 3. The de novo GABAA receptor β subunit mutations found in IS patients altered GABAA receptor current kinetic properties.

Representative traces showing rise times of GABA-evoked currents produced by 4 s (A, top panel) or 10 ms (B) applications of 1 mM GABA to wt receptors or receptors containing β3(N110D) or β1(F246S) subunits expressed in the hom condition. Bar graphs in the bottom panels of (A) show average rise times from the cells expressing wt GABAA receptors or receptors containing β3(N110D) or β1(F246S) subunits expressed in the hom condition. Representative current traces showing deactivation or current relaxation at the end of 4 s (C) or 10 ms (D) GABA application (1 mM) to wt receptors or receptors containing the β3(N110D) or β1(F246S) subunits. Bar graphs in the bottom panel of (C) show average current deactivation time constants from the cells expressing GABAA receptors containing β3(N110D) or β1(F246S) subunits expressed in the hom condition. All traces were normalized for clarity. Values are expressed as mean ± SEM (See Table 1 for details). One-way ANOVA with Dunnett’s post-test was used to determine significance. * and # represents significant difference compared to the wt and het condition, respectively. */# = p < 0.05, **/## = p < 0.001, ***/### = p < 0.0001.
None of the mutations in the hom or het expression conditions altered current desensitization, except for the β(E180G) subunit mutation, which produced strong desensitization of currents when expressed only in the hom condition (Table 1). Further, we determined current deactivation by measuring current decay after termination of GABA application. In hom and het conditions, the β3(N110D, D120N, Y302C) subunit mutations increased the current deactivation rate (reduced weighted deactivation rate constant; Figure 3C left panels; Table 1). For β3(E180G) subunit-containing GABAA receptors, removal of GABA led to a positive overshoot of the current from the baseline, which prevented meaningfully fitting exponential functions to determine deactivation rate constants. In the het condition, the β3(E180G) subunit mutation did not affect current deactivation (Table 1). However, the current kinetic changes were hard to interpret for β3(D120N, E180G and Y302C) subunit mutations due to the small current size and seemed to be less significant than the substantial reduction (76.1–98.9%) of peak GABA-evoked currents. In contrast to the β3 mutations, the β1(F246S) subunit mutation reduced the deactivation rate (increased weighted deactivation rate constant) compared to wt currents (Figure 3C, right panels; Table 1). Similar results were obtained for current deactivation following rapid application of brief 10 ms GABA pulses (Figure 3D; a more accurate method to determine current deactivation).
De novo GABAA receptor β3 subunit mutations did not reduce surface levels of α, β or γ subunits
Since the LGS-associated mutations reduced GABA-evoked currents, we determined if this resulted from reduced expression of β3 subunits leading to loss of surface GABAA receptors. Surprisingly, none of the mutations reduced total or surface levels of α1, β3 or γ2LHA subunits in the hom condition (Figure 4A, B, C). Similar results were obtained for the IS-associated GABRB3(N110D) mutation, suggesting that none of the β3 subunit mutations impaired α1β3γ2L GABAA receptor synthesis or trafficking. Given that we did not observe reduced total or surface levels for α1, β3 or γ2LHA subunits in the hom condition, we did not extend these experiments to the het condition.
Figure 4. The β subunit mutations did not reduce surface and total levels of GABAA receptor subunits.

Flow cytometry was used to determine surface (A, B, D) and total (C, E) levels of α1, β1HA/ β3 and γ2LHA subunits in HEK293T cells. (A, D left most panel) Representative fluorescence intensity (FI) histograms showing the surface β3/β1HA subunit levels from cells expressing α1mutant β3/β1HAγ2L subunits (shaded), α1wt β3/β1HAγ2L subunits (unfilled with solid black line) and empty vector (unfilled with black line). The bar graphs represent FI of the Alexa 674 fluorophore for each condition normalized to the intensity of the wt condition (Relative FI). Surface (B,D) and total (C,E) relative FI levels of α1, β3/β1HA and γ2LHA subunits in cells expressing only α1, β3/β1HA or γ2LHA subunits (used as antibody controls), as well as co-expressing α1, γ2LHA, wt or mutant β3/β1HA subunits (hom condition). In the hom condition the IS-associated β3(N110D) and LGS-associated β3(E180G) subunit mutant subunits had 42 % and 25 % higher surface levels, respectively, than β3 subunits in the wt condition. Values are expressed as mean ± SEM. One-way ANOVA with Dunnett’s post-test was used to determine significance. * represents significant difference compared to the wt condition, * = p<0.05, ** = p<0.001, *** = p<0.0001. (A) and (D) share same legends.
Also surprisingly, the IS-associated β3(N110D) subunit mutation significantly increased surface β3 subunit levels (142.3 ± 0.08 % of wt condition; Figure 4A, B), without increasing α1 or γ2LHA subunit levels (Figure 4B; Table 1). The surface levels of β3(E180G) subunits were increased by 24.9 ± 0.08 % in the hom condition but were not significantly higher than the wt condition. Moreover, there were significant increases in the total levels of both β3(Y302C) and β3(N110D) subunits (Figure 4C, middle panel). It is noteworthy that even in the absence of α or γ subunits, wt β3 subunits were expressed on the cell surface at levels similar to that of wt β3 subunits in the α1β3γ2L receptor condition (Figure 4B, middle panel), suggesting the presence of homomeric receptors as previously described 18, 19. When β3(E180G) and β3(N110D) subunits were expressed alone, they had 37.3% and 17.6% higher surface levels as compared to when wt β3 subunits were expressed alone (data not shown). Thus, β3(E180G) and β3(N110D) subunit mutations may favor either the formation of homomeric β3 receptors or GABAA receptors with a different subunit stoichiometry than wt receptors.
In addition we co-expressed α1, γ2LHA and either wt β1 or mutant β1(F246S) HA-tagged (β1HA, β1(F246S) HA) subunits in HEK293T cells (hom condition). The β1(F246S)HA subunit expression levels were slightly, but significantly, reduced compared to wt β1HA levels, without any alteration in α1 or γ2LHA levels (Figure 4D). Total expression levels of α1, β1HA or γ2LHA were not changed (Figure 4E). Reduced surface β1(F246S) levels without a reduction in the total levels suggest that mutant subunits affected the assembly and/or trafficking of GABAA heteromeric or β1 homomeric receptors but not the biogenesis of the mutant subunits.
