

Summary
Chromatin is thought to carry epigenetic information from one generation to the next, although it is unclear how such information survives the disruptions of nucleosomal architecture occurring during genomic replication. Here, we measure a key aspect of chromatin structure dynamics during replication—how rapidly nucleosome positions are established on the newly replicated daughter genomes. By isolating newly synthesized DNA marked with 5-ethynyl-2′-deoxyuridine (EdU), we characterize nucleosome positions on both daughter genomes of S. cerevisiae during chromatin maturation. We find that nucleosomes rapidly adopt their mid-log positions at highly transcribed genes, which is consistent with a role for transcription in positioning nucleosomes in vivo. Additionally, experiments in hir1Δ mutants reveal a role for HIR in nucleosome spacing. We also characterized nucleosome positions on the leading and lagging strands, uncovering differences in chromatin maturation dynamics at hundreds of genes. Our data define the maturation dynamics of newly replicated chromatin and support a role for transcription in sculpting the chromatin template.
Graphical Abstract
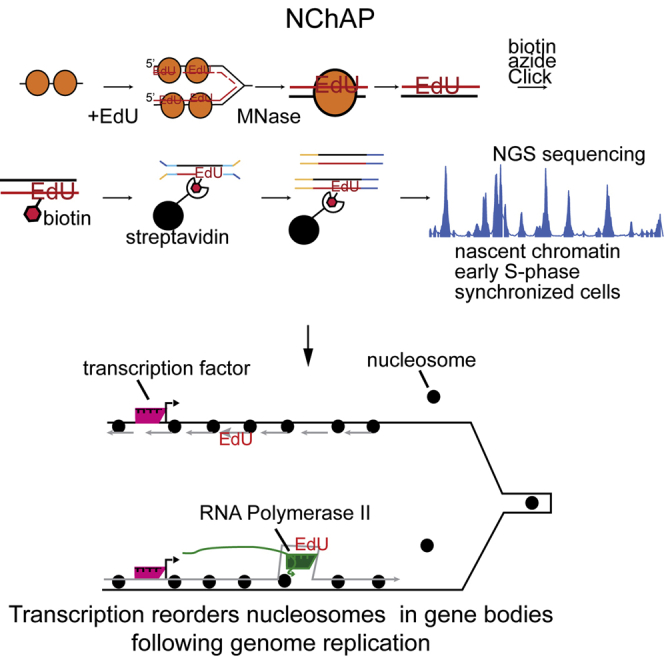
Highlights
-
•
Nucleosome positions are determined on newly replicated DNA
-
•
Transcription reorders nucleosomes in gene bodies after DNA replication
-
•
The HIR complex tightens nucleosome spacing in gene bodies following replication
-
•
Nucleosome positions on leading and lagging strands depend on genes’ orientation
Vasseur et al. present a method for mapping nucleosome positions on nascent DNA shortly after the passage of the replication fork in S. cerevisiae. They show that transcription is involved in reordering nucleosomes after DNA replication.
Introduction
Chromatin is the complex of DNA and histone proteins that packages eukaryotic DNA into chromosomes. The nucleosome is the repeating structural subunit of chromatin and consists of 147 bp of DNA wrapped around a histone octamer core. Translational positioning of nucleosomes along the DNA sequence influences the accessibility of regulatory sequences to the transcriptional machinery and can thereby regulate gene expression levels (for review, see Hughes and Rando, 2014, Radman-Livaja and Rando, 2010).
The average nucleosome-positioning profile over all yeast genes consists of a nucleosome-depleted region (NDR) of ∼150 bp, with well positioned −1 and +1 nucleosomes upstream and downstream of the NDR, respectively. The transcription start site (TSS) is located in the +1 nucleosome, there is a regularly spaced nucleosomal array over the first kb of the gene body, and nucleosome positions become fuzzier toward the middle and end of the coding sequence (Brogaard et al., 2012, Tsankov et al., 2010, Weiner et al., 2010, Vaillant et al., 2010). Because NDRs are thought to facilitate transcriptional activation by enabling access of regulatory proteins to their binding sequences, NDR formation or loss can lead to gene activation or silencing, respectively. The distribution of nucleosomes along the genome depends in part on the underlying DNA sequence, with promoter regions enriched in poly A tracts mostly excluding nucleosomes (Kaplan et al., 2009, Yuan et al., 2005). In addition to poly A tracts that passively disfavor nucleosome assembly, NDRs can also be formed through active nucleosome removal from promoter regions by remodelers, such as RSC (Parnell et al., 2008), or nucleosome displacement by general transcription factors (TFs), such as Abf1 and Rap1 (Yarragudi et al., 2004, Yarragudi et al., 2007).