LGS-associated mutations reduced GABA-activated currents by reducing GABA potency or efficacy
Our results demonstrated that the LGS-associated GABRB3(D120N, E180G, Y302C) mutations reduced GABA-evoked current amplitudes, slowed activation and accelerated deactivation. Similar decreased peak currents and kinetic changes have been described even for wt GABAA receptors when sub-saturating GABA concentrations were used 20, 21. Additionally, the β3(D120N, E180G, Y302C) subunit mutations were located in loop A and loop B (β7 sheet) of the GABA binding pocket and M2–M3 loop (involved in the ligand binding-channel gating coupling mechanism), respectively (Figure 1B, residues in orange). Therefore, it is likely that these mutations disrupted coupling of GABA binding to channel gating leading to reduced GABA potency and/or efficacy.
To test this possibility we measured GABA-evoked current responses to a supersaturating concentration of 10 mM GABA. We found that cells expressing β3(D120N) subunit-containing receptors had current amplitudes evoked by 10 mM GABA that were similar to those evoked by 1 mM GABA from cells expressing wt receptors (Figure 5A, left panel and 5B). However, GABA-evoked currents from β3(E180G) and β3(Y302C) subunit-containing receptors only minimally recovered when 10 mM GABA was applied (Figure 5A, middle and right panels, and 5B). For receptors containing β3(D120N) subunits, the current response increased from 23.9 ± 3.6% of the wt current with 1 mM GABA to 83.6 ± 7.3% with 10 mM GABA. These results suggest a reduction of GABA potency with no changes in GABA efficacy. In contrast, for both β3(E180G) and β3(Y302C) subunit mutations, the maximal fractional response at 1 and 10 mM GABA was not very different (1.2 ± 0.3% and 5.1 ± 1.5% of wt in 10 mM GABA, respectively), suggesting a major reduction in GABA efficacy.
Figure 5. GABAA receptors containing mutant βsubunits identified in LGS patients reduced GABA potency or efficacy.
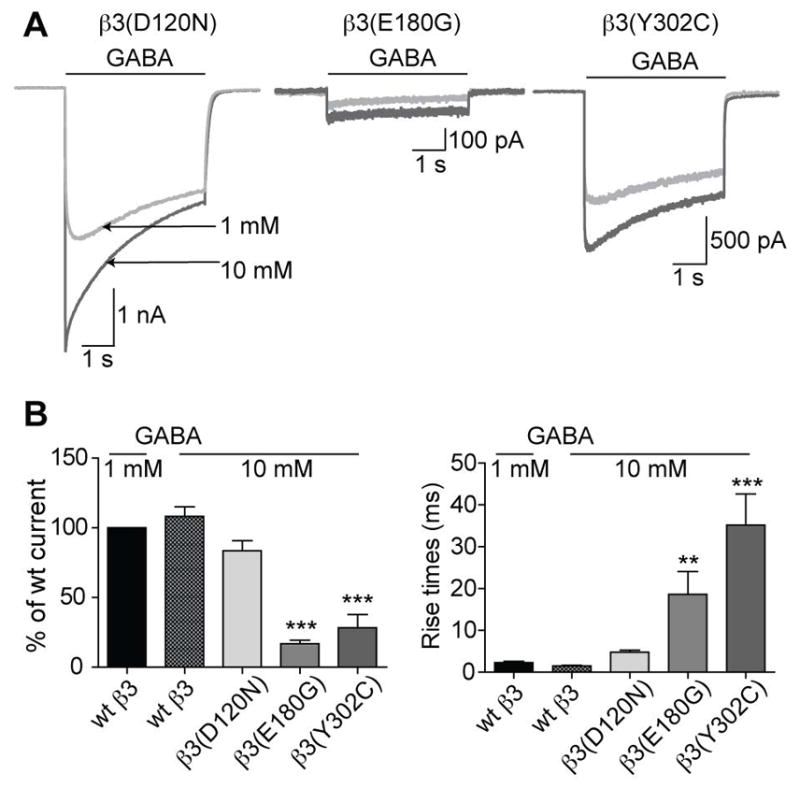
(A) Representative whole cell current responses following GABA application from cells expressing wt or mutant receptors (hom condition). The current traces with 1 mM GABA application (light grey) were overlaid with current traces with 10 mM GABA application (dark grey). (B, left) Bar graphs show average peak current responses to 10 mM GABA application as % of wt response to 1 mM GABA. 10 mM GABA-evoked currents from β3(D120N), β3(E180G), and β3(Y302C) subunit-containing receptors were 83.6 ± 7.3%, 16.9 ± 2.5%, and 28.3 ± 9.6% of the wt current, respectively, with 1 mM GABA. (B, right) Bar graphs show the average rise times of GABA-evoked currents to 1 and 10 mM GABA application from cells with wt or hom expression. Rise times for β3(D120N), β3(E180G), and (β3(Y302C) subunit-containing receptors were 4.8 ± 0.5 ms, 18.6 ± 5.5 ms, and 35.2 ± 7.4 ms, respectively. Values were expressed as mean ± SEM. One-way ANOVA with Dunnett’s post-test was used to determine significance. * represents significant difference compared to the wt condition with 1 mM GABA application, * = p < 0.05, ** = p < 0.001, *** = p < 0.0001.
Additionally, the rise times for the β3(D120N) subunit-containing receptors were similar to those of the wt condition with 1 mM GABA application, while for β3(E180G) and β3(Y302C) subunit-containing receptors rise times were significantly longer (Figure 5B right panel). Although, with 10 mM GABA current activation was ~14-fold, ~3-fold, and ~3-fold faster from cells expressing the β3(D120N), β3(E180G), and β3(Y302C) subunits, respectively, (Figure 5B, right panel) than those activated by 1 mM GABA (Table 1). These results further support the finding that the β3(D120N) subunit mutation reduced GABA potency, but the β3(E180G) and β3(Y302C) subunit mutations reduced GABA efficacy.
β subunit mutations impaired the single channel gating properties of GABAA receptors
Gating properties of GABAA receptors were determined by analyzing microscopic single channel currents of wt and mutant α1β1/3γ2L receptors. In response to 1 mM GABA, wt α1β3γ2L and α1β1γ2L receptors opened into brief bursts and frequent prolonged (> 500 ms) clusters of bursts to a main conductance level of ~26 pS 22 (Figures 6A, 7A, D; Tables 2 and 3). Open time distributions were fitted best by three weighted (ao1, ao2 and ao3) exponential functions with three open time constants (τo1, τo2 and τo3) (Figures 6B, 7B, E, left top panels), suggesting openings to at least three different open states.
Figure 6. Single channel properties of GABAA receptors with LGS-associated β subunit de novo mutations.

(A) Representative single-channel current traces from cell attached patches expressing wt or mutant GABAA receptors (hom condition). (B) Mean open time (left panels) and burst duration (right panels) histograms for wt and mutant receptors were fitted to three and two exponential functions, respectively. The open and burst duration histograms are sums of multiple exponential functions. Average time for each exponential function is marked with a square. (C) Bar graphs summarize the effects of wt and LGS-associated β3 subunit mutations on the kinetic properties of the receptor. Values represent mean ± S.E.M. Statistical differences were determined using One-way ANOVA with Dunnett’s multiple comparisons test (see Table 2 for details). **, *** and **** indicate p < 0.01, p<0.001 and p < 0.0001, respectively.
Figure 7. Single channel properties of GABAA receptors with β subunit de novo mutations from IS patients.

(A, D) Representative single-channel current traces from cell attached patches expressing wt and mutant GABAA receptors (hom condition). (B, E) Open time (right panels) and burst duration (left panels) histograms for wt and mutant receptors were fitted to three and two exponential functions, respectively. (C, F) Bar graphs summarize the effects of wt and β1(F246S) mutation on the kinetic properties of the receptor. Values represent mean ± S.E.M. Statistical differences were determined using unpaired t-test (see Table 3 for details). **, *** and **** indicate p < 0.01, p<0.001 and p < 0.0001, respectively.
Table 2.
Single channel properties of the de novo GABRB3 mutations associated with Lennox-Gastaut Syndrome.
| α1β3γ2L (n)
|
||||
|---|---|---|---|---|
| wt β3 (6) | β3(D120N) (5) | β3(E180G) (3) | β3(Y302C) (3) | |
| Channel conductance (pS) | 24.79 ± 1.62 | 21.77 ± 2.18 | 22.55 ± 2.39 | 18.88 ± 2.47** |
| Mean open time (ms) | 6.60 ± 0.73 | 7.17 ± 0.04 | 2.56 ± 0.08*** | 6.58 ± 0.23 |
| Opening frequency (S−1) | 49 ± 2 | 21 ± 3** | 64 ± 8 | 12 ± 1*** |
| Open probability (Po) | 0.62 ± 0.05 | 0.12 ± 0.03**** | 0.23 ± 0.05*** | 0.035 ± 0.001**** |
| Open time constants: | ||||
| τo1 (ms) | 3.18 ± 0.28 | 4.37 ±0.11* | 1.90 ± 0.06* | 3.75 ±0.26 |
| τo2 (ms) | 9.77 ± 0.84 | 9.94 ± 0.84 | 4.97 ± 1.55** | 9.99 ± 0.29 |
| τo3 (ms) | 20.8 ± 3.4 | 21.1 ± 1.6 | 9.98 ± 1.92 | 16.4 ± 3.1 |
| ao1 (%) | 67 ± 1 | 59 ± 8 | 88 ± 5* | 63 ± 2 |
| ao2 (%) | 28 ± 2 | 37 ± 8 | 5 ± 2* | 30 ± 5 |
| ao3 (%) | 5 ± 2 | 5 ± 0.3 | 7 ± 3 | 8 ± 4 |
| Burst duration (ms) | 20.13 ± 0.72 | 13.11 ± 0.86*** | 5.78 ± 0.38**** | 10.04 ± 0.52**** |
| Openings/burst | 3.12 ± 0.19 | 1.64 ± 0.09**** | 1.65 ± 0.06**** | 1.40 ± 0.02**** |
| Burst time constants: | ||||
| τ1(ms) | 2.54 ± 0.20 | 6.67 ± 0.36*** | 2.28 ±0.13 | 4.40 ±0.49** |
| τ2(ms) | 35.4 ± 2.9 | 23.5 ± 3.7* | 10.7 ± 0.8*** | 14.2 ± 0.1*** |
| a1 (%) | 37 ± 6 | 61 ± 4* | 59 ± 8* | 43 ± 4 |
| a2 (%) | 63 ± 6 | 39 ± 4* | 41 ± 8* | 57 ±4 |
Values represent mean ± S.E.M.
indicate p < 0.05, p < 0.01, p<0.001 and p < 0.0001 (one-way ANOVA with Dunnett’s multiple comparisons test) statistically different from wt, respectively.
Table 3.
Single channel properties of the de novo GABRB mutations associated with infantile spasms.
| α1β3γ2L (n) | α1β1γ2L (n) | |||
|---|---|---|---|---|
|
| ||||
| wt β3 (6) | β3(N110D) (5) | wt β1(6) | β1(F246S) (3) | |
| Channel conductance (pS) | 24.79 ± 1.62 | 23.66 ± 0.97 | 25.72 ± 1.09 | 22.15 ± 0.75* |
| Mean open time (ms) | 6.60 ± 0.73 | 7.00 ± 0.36 | 4.30 ± 0.78 | 16.13 ± 0.14*** |
| Opening frequency (S−1) | 49 ± 2 | 48 ± 4 | 30 ± 3 | 14 ± 1** |
| Open probability (Po) | 0.62 ± 0.05 | 0.11 ±0.03**** | 0.48 ±0.03 | 0.55 ±0.02 |
| Open time constants: | ||||
| τo1 (ms) | 3.18 ± 0.28 | 3.98 ± 0.44 | 2.39 ± 0.14 | 4.49 ± 0.05*** |
| τo2 (ms) | 9.77 ± 0.84 | 8.92 ± 1.17 | 8.33 ±1.01 | 16.6 ± 0.3** |
| τo3 (ms) | 20.8 ± 3.4 | 21.6 ± 5.1 | 17.4 ± 5.3 | 47.9 ± 3.5** |
| ao1 (%) | 67 ± 1 | 66 ± 8 | 72 ± 6 | 19 ± 3*** |
| ao2 (%) | 28 ± 2 | 29 ± 8 | 25 ± 5 | 75 ± 4*** |
| ao3 (%) | 5 ± 2 | 5 ± 1 | 6 ± 2 | 6 ± 1 |
| Burst duration (ms) | 20.13 ± 0.72 | 14.34 ± 1.17** | 13.33 ± 2.38 | 42.95 ± 2.61** |
| Openings/burst | 3.12 ± 0.19 | 2.21 ± 0.16* | 2.42 ± 0.11 | 2.49 ± 0.13 |
| Burst time constants: | ||||
| τ1(ms) | 2.54 ± 0.20 | 4.85 ± 0.60* | 2.42 ± 0.24 | 5.62 ± 1.04* |
| τ2(ms) | 35.4 ± 2.9 | 24.6 ± 3.4* | 21.2 ± 3.1 | 52.0 ± 1.6*** |
| a1 (%) | 37 ± 6 | 55 ± 3* | 54 ± 3 | 20 ± 3*** |
| a2 (%) | 63 ± 6 | 45 ± 3* | 49 ± 2 | 80 ± 3*** |
Values represent mean ± S.E.M.