Whereas DNA sequence composition contributes to nucleosome occupancy in yeast, it is the action of chromatin remodelers and the transcriptional machinery that establishes precise nucleosome positioning over genes (Gkikopoulos et al., 2011, Hughes et al., 2012, Lieleg et al., 2015, Pointner et al., 2012, Weiner et al., 2010). Indeed, in vitro assembly of nucleosomes onto purified yeast genomic DNA results only in nucleosome depletion over poly A tracts but little evidence for nucleosome positioning, whereas addition of yeast extract to such reconstitutions yields a more-accurate recapitulation of nucleosome positioning patterns observed in vivo (Zhang et al., 2011). Purified ATP-dependent remodelers, such as CHD1 and SWI/SNF family members, can generate NDRs at promoters and regularly spaced nucleosomal arrays over gene bodies similar to those seen in vivo, even in the absence of transcription (Lieleg et al., 2015). However, such in vitro nucleosome reconstitutions do not perfectly match nucleosome positions observed in vivo (Hughes et al., 2012). In vivo, the process of transcription plays a key role in nucleosome positioning, due both to the direct effects of RNA polymerase on nucleosomes and to the effects of remodelers that are recruited to target genes during transcriptional activation or elongation (Bintu et al., 2011, Radman-Livaja et al., 2011, Studitsky et al., 1997, Weiner et al., 2010).
Contrary to the steady-state landscape of nucleosome positioning, chromatin structure dynamics over the cell cycle, during which chromosomes are subject to dramatic perturbations caused by replication and mitosis, are not well characterized. DNA replication initiates the disassembly of maternal nucleosomes ahead of the replication fork and their reassembly in its wake on one or the other daughter chromatid (Alabert and Groth, 2012). As nucleosomes can influence transcription, depending on their precise locations, replication provides an opportunity for the cell either to re-establish the same nucleosome-positioning profiles or to rearrange the nucleosomal landscape and thereby maintain or change its gene expression program, respectively. The process of nucleosome re-positioning after the disruption caused by replication is not well understood. Specifically, it is not known how and where nucleosomes re-position themselves on newly replicated DNA and how long it takes them to reconstitute the canonical mid-log positioning pattern. This is related to the questions of when transcription resumes after the disruption caused by DNA replication, whether both new gene copies are transcribed, and whether transcription re-activation is a cause or a consequence of nucleosome positioning maturation.
In order to address these questions, we have developed a method for genome-wide mapping of nucleosome positions on recently replicated DNA in budding yeast: nascent chromatin avidin pull-down (NChAP). In this method, we isolate newly synthesized DNA at varying times after a pulse of the nucleotide analog 5-ethynyl-2′-deoxyuridine (EdU), which, along with micrococcal nuclease (MNase) digestion, allows us to follow genome-wide nucleosome-positioning dynamics after the passage of the replication fork on both leading and lagging DNA copies. We find that nucleosomes assume their mid-log positions with varying rates at different genomic loci. Because we find that highly transcribed genes exhibit a rapid return to their canonical chromatin architecture, we hypothesized that transcription participates in the regular phasing of nucleosomes in the gene body minutes after the passage of the fork. Consistent with this, treatment with the RNA polymerase inhibitor thiolutin interfered with chromatin maturation over coding regions. Experiments in deletion mutants reveal a role for CHD1 and ISW1a in nucleosome phasing relative to the TSS and a role for HIR in determining the linker length between nucleosomes. In contrast to transcription-dependent maturation of gene body chromatin, aspects of promoter packaging, such as the NDR midpoint and the position of the +1 nucleosome, appear to be determined earlier, possibly in the absence of transcription elongation. Together, our data illuminate the genomic landscape of chromatin maturation following replication, and our methodology enables future genetic interrogation of the mechanisms responsible for chromatin maturation.