indicate p < 0.05, p < 0.01, p<0.001 and p < 0.0001 (unpaired t-test) statistically different from wt, respectively.
The mutations that reduced GABA-evoked currents (GABRB3(D120N, E180G, Y302C)) reduced channel Po compared to wt receptors (Figure 6A, C). Receptors containing the GABRB3(D120N, Y302C) mutations had reduced single channel opening frequency without affecting the single channel mean open time (Figure 6C; Table 2). Consistent with this, there were minimal differences among the three open time distributions for β3(D120N) and β3(Y302C) containing receptors (Figure 6B; Table 2). In contrast, receptors containing the GABRB3(E180G) mutation had unaltered single channel opening frequency but had reduced single channel mean open time (Figure 6B; Table 2), resulting in reduced open time constants and a significant increase (88 ± 5% of the relative proportion (ao1)) in the relative occurrence of the shortest open state (τo1; Table 2). Additionally, the β3(Y302C) subunit mutation reduced single channel conductance (~21 pS; Table 2) while β3(D120N) and β3(E180G) subunit mutations did not. The GABRB3 mutations caused additional defects in channel gating properties of bursts. All three mutations reduced mean burst durations and number of openings per burst (Figure 6C; Table 2). In general, receptors containing the mutant subunits had increased duration and relative frequency of short bursts, and decreased duration and relative frequency of long bursts (Figure 6B, right panel; Table 2).
The β subunit mutations that produced minor or no loss of GABA-evoked currents also altered single channel properties (Figure 7). The GABRB3(N110D) mutation also reduced channel Po but without changing the single channel opening frequency or mean open time (Figure 7C; Table 3). Consistent with this, the open time constants and relative areas were unaffected. Similar to the GABRB3(D120N) mutation, the β3(N110D) subunit mutation reduced channel Po mainly by reducing burst duration (to 70% of control), due to a shift to briefer bursts (Figure 7B, C; Table 3). Reduced Po, number of openings per burst and burst duration accounted for the slow rise time and fast current deactivation from β3(N110D) containing receptors. Unlike the LGS-associated mutations, the D110 residue is located in the β subunit interface far from the GABA binding site, and thus did not affect the whole cell peak currents. In contrast, the IS-associated β1(F246S) subunit mutation did not alter channel Po despite decreasing opening frequency. Additionally, for the β1(F246S) subunit mutation, a gain of function resulted in increased mean open time (~3.75 fold) and burst duration (~3.2 fold) from prolonged openings. This is consistent with a significant shift in the distribution of the longest open states (Figure 7D, E; Table 3) and may account for the prolonged macroscopic current deactivation. Furthermore, reduced conductance level of ~21 pS accounted for a small reduction of whole cell GABA-evoked currents in the β1(F246S) hom condition.
De novo β3(E180G) and β1(F246S) subunit mutations produced spontaneous current
Both LGS-associated GABRB3(E180G) and IS-associated GABRB1(F246S) mutations significantly increased (~5 fold ) holding currents recorded from α1βγ2L receptors, suggesting spontaneous channel openings in hom, but not het, conditions (Table 1). In general, holding currents for receptors containing β3(N110D, Y302C) subunits were reduced, and for receptors containing β3(D120N) subunits were similar to those of wt receptors. We examined whether the increased holding currents observed for β3(E180G) or β1(F246S) subunit-containing receptors could be due to formation of homomeric β3 or β1 GABAA receptor channels. Zinc blocks spontaneous GABAA receptor “leak” current leading to a positive shift in the baseline current from cells expressing only β1 or β3 subunits (subunits known to form homomeric GABAA receptors) 18, 19, 23. Surprisingly with hom expression of β3(E180G) or β1(F246S) subunits about, 10–15% of the holding current was blocked by 10 μM zinc (Figure 8A, B; Table 1). The holding current was blocked to similar extent by 100 μM zinc (data not shown). These results are consistent with increased tendency of β3(E180G) subunits to assemble and traffick to the cell surface without an increase in surface levels of α1 and γ2L subunits, suggesting that β3(E180G) subunits form zinc-sensitive homomeric β3 subunit receptors. In contrast, the IS-associated β1(F246S) subunit mutation produced a slight but significant decrease in surface levels of β1(F246S) subunits (Figure 4D), suggesting that the β1(F246S) subunit-containing heteropentameric GABAA receptors opened spontaneously (as previously reported 24).
Figure 8. Both LGS-associated GABRB3(E180G) and IS-associated GABRB1(F246S) de novo mutations produced spontaneously gated GABAA receptors.

(A) Representative traces from whole cell recordings showing a shift in the baseline current (seen as an outward current) with 10 μM Zn2+ application from cells expressing GABAA receptors with β3(E180G) and β1(F246S) subunits (grey traces), but minimal or absent in cells expressing wt receptors (black traces). (B) Bar graphs are presented showing average outward current responses to 10 μM Zn2+ application. Representative spontaneous single-channel current traces recorded from cells expressing wt β3 or β3(E180G) (C), or wt β1 or β1(F246S) (E) subunit-containing GABAA receptors in absence (upper panels) and presence (bottom panels) of 100 μM Zn2+. (D, F) Bar graphs show single-channel amplitude and Po of wt (black bars) and spontaneously activated mutant (gray bars) receptors. For wt β3 subunit-containing receptors the low and high conductance openings were 12.5 pS (1.1 ± 0.07 pA, n = 7) and 21 pS (1.8 ± 0.10 pA, n = 4) with Po of 0.13 ± 0.02 (n=7) and 0.06 ± 0.01 (n = 4), respectively. The β3(E180G) subunit mutation significantly increased the Po of low conductance openings (0.34 ± 0.05, 1.1 ± 0.08 pA, n = 10), without altering high conductance openings (0.10 ± 0.03, 1.7 ± 0.03 pA, n = 4, p > 0.05). The wt β1 subunit-containing receptors had conductance level 0.99 ± 0.06 pA, (n = 5) with Po of 0.03 ± 0.01 (n = 5). The β1(F246S) subunit mutation increased Po to 0.13 ± 0.02 (n = 8) but did not alter single channel conductance (1.1 ± 0.04 pA n = 8, p > 0.05). Values represent mean ± S.E.M. Two-way ANOVA with Tukey’s multiple comparisons (D) test or unpaired t-test (F) was used to determine statistical significance. ** indicate p < 0.01.
De novo β3(E180G) and β1(F246S) subunit mutations resulted in spontaneous opening of GABAA receptor channels
We further investigated the mechanisms by which these de novo GABRB mutations increased holding currents by analyzing their spontaneous single-channel openings in the absence of GABA (Figure 8C–F). We found that even cells expressing wt α1β3γ2L receptors displayed spontaneous openings with two conductance levels of ~12.5 pS (low) and ~21 pS (high) (Figure 8C, D, upper panels), which were different from the main conductance level evoked by GABA of ~26 pS for wt α1β3γ2L receptors. The spontaneous wt α1β3γ2L openings occurred as frequent isolated single openings and brief bursts, but no prolonged clusters of bursts were evident. There were no differences in the Po between the two spontaneously conducting states (Figure 8D, lower panel). In contrast, GABAA receptors with β3(E180G) subunits significantly increased spontaneous activation of the channel, with more brief bursts and frequent prolonged clusters of bursts (>1 s) that opened to two conductance levels resembling to those of GABA-evoked currents from wt receptors (Figure 8C, lower panel and 8D, upper panel). Moreover, the increased spontaneous bursting was the result of an increased Po (up to ~3-fold) of the low-conductance openings (Figure 8D, lower panel).
Similar to wt α1β3γ2L receptors, wt α1β1γ2L receptors displayed spontaneous single channel currents (Figure 8E, upper panel). However, the spontaneous openings of wt α1β1γ2L receptors were very brief with isolated low-conductance openings (Figure 8E, upper panel and F, left panel) and with a Po that was only ~25% of that of wt α1β3γ2 receptors (Figure 8C–F). GABAA receptors containing the β1(F246S) subunit mutation increased spontaneous Po by 4-fold (Figure 8F, right panel) but did not alter single-channel conductance. Further, spontaneous currents from wt and mutant β3(E180G) or β1(F246S) subunit-containing receptors were inhibited by 100 μM zinc (Figure 8C, E). These results are consistent with the holding currents observed during whole cell recordings from receptors containing mutant subunits.
GABAA receptor containing LGS-associated GABRB3(D120N, Y302C) and IS-associated GABRB3(N110D) mutations also displayed spontaneous openings, although with smaller conductance than wt receptors. Thus, whereas the β3(D120N) subunit-containing GABAA receptors displayed low-conductance spontaneous openings that occurred with a similar Po compared to wt β3 subunit-containing receptors (1.2 ± 0.12 pA, n = 4 and 0.08 ± 0.01, n = 4), receptors with β3(Y302C) or β3(N110D) subunits displayed rare, brief, low-conductance spontaneous openings (1.0 ± 0.18 pA, n = 3 and 0.001 ± 0.003, n = 3; 1.1 ± 0.01 pA, n = 3 and 0.001 ± 0.002, n = 3, respectively). The spontaneous activation of GABAA receptors with β3 and β1 mutant subunits was blocked by picrotoxin (100 μM) in a similar fashion to that for receptors containing wt β3 or β1 subunits (data not shown). Overall de novo GABRB3 and GABRB1 mutations altered the spontaneous activation of GABAA receptors.
De novo β subunit mutations rearrange conserved structural domains related to GABAA receptor function
To understand the structural changes induced by the GABRB mutations, we generated wt and mutant pentameric αβγ GABAA receptor simulations (Figure 1B) using solved structures of both the C. Elegans GluCl channel and the human GABAA receptor β3 subunit homopentamer as templates (see Materials and Methods for details). We computed rearrangements of the subunit’s secondary structure and side chain conformational changes by computing the root mean squared (RMS) deviation between wt and mutant receptors (Figure 9). We evaluated the effects of β subunit mutations based on their location. The major structural changes induced by the LGS-associated GABRB3 mutations occurred at the interface between the principal (+) side of the β3 subunit and the complementary (−) side of the α1 subunit (the β+/α− interface) (Figure 9A), outlining important domains within the ligand-binding pocket. In contrast, the IS-associated mutations occurred at two other interfaces, between the (+) side of γ and α subunits and the (−) side of the β subunit (the γ+/β−, and α+/β− interfaces, respectively), in close contact with the ligand binding-channel gating coupling zone and structural assembly motifs 16, 17.
Figure 9. De novo GABRB mutations induced a wave of structural rearrangements in conserved structural domains important for GABAA receptor function.