Results
A Method for Mapping Nucleosome Positions on Recently Replicated DNA
In order to map the positions of nucleosomes on recently replicated DNA, we developed NChAP, which combines EdU labeling of nascent DNA (Sirbu et al., 2011, Wirges et al., 2007) with MNase digestion of chromatin. EdU is a thymidine analog that is incorporated during the synthesis of new DNA, and MNase is an endonuclease that preferentially cleaves DNA in the linker regions between nucleosomes. Figure 1A outlines NChAP steps for an experiment in a synchronized culture. Cells are arrested in G1 with α factor and released into S phase in the presence of EdU. Aliquots are taken at regular intervals following release into S phase, and cells are fixed with formaldehyde. Fixed cells are then treated with MNase, and nucleosome-protected DNA is isolated after cross-link reversal. EdU from purified ∼150-bp fragments is conjugated to biotin azide in a click reaction, and biotinylated DNA fragments are isolated using streptavidin-coated magnetic beads. Illumina paired end sequencing libraries are prepared (adapted from Borodina et al., 2011) from the nascent DNA attached to beads. In order to differentiate between the lagging and the leading strand copies, the EdU-containing nascent strand is separated from its template using primer extension from one end of the adaptor-ligated fragment (Figure 1A). Consequently, sequencing reads that map to the Watson strand “upstream” of efficient replication origins will originate from the lagging strand copy, whereas the complementary Crick reads will be from the leading copy. The opposite is true for reads located downstream of efficient origins.
Figure 1.

Nascent Chromatin Avidin Pull-Down
(A) Diagram of nascent chromatin avidin pull-down (NChAP). For synchronized cells, after arrest in G1, cells are released into fresh media in the presence of EdU and aliquots are fixed at regular time intervals. In asynchronous populations, cells are pulsed with EdU, followed by a thymidine (T) chase. Chromatin is digested with MNase, and the isolated DNA fragments are subject to a click reaction that adds biotin to the incorporated EdU. Biotinylated DNA is purified with streptavidin-conjugated magnetic beads, and NGS libraries are constructed on DNA fragments attached to the beads. cDNA strands are separated with primer extension in the presence of dUTP. The dUTP-containing strand is then digested with USER enzymes prior to PCR. This ensures that only nascent strands are sequenced.
(B) Density distribution of DNA content measured by flow cytometry before arrest (mid-log) in G1 and at indicated times after release from G1 arrest (left panel). Nascent chromatin Watson (W) strand read distribution on chromosome 2 at indicated times after release (blue bars) and total chromatin input are shown (total MNase-digested chromatin isolated prior to the click reaction; 32.5-, 40-, and 55-min time points, pink bars). Replication origins (ARS) are shown in the two bottom rows: ARS from this study (first) and previously documented ARS (second) are shown. Read counts were grouped in 400-bp bins and first normalized to the genome average read count and then to the highest peak value in each chromosome.
Several controls validate the ability of NChAP to identify newly replicated DNA in relatively unperturbed cells. First, flow cytometry profiling of DNA content shows that S phase progression is not impaired in the presence of EdU (Figure S1A). Second, we tested the ability of our protocol to specifically capture the single EdU-bearing strand of DNA, using in-vitro-generated fragments that have incorporated EdU in only one strand (Figure S2). EdU was incorporated into one strand of a PCR fragment. This fragment was then subjected to the same procedure that we used to generate NChAP libraries as outlined in Figures 1A, S2A, and S2B. Following streptavidin pull-down of these test libraries, qPCR with strand-specific primers showed that the fraction of fragments with the EdU-containing strand in the expected orientation was 70%–85%. Finally, we also generated DNA fragments in which only one strand has incorporated EdU in vivo, taking advantage of the 5′–3′ resection and gap-filling steps that occur during the repair of a double-strand break at the MATalpha1 locus (reviewed in Haber, 2012), which we induced in the presence of EdU (Figure S2C). Here, we observe a ∼10-fold enrichment of the EdU-containing strand in the expected orientation. Overall, our tests show that our strand-specific library construction protocol can efficiently isolate the nascent DNA strand and thus differentiate between leading and lagging strand copies of the genome.