(A) Extracellular view of the N-terminal domain in a structural model of pentameric αβγ GABAA receptor (as seen from the synaptic cleft) displaying LGS- (in orange) and IS-associated (in green) GABRB mutations on β (blue ribbons) subunits. α and γ subunits are represented as gray and yellow ribbons, respectively. The principal (+) and complementary (−) interfaces of each subunit are shown. Bottom panel lists the location of the mutations in their respective interfaces. (B and C) Enlarged views of the domains that had structural rearrangements caused by the LGS-associated β3(D120N, E180G, Y302C) and IS-associated β3(N110D) and β1(F246S) subunit mutations. The perturbations of the secondary structures that differ among the wt (in gray) and mutant (in rainbow) structures are indicated by solid black lines (Left panels). Box plots show perturbations (as root mean square deviation (RMS)) caused by the mutations in the sidechain residues that are propagated through β-sheets, loops and TM helices (right panels). RMS values for up to 10 simulations are represented as interleaved box and whiskers plots (25–75% percentile, median, and minimum and maximum). The secondary structure containing the mutation is highlighted in red.
Perturbations of the secondary structure (mutation-associated alternative ribbon in rainbow when RMS > 0.03 Å) and side chain residues (box plots) through neighborhood structural domains at the β+/α−, γ+/β−, and α+/β− interfaces were predicted for both LGS- and IS-associated mutations (Figure 9B, C). It is noteworthy that both LGS- and IS-associated GABRB mutations caused local effects (intra-subunit) confined to structural domains of the subunit, and global effects (inter-subunit) propagated to the nearest subunit through rearrangements of nearby residues and structural domains. Thus, at the β+/α− interface, the LGS-associated β3(D120N) and β3(E180G) subunit mutations induced mainly structural perturbations in loops A, B and C of the β3 subunit, which are crucial domains within the GABA binding pocket. The β3(Y302C) subunit mutation lies in the M2–M3 loop, and disrupted the Cys loop, β1–β2 loop and M2–M3 loop, which are part of the ligand binding-channel gating coupling zone of the receptor (Figure 9B). Thus it is not surprising that the LGS-associated mutations located at the β+/α− interface within the binding-coupling pathway mainly reduced whole cell GABA-evoked currents by decreasing the functional response to GABA.
Similar to the β3(Y302C) subunit mutation, the β1(F246S) subunit mutation caused structural rearrangements mainly restricted to the coupling zone domains and propagated to the TM domains at both γ+/β− and α+/β− interfaces (Figure 9C). The β3(N110D) subunit mutation caused structural changes similar to those caused by the β3(D120N) subunit mutation, but in the principal (+) side of both α and γ subunits. Interestingly only at the γ+/β− interface, the β3(N110D) subunit mutation predicted changes that were extended across the α−β1 loop of the γ2 subunit, a motif previously established to impair receptor gating and βγ subunit interaction 15, 16, 25. The β3(N110D) subunit mutation lies in the inner β3-sheet and it seems unlikely that being on the opposite side of the ligand binding-channel gating coupling interface would reduce the gating of the channel, but similar results were found after glycine insertions in the inner β4-β5 sheets at the β3− interface with decreased GABAA receptor activation 26. In line with this, IS-associated mutations mainly affected the kinetic properties of the channel and altered similar domains at the homologous γ+/β− and α+/β− interfaces required for receptor expression and function. Thus all five de novo GABRB mutations induced a wave of structural rearrangements of conserved domains important for translating ligand binding to channel gating of GABAA receptors 3, 4.
Discussion
Both LGS and IS are severe EEs with early onset and developmental delays, although the temporal course and semiology of the seizures are different. By analysis of triads with LGS and IS, three GABRB3 mutations were associated with LGS and one GABRB1 and one GABRB3 mutations were associated with IS 2. In this study we characterized the pathophysiological effects of these mutations in transfected cells. Our functional studies provide strong evidence that while none of the mutations reduced surface expression of GABAA receptors, all five GABRB mutations associated with the EEs LGS and IS disrupt GABAA receptor functions. Surprisingly our data also revealed two different modes of action of LGS- and IS-associated mutations to impair GABAA receptor functions, consistent with their suggested contribution to different EEs.
The most consequential actions of LGS-associated mutations were reductions of peak GABA-evoked whole cell currents with reduced GABA potency or efficacy. The current reduction resulted generally from disruption of the GABA binding site (β3(D120N, E180G)) and binding-channel gating coupling domain (β3(Y302C)), which reduced single channel currents by reduction of single channel Po, open frequency, burst duration and openings per burst. These changes would reduce amplitudes of inhibitory post synaptic currents (IPSCs).
Conversely, the major effects of IS-associated mutations were altered rise time and deactivation of GABA-evoked whole cell currents. The GABRB3(N110D) mutation reduced single channel Po resulting from decreased single channel burst duration and openings per burst. In contrast, the GABRB1(F246S) mutation did not change Po despite increasing channel mean open time and burst duration likely due to a decrease in opening frequency, but did decrease single channel conductance. These single channel deficits accounted for both whole cell current rise time and deactivation changes produced by the IS-associated mutations. The slow rise times and fast deactivation of β3(N110D) subunit-containing GABAA receptors could lead to brief IPSCs and reduced charge transfer. In contrast, faster rise time and slow current deactivation due to the β1(F246S) subunit mutation would increase the IPSC duration and the charge transfer during the first IPSC, but reduce charge transfer during subsequent IPSCs by reducing the number of unbound GABAA receptors, especially during high frequency neuronal firing. Counter intuitively, both faster and slower deactivation would result in the net loss of GABAergic inhibition, as previously reported for γ2(K289M) and γ2(L313S/L9′S) subunit mutations, respectively 14, 27. Despite these striking differences in GABAA receptor currents among these five LGS- and IS-associated mutations, it is very likely that additional GABR mutations associated with EEs (with highly heterogeneous phenotypes) may have overlapping functional deficits. Moreover, about 20–50% of children with IS progress to LGS and share common therapies 28. Patients with GABRB3(D120N) and GABRB3(E180G) mutations had IS as the initial seizure type which progressed to LGS (trio id jw and jr respectively; supplementary Table 13 of Allen AS et al. 2). Thus the contributions of these mutations to the epilepsy phenotype are difficult to assess in light of in-vitro data alone.
Both β3 and β1 subunits are widely expressed in the developing and adult brain. The β3 subunits are abundant during early development, whereas expression of β1 subunits begins after birth and undergoes down-regulation until reaching stable levels in mature neurons 10, 11. Both β3 and β1 subunits are highly expressed in circuits involved in seizure generation such as cortex, hippocampus, and thalamic reticular nucleus 10, 29, where they mediate phasic and tonic inhibition. Therefore, the loss of or altered depolarizing drive via mutated GABAA receptors would hamper formation of appropriate neuronal circuits during critical periods of central nervous system development. Based on the effects we observed on GABA-evoked currents it could be speculated that the IS-associated mutations could have more prominent effects on phasic inhibition while the LGS-associated mutations could affect both phasic and tonic inhibition.
Our results also demonstrated a structure-dysfunction correlation with the location of the mutation in the receptor. The LGS-associated GABRB3 mutations are at the β+/α− interface, which is directly coupled with the ligand binding-channel gating pathway of the receptor 30, 31. These mutations could be more disruptive to channel function than the IS-associated mutations at the α+/β− and γ+/β− interfaces (that are indirectly coupled by rearrangements throughout the β-sheets/α-helices of the receptor 26). For the mutations located in the signal peptide, GABRB3 (P11S, S15F) 6, and at the γ+/β− interface, GABRB3(G32R) 7 and GABRG2(R82Q, P83S) 32, the reductions in GABAA receptor currents were smaller (reduced to ~42, ~48, ~50–62, ~34 and ~12% of the wt currents, respectively) than those caused by the LGS-associated GABRB3(D120N, E180G, Y302C) mutations (reduced to ~24, ~1, ~5% of the wt currents, respectively) located at the β+/α− interface. Similarly, we found small (reduced to ~75% of the wt current) or no effects on current amplitudes for the IS-associated mutations at the γ+/β− interface. Moreover, the GABRA1(D219N) mutation also located at the β+/α− interface was found to reduce up to 70% of receptor currents 33. In line with this assumption, recent whole-exome sequencing studies have associated severe developmental disorders such as Dravet syndrome and intellectual disability with novel GABR missense mutations (GABRA1(R112Q, G251S)34, GABRB2(M79T)35), again at the β+/α− interface, rather than the previously described nonsense mutations. Thus, it seems that mutations at the β+/α− interface that cause major rearrangements of structural domains crucial for translating ligand binding to channel gating of the GABAA receptor. This may explain, at least in part, how different mutations in GABRB3 could be associated with increased severity of channel dysfunction and with both mild (childhood absence epilepsy) and severe (IS, LGS) epilepsy syndromes.
Recent genomic studies have contributed enormously to identification of genetic mutations in patients especially in the absence of a family history of epilepsy. Functional validation remains the next important step since the contributions of the mutations to epilepsy are not always clear, and their mechanisms of action cannot be predicted solely from in silico approaches (such as PolyPhen and SIFT scores) 36. Our data provides strong functional evidence that the GABRB mutations identified in LGS and IS patients by the Epi4K consortium disrupt GABAA receptor function but by different mechanisms. Even though heterologous systems allow evaluation of the impact of specific mutations on GABAA receptor functions, the results cannot be extrapolated directly to predict the impact of the mutations on neuronal systems or animal models of epilepsies. In neurons GABAA receptor expression is dynamic, activity dependent and cell type specific. Even within a single neuron GABAergic inhibition varies depending on the expression of other partnering subunits and the neuronal compartment. Additionally, if the mutant subunits are preferentially expressed in interneurons, loss of function could produce hypo-excitable networks. Thus, further analysis of the impact of the mutation in neuronal preparations and mouse models will be crucial to understand the mechanisms of action of these mutations and improve treatments.
Acknowledgments
Acknowledgement statement (including conflict of interest and funding sources): Supported by NIH R01 NS33300 to RLM.
This work was supported by the NIH RO1 NS 33300 grant to RLM. We thank the following Epi4K and Epilepsy Phenome/Genome Project investigators who contributed to the discovery of the GABRB3 and GABRB1 mutations examined in this manuscript. Drs. Dlugos, Dennis, MD, MCS, The Children’s Hospital of Philadelphia; Geller, Eric, MD, St. Barnabas Health Care System; Kirsch, Heidi, MD, MS, University of California, San Francisco; Kossoff, Eric, MD, The Johns Hopkins University School of Medicine; Lowenstein, Daniel, MD, University of California, San Francisco; Sherr, Elliott, MD, PhD, University of California, San Francisco; Sullivan, Joe, MD, University of California, San Francisco; Venkat, Anu, MD, The Children’s Hospital of Philadelphia; Vining, Eileen, MD, The Johns Hopkins University School of Medicine; and Widdess-Walsh, Peter, MB, FRCPI, St. Barnabas Health Care System.
Footnotes
Author Contributions: VJS, CCH and RLM were involved in conception and design of the study and manuscript preparation. VSJ, CCH acquired and analyzed electrophysiology data; KMV, VSJ acquired and analyzed flow cytometry data; and NH made and sequenced cDNA constructs. VSJ and CCH drafted the figures and the manuscript.
Potential Conflicts of Interest: Authors have no conflict of interest to report.