We next applied NChAP to cells released from G1 arrest into S phase for varying lengths of time. The results are shown in Figure 1B. NChAP data at early time points reveal strong peaks surrounding known origins of replication (autonomously replicating sequence [ARS]; Nieduszynski et al., 2007, Yabuki et al., 2002), validating the ability of our protocol to specifically map nucleosomes assembled on newly synthesized DNA. The enrichment of NChAP data (blue peaks) around origins is not due to MNase bias toward replicated regions, because read density distributions from MNase-digested input fractions (before biotinylation and streptavidin pull-down) exhibit the relatively even occupancy expected of the nucleosome landscape in yeast (pink peaks). Average nucleosome profiles from the nascent chromatin fraction resemble classic average nucleosome profiles, indicating that EdU incorporation does not interfere with MNase digestion or nucleosome assembly (Figure S3A). Together, these data validate the ability of NChAP to accurately identify nucleosome-protected regions of recently replicated DNA in a strand-specific manner.
Pulse-Chase Strategy to Characterize Chromatin Maturation Dynamics
During cell-cycle arrest and release, asynchrony among individual cells in the timing of G1 release and entry into S phase results in heterogeneity in the location of the replication fork in any given cell, meaning that, even for early firing regions of the genome, NChAP will capture DNA that has been replicated from ∼1 to as many as ∼20 min prior. Consequently, in order to characterize the dynamics of chromatin maturation following replication, we carried out NChAP across a time course in which asynchronous yeast was subject to a brief pulse of EdU followed by a thymidine chase for varying lengths of time. An asynchronous population by definition contains cells at all stages of the cell cycle, including ∼15%–40% of cells that are in S phase and which, as a population, will have replication forks located at every location along the genome. As a result, a relatively short pulse of EdU will label short stretches of replicating DNA covering the entire genome over the whole cell population. The thymidine chase stops further incorporation of EdU, and subsequent fixation at regular time intervals provides snapshots of simultaneous nucleosome positioning changes shortly after replication in all replicating cells.
In order to capture times as close as possible to the moment immediately after the passage of the replication fork, we sought to identify the minimal duration of the EdU pulse that provided appreciable incorporation into replicated DNA. We used flow cytometry profiling of cells labeled with fluorescein (FAM)-conjugated EdU to monitor the kinetics of EdU incorporation in asynchronous cells (Figure S4). EdU labeling is detectable within 15 min of its addition to the culture (Figure S4A), and the majority of replicating cells have incorporated EdU after ∼25–30 min (Figure S4). EdU incorporation could be delayed and slowed by growing yeast at suboptimal temperatures to extend the length of the cell cycle (Figure S4B). At 30°C, an initial lag phase of ∼15 min is followed by a gradual increase in the numbers of cells that have incorporated EdU, as well as an increase in EdU-FAM fluorescence intensity per cell as genome replication progresses and more EdU is incorporated in each S phase cell (Figure S4C). A slower increase in the average cellular EdU-FAM intensity over time at 25°C and 37°C compared to 30°C is consistent with slower or stalled replication forks, delayed S phase entry, and/or slower EdU uptake and processing (Figure S4C). Because the fraction of EdU-positive cells in the asynchronous cell population increases gradually and cells need to be incubated with EdU for 25, 40, or 50 min (if grown at 30°C, 37°C, or 25°C, respectively) before all cells that were in S phase at the moment of EdU addition become EdU positive (Figure S4B), we conclude that the rates of EdU import and processing can vary widely among different cells in the population, possibly due to variable expression of the EdU transporter (hENT1) and thymidine kinase (TK) that were introduced into our yeast strain. All subsequent experiments were performed at 30°C.
To assess whether we could effectively halt EdU incorporation using an excess of cold thymidine, we pulsed asynchronous yeast with EdU for 20 or 5 min and then assayed EdU-FAM at varying times after thymidine addition (Figure S5). Both the levels of EdU-FAM across the population and the fraction of EdU-FAM-positive cells stay constant up to 25 min after the thymidine chase (Figures S5B and S5C), and then as cells enter a new round of replication, the fraction of EdU-positive cells decreases, as expected for a successful thymidine chase. Moreover, the average EdU-FAM intensity per cell remains low throughout the chase, consistent with EdU being incorporated into only a small fraction of the genome, as intended (Figure S5E).
Nucleosome Positioning Maturation Indices Correlate with Global RNAPol2 Occupancy
We carried out two separate pulse-chase time course experiments: one with a 20-min and another with a 5-min EdU pulse (Figure S5).
We calculated the Pearson correlation between the nucleosome-positioning profile for each gene in the yeast genome (from 500 bp upstream of the TSS to the stop codon; Xu et al., 2009) in our NChAP data and the corresponding profile from a mid-log total chromatin standard (Weiner et al., 2010) for each time point (Figure 2A; Table S1). For each gene, we define its “maturation index” as the average correlation over the time course between the nucleosome profiles from nascent chromatin and the total chromatin standard, with individual genes exhibiting a wide range of maturation indices (Figures S3B and S3C). Importantly, data for total chromatin consistently exhibit higher correlations to the standard than do nascent chromatin data (Figure 2A; plot below the heatmap), demonstrating that the wide range of maturation indices in nascent chromatin is not an artifact of variability in MNase digestion across the time course. Progressive changes in nucleosome positioning on nascent DNA are also evident from whole-genome pairwise correlation analysis (Figure S6A; Table S2).
Figure 2.

Transcription Influences Nucleosome Positioning Maturation Rates
(A) Heatmap of Pearson correlations between nucleosome profiles from nascent chromatin (5- and 20-min EdU pulse) or total chromatin input (5-min EdU pulse) and total chromatin from log phase cells (mid-log standard) for every yeast gene (rows) at indicated time points after the thymidine chase (columns). 0.5 was subtracted from the actual correlation to obtain higher contrast. Correlation values were corrected for variability in total sequencing read numbers between time points (Supplemental Experimental Procedures).Correlation profiles were sorted by maturation index (increasing average correlation over the time course) in the 20-min EdU pulse. Mid-log RNA Pol2 occupancy is shown as a 50-gene moving window average (middle graph) and as an average for each correlation quintile (right bar graph). Only Watson strand reads analysis is shown (Crick reads analysis is comparable). The plot below the heatmap shows the evolution of median correlations for each time point over time, indicating that nascent chromatin from both datasets (5- and 20-min EdU pulse) mature at similar rates whereas total chromatin does not change.
(B) Scatterplot of maturation indices for the 20-min EdU versus the 5-min EdU pulse datasets. All genes (6,226): red. Genes within a deviation of 0.25 or 0.1 from the regression line (y = 0.85x): green (82% of all genes) and blue (41% of all genes), respectively. Gene maturation indices from these two datasets are overall correlated. Variance from the regression line for individual genes likely reflects experimental technical variability, to which the Pearson correlation metric is sensitive. As shown in the corresponding correlation heatmaps on the right, the correlation between maturation indices and RNA Pol2 occupancy is preserved in robustly correlated genes from the two time courses.
(C) Average nucleosome profiles from the 20-min EdU pulse dataset at indicated time points after thymidine incubation: nascent chromatin (blue); mid-log standard (pink), for the slow- and fast-maturing first and fifth quintiles (1,245 genes each), respectively (as defined in A).
(D) The change in the average peak/trough ratio (diagram on top) for nucleosomes +2 to +7 in the average nascent and total chromatin profiles of the two quintiles from (C) (top) and (E) (bottom).
(E) As in (C), but for the 5-min EdU pulse dataset. Nascent and total input chromatin fractions are shown in the top and bottom panels, respectively. The seemingly faster stabilization of the average peak/trough ratio compared to the 20-min EdU pulse experiment is likely due to heterogeneous EdU incorporation rates in the population. In the 5-min EdU pulse experiment, we are detecting a subpopulation of cells that incorporates EdU very rapidly after addition (Figure S5), and consequently, our time course somewhat counterintuitively detects chromatin maturation events that are taking place later after EdU incorporation than in the 20-min EdU pulse experiment, in which most cells have incorporated EdU much later after addition.
What distinguishes genes that rapidly adopt their mature chromatin state following replication? Sorting genes according to their maturation index from the 20-min EdU pulse experiment, we find that genes that have the highest maturation index are also generally highly transcribed during active growth in rich medium (Kim et al., 2010). This trend is shown via a 50-gene running window average of RNA Pol2 occupancy and is even more evident when genes are divided into quintiles (1,245 genes each) of maturation indices: the average RNA Pol2 occupancy in the highest quintile (5) is ∼10-fold higher than in the lowest quintile (1; Figure 2A, top middle and right panels). This suggests that the process of transcription plays a role in re-establishing nucleosome positions over genes after chromatin disruptions caused by DNA replication. Box plot and pairwise t test analysis of correlations to the standard for quintiles 1 and 5 from two biological replicates of the 20-min EdU pulse experiment show that the two quintiles are significantly different in all time points from both replicates (p value of t test ≤ 0.05) and that most of the variability between replicates comes from early time points in the first quintile (Figures S6B and S6C). Although data from the 5-min EdU time course differ quantitatively from data from the 20-min EdU time course (Figure 2B), in both datasets, nascent chromatin increasingly matches the mid-log standard as the time course progresses (Figures 2A, bottom, and S7), and this trend is also replicated in the second 20-min EdU pulse experiment (Figure S7). Moreover, the correlation of maturation indices in each quintile (defined above) with average RNA Pol2 occupancy is also preserved in both time courses when focusing on robustly measured genes (Figure 2B).
To examine the process of chromatin maturation more closely, we compared the average TSS-aligned nucleosome profiles from nascent chromatin to the same profiles obtained from mid-log yeast, averaging data according to quintiles of genes grouped by maturation index. Visual inspection of these averaged profiles reveals that nucleosomes become better defined over the first kb of the gene body over time (Figures 2C–2E). This improvement in nucleosome phasing can be quantitated using a measure of peak to trough for nucleosomes—a low peak-to-trough ratio can either be due to a low average nucleosome occupancy at that position across the cell population or fuzzy positioning (nucleosomes are not placed at the same distance from the TSS in all genes and in all cells). The average peak-to-trough ratios for nucleosomes +2 to +7 on nascent chromatin after a 20-min EdU pulse reach a plateau between the 4- and 8-min time points for rapidly maturing (fifth quintile) genes, whereas, in the first quintile, the ratios are stabilized only ∼10 min later (Figure 2D, top). Similar rates of average peak/trough increase were confirmed in a second biological replicate of the 20-min EdU pulse experiment (Figure S8). A quadratic fit to the curves from Figure 2D (top) reveals that genes in the first and fifth quintiles reach their half-maximal peak/trough 20 and 7 min after the chase, respectively. Consistent results were obtained using the data from the 5-min EdU pulse (Figure 2E), albeit with somewhat more-rapid apparent rates of nucleosome phasing over gene bodies (Figures 2D, bottom, and 2E). As the genes that exhibit comparatively rapid phasing of nucleosomes over gene bodies (the fifth quintile) are relatively highly transcribed, we propose that the regular phasing of nucleosomes downstream of the TSS is a consequence of transcription elongation.
In contrast to the relatively slow chromatin maturation observed over gene bodies, the +1 nucleosome and the midpoint of the NDR are already in place at the beginning of our time course for the majority of genes (Figures 2C and 2E). This is consistently observed for both the 20-min and 5-min EdU experiments. Thus, the positions of the +1 nucleosome and the NDR are determined early after the passage of the replication fork. Indeed, promoter chromatin architecture is established so rapidly that it is impossible to pinpoint its exact kinetics using our assay, as even for the 5-min EdU pulse time course, there is a 10-min window between EdU addition and the first recorded time point, during which time EdU is incorporated into DNA at different moments in different cells (Figure S4).
Nucleosome Phasing over Coding Regions Is Transcription Dependent
To directly test the involvement of transcription in nucleosome positioning maturation, we treated cells with the RNA polymerase inhibitor thiolutin in a parallel experiment with the 5-min EdU pulse time course shown in Figure 2 (Figure S9). Chromatin maturation is greatly impaired upon treatment with the inhibitor (Figures 3, S6, and S7), thus providing experimental support to our hypothesis that transcription elongation is involved in the reordering of nucleosomes on nascent DNA. Importantly, thiolutin specifically affects nascent chromatin maturation, as total chromatin fractions from thiolutin-treated and untreated cells are nearly indistinguishable (Figures 3, S6, and S7). Note that thiolutin has been added after the EdU pulse to avoid negative effects of the inhibitor on replication and EdU incorporation. As expected, the average peak/trough ratios in thiolutin-treated cells are lower than in non-treated cells. We can still detect slow nucleosome phasing maturation, with peak/trough ratios increasing at a similar rate in the first and fifth quintiles, even in the absence of transcription (Figure 3C), suggesting that there is also a transcription-independent mechanism responsible for nucleosome reorganization. Still, we cannot exclude the possibility that some residual transcription occurs in thiolutin-treated cells and accounts for the observed slow chromatin maturation. In any case, we conclude that transcription plays a central role in establishing chromatin architecture over gene bodies.
Figure 3.

Transcription Inhibition Impairs Chromatin Maturation
(A) Heatmap of Pearson correlations between nucleosome profiles of the mid-log standard (as in Figure 2) and nascent chromatin or total chromatin input (5-min EdU pulse) from thiolutin-treated and untreated cells (lines) at indicated time points after EdU addition (columns). 0.5 was subtracted from actual correlations for higher contrast. Correlation profiles were sorted by increasing maturation index from the 20-min EdU pulse experiment as in Figure 2. Only Watson reads are shown.
(B) Evolution of median correlations over time. Maturation indices are on average 35% lower in thiolutin-treated cells.
(C) Change in average peak/trough ratios for nucleosomes +2 to +7 in the fifth and first quintiles from Figure 2.
(D) Scatterplot of maturation indices for the 20-min EdU versus the 5-min EdU pulse in thiolutin-treated (red) or untreated (blue) cells. Plots are shown for all genes from nascent chromatin (top left), robustly measured genes from nascent chromatin (top right), and total chromatin input (bottom right). The difference between maturation indices of −thiolutin and +thiolutin cells is at least +0.1 in 73% of genes (top left) or 70% of genes (top right) represented on the plots. There is no difference in maturation indices in total chromatin between thiolutin-treated and untreated cells, suggesting that transcription-governed chromatin maturation is specific for newly replicated chromatin (bottom right).
(E) Maturation indices of highly transcribed genes are more affected by thiolutin. The plot shows the 25-gene moving window average of the difference between maturation indices in non-treated and thiolutin-treated cells (red; Δcorrelation((−thiolutin) − (+thiolutin))) for 551 genes ordered by log2 (RNA pol2 occupancy; blue). Only genes with log2(RNA pol2) ≤ −1 (poorly transcribed genes) or log2(RNApol2) ≥ 1 (highly transcribed genes) were used in the analysis.
HIR Is Involved in Nucleosome Spacing Readjustment after Replication
In order to better understand the mechanisms involved in nucleosome positioning maturation, we repeated the 5-min EdU pulse-chase experiment in mutants with deletions of hir1, chd1, or ioc3 (Isw1a; Figure 4A). CHD1 and ISW1a are both ATP-dependent chromatin remodelers that associate with the gene body during transcription and are involved in nucleosome array organization over coding sequences (Gkikopoulos et al., 2011, Radman-Livaja et al., 2012, Smolle et al., 2012). Hir1 is a subunit of the HIR nucleosome assembly complex, which participates in histone turnover and replication-independent nucleosome assembly (Green et al., 2005, Lopes da Rosa et al., 2011, Ray-Gallet et al., 2002). Nucleosome phasing is globally reduced in all three mutants (although to a somewhat lesser extent in hir1Δ) compared to wild-type (WT) profiles (Figure 4B, left). The level of disorganization of nucleosomal arrays in gene bodies is comparable between nascent and total chromatin profiles, suggesting that perturbations caused by DNA replication persist after S phase in the absence of these chromatin remodelers. More striking, however, is the difference in linker lengths between nascent and total chromatin profiles (Figure 4B, right). Nucleosomes appear to be less densely packed shortly after replication, with an average linker length of ∼20 bp compared to 13 bp in total chromatin in WT, chd1Δ, and ioc3Δ cells alike. In hir1Δ cells, total and nascent chromatin have the same ∼20-bp linker length, suggesting that HIR activity tightens the spacing between nucleosomes after replication, consistent with a recent report on hir1Δ effects on nucleosome positioning in nascent chromatin (Fennessy and Owen-Hughes, 2016).
Figure 4.

Nucleosome Positioning Maturation in Chromatin Remodeler Mutants
(A) Average TSS-aligned nucleosome profiles for all yeast genes in WT and mutant backgrounds (blue lines) from nascent (top row) and total chromatin (bottom). Profiles for the earliest and latest time point after the 5-min EdU pulse and 5-min thymidine chase (marked in green) are shown for nascent profiles. The last time point from the corresponding total chromatin input fraction is shown for replicate 1 of WT and replicates 1 and 2 of hir1Δ cells. The total chromatin profiles for chd1Δ and ioc3Δ are from asynchronous log phase cultures without EdU. The WT mid-log standard profile (pink) is the same as in Figure 2. The WT replicate 1 profiles are from the 5-min EdU pulse dataset from Figures 2 and 3. WT replicate 2 is a repeat of the 5-min EdU pulse experiment.
(B) Average peak/trough ratios (for nucleosomes +2 to +7; left) and average linker length (values in the center of the bar; between nucleosomes +1 and +2, +2 and +3, and +3 and +4; right). The error bars represent the SD between time points in the EdU pulse-chase experiment: hir1Δ replicate 1 nascent (0, 2, 4, 6, 15, and 25 min); hir1Δ replicate 2 nascent (0, 2, 4, 6, 8, 15, and 25 min); hir1Δ replicate 1 total (0, 8, 15, and 25 min); hir1Δ replicate 2 total (2, 6, 8, and 25 min); ioc3Δ nascent (0, 2, 4, 6, 8, 15, and 25 min); chd1Δ replicates 1 and 2 nascent (0, 2, 4, 6, 8, 15, and 25 min); WT replicate 1 nascent and total (4, 8, 12, and 16 min); and WT replicate 2 nascent (0, 2, 4, 6, 8, 15, and 25 min).
Differential Nucleosome Positioning on Leading and Lagging Strand Copies Is Linked to Genic Orientation
As detailed above, the strand separation step in our library-generation protocol enables us to distinguish between nucleosome-positioning profiles on the leading and lagging daughter chromatids. The leading or lagging copy annotation is assigned according to the position of a gene relative to its closest replication origin and whether its reads map to the Watson or the Crick strand—Watson reads upstream and downstream of an origin will be lagging and leading copies, respectively. The converse applies to Crick reads. However, due to varying efficiencies of yeast origins (every origin is not activated in every S phase cell; Yang et al., 2010), leading and lagging annotations can only be unambiguously assigned to genes located near efficient origins of replication, leaving us with a set of 1,064 genes (Figures S10A and S10B; Experimental Procedures).
Nucleosome profiles of lagging and leading copies of each gene in this set and from every time point were compared to the corresponding profiles from the final (22 min) time point in the 20-min EdU pulse-chase experiment (Figure 2). The resulting correlation profiles were ordered according to the average difference (for all three experiments) in maturation indices between the lagging and the leading strand copies of each gene (Figure 5A; Table S3). We could detect comparable differences in nucleosome positioning maturation on leading and lagging copies in the three datasets at 433 genes out 1,064, with ∼200 genes showing significant differences between leading and lagging copies (Figures 5B and S11), suggesting that nucleosome re-positioning can occur independently on the two daughter chromatids. There is no global effect of leading or lagging strand replication on nucleosome re-positioning because maturation indices are higher on the leading or the lagging copy in equal proportions. Lagging and leading profiles from total chromatin also show asymmetry in their correlations, albeit to a lesser extent than the nascent profiles (Figures 5A and 5E). This is not due to MNase sequence bias toward the Watson or the Crick strands of individual genes, as all genes were compared to the corresponding Watson or Crick profiles from the 22-min time point standard. It is more likely that the asymmetry we observe in total chromatin profiles comes from the substantial fraction of S phase cells, which in this experiment represents ∼40% of the population (Figure S9B).
Figure 5.

Nucleosome Positioning Maturation Is Impaired if the Nascent Strand Is the Transcription Template Strand
(A) Heatmap of Pearson correlations between nascent or total chromatin profiles and the 22-min time point of the 20-min EdU pulse experiment (Figure 2) for the lagging and leading strand copies of 433 genes replicated from efficient origins. Genes (lines) and time points (columns) are shown. 0.5 was subtracted from the correlations as in Figures 2 and 3. Correlation profiles were sorted by the increasing average difference between the nascent chromatin maturation indices (Δcorrelation) of the lagging and the leading copies from three experiments—one 5-min EdU and two 20-min EdU pulse experiments