References
- 1.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010 Apr;51(4):676–85. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 2.Epi KC, Allen AS, et al. Epilepsy Phenome/Genome P. De novo mutations in epileptic encephalopathies. Nature. 2013 Sep 12;501(7466):217–21. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Althoff T, Hibbs RE, Banerjee S, Gouaux E. X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature. 2014 Aug 21;512(7514):333–7. doi: 10.1038/nature13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller PS, Aricescu AR. Crystal structure of a human GABA receptor. Nature. 2014 Jun 8; doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010 Jun 1;588(Pt 11):1861–9. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008 Jun;82(6):1249–61. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gurba KN, Hernandez CC, Hu N, Macdonald RL. GABRB3 mutation, G32R, associated with childhood absence epilepsy alters alpha1beta3gamma2L gamma-aminobutyric acid type A (GABAA) receptor expression and channel gating. J Biol Chem. 2012 Apr 6;287(15):12083–97. doi: 10.1074/jbc.M111.332528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Homanics GE, DeLorey TM, Firestone LL, et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci U S A. 1997 Apr 15;94(8):4143–8. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Handforth A, Delorey TM, Homanics GE, Olsen RW. Pharmacologic evidence for abnormal thalamocortical functioning in GABA receptor beta3 subunit-deficient mice, a model of Angelman syndrome. Epilepsia. 2005 Dec;46(12):1860–70. doi: 10.1111/j.1528-1167.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 10.Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci. 1992 Nov;12(11):4151–72. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fillman SG, Duncan CE, Webster MJ, Elashoff M, Weickert CS. Developmental co-regulation of the beta and gamma GABAA receptor subunits with distinct alpha subunits in the human dorsolateral prefrontal cortex. Int J Dev Neurosci. 2010 Oct;28(6):513–9. doi: 10.1016/j.ijdevneu.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6beta22 and 6beta2 GABA(A) receptor channel gating and expression. J Physiol. 2011 Dec 1;589(Pt 23):5857–78. doi: 10.1113/jphysiol.2011.218883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang X, Hernandez CC, Macdonald RL. Modulation of spontaneous and GABA-evoked tonic alpha4beta3delta and alpha4beta3gamma2L GABAA receptor currents by protein kinase A. J Neurophysiol. 2010 Feb;103(2):1007–19. doi: 10.1152/jn.00801.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002 Jul 1;22(13):5321–7. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo WY, Lagrange AH, Hernandez CC, et al. Glycosylation of {beta}2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem. 2010 Oct 8;285(41):31348–61. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klausberger T, Fuchs K, Mayer B, Ehya N, Sieghart W. GABA(A) receptor assembly. Identification and structure of gamma(2) sequences forming the intersubunit contacts with alpha(1) and beta(3) subunits. J Biol Chem. 2000 Mar 24;275(12):8921–8. doi: 10.1074/jbc.275.12.8921. [DOI] [PubMed] [Google Scholar]
- 17.Sarto I, Wabnegger L, Dogl E, Sieghart W. Homologous sites of GABA(A) receptor alpha(1), beta(3) and gamma(2) subunits are important for assembly. Neuropharmacology. 2002 Sep;43(4):482–91. doi: 10.1016/s0028-3908(02)00160-0. [DOI] [PubMed] [Google Scholar]
- 18.Dunne EL, Hosie AM, Wooltorton JR, et al. An N-terminal histidine regulates Zn(2+) inhibition on the murine GABA(A) receptor beta3 subunit. Br J Pharmacol. 2002 Sep;137(1):29–38. doi: 10.1038/sj.bjp.0704835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wooltorton JR, McDonald BJ, Moss SJ, Smart TG. Identification of a Zn2+ binding site on the murine GABAA receptor complex: dependence on the second transmembrane domain of beta subunits. J Physiol. 1997 Dec 15;505( Pt 3):633–40. doi: 10.1111/j.1469-7793.1997.633ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald RL, Rogers CJ, Twyman RE. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol. 1989 Mar;410:479–99. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng HJ, Macdonald RL. Multiple actions of propofol on alphabetagamma and alphabetadelta GABAA receptors. Mol Pharmacol. 2004 Dec;66(6):1517–24. doi: 10.1124/mol.104.003426. [DOI] [PubMed] [Google Scholar]
- 22.Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits: alpha 1 beta 1 and alpha 1 beta 1 gamma 2S subunits produce unique ion channels with dissimilar single-channel properties. J Neurosci. 1993 Apr;13(4):1429–40. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishek BJ, Moss SJ, Smart TG. Homomeric beta 1 gamma-aminobutyric acid A receptor-ion channels: evaluation of pharmacological and physiological properties. Mol Pharmacol. 1996 Mar;49(3):494–504. [PubMed] [Google Scholar]
- 24.Anstee QM, Knapp S, Maguire EP, et al. Mutations in the Gabrb1 gene promote alcohol consumption through increased tonic inhibition. Nat Commun. 2013;4:2816. doi: 10.1038/ncomms3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo WY, Lagrange AH, Hernandez CC, Gurba KN, Macdonald RL. Co-expression of gamma2 subunits hinders processing of N-linked glycans attached to the N104 glycosylation sites of GABAA receptor beta2 subunits. Neurochem Res. 2014 Jun;39(6):1088–103. doi: 10.1007/s11064-013-1187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkatachalan SP, Czajkowski C. Structural link between gamma-aminobutyric acid type A (GABAA) receptor agonist binding site and inner beta-sheet governs channel activation and allosteric drug modulation. J Biol Chem. 2012 Feb 24;287(9):6714–24. doi: 10.1074/jbc.M111.316836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bianchi MT, Macdonald RL. Mutation of the 9′ leucine in the GABA(A) receptor gamma2L subunit produces an apparent decrease in desensitization by stabilizing open states without altering desensitized states. Neuropharmacology. 2001 Nov;41(6):737–44. doi: 10.1016/s0028-3908(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 28.Trevathan E. Infantile spasms and Lennox-Gastaut syndrome. J Child Neurol. 2002 Feb;17( Suppl 2):2S9–2S22. doi: 10.1177/08830738020170021201. [DOI] [PubMed] [Google Scholar]
- 29.Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013 Apr 16;236:345–72. doi: 10.1016/j.neuroscience.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldschen-Ohm MP, Wagner DA, Jones MV. Three arginines in the GABAA receptor binding pocket have distinct roles in the formation and stability of agonist- versus antagonist-bound complexes. Mol Pharmacol. 2011 Oct;80(4):647–56. doi: 10.1124/mol.111.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett WM, Mela-Riker LM, Houghton DC, Gilbert DN, Buss WC. Microsomal protein synthesis inhibition: an early manifestation of gentamicin nephrotoxicity. Am J Physiol. 1988 Aug;255(2 Pt 2):F265–9. doi: 10.1152/ajprenal.1988.255.2.F265. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Hernandez CC, Hu N, Macdonald RL. Three epilepsy-associated GABRG2 missense mutations at the gamma+/beta- interface disrupt GABAA receptor assembly and trafficking by similar mechanisms but to different extents. Neurobiol Dis. 2014 Aug;68:167–79. doi: 10.1016/j.nbd.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lachance-Touchette P, Brown P, Meloche C, et al. Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011 Jul;34(2):237–49. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- 34.Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014 Apr 8;82(14):1245–53. doi: 10.1212/WNL.0000000000000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava S, Cohen J, Pevsner J, et al. A novel variant in GABRB2 associated with intellectual disability and epilepsy. Am J Med Genet A. 2014 Nov;164A(11):2914–21. doi: 10.1002/ajmg.a.36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011 Jun 24;145(7):1036–48. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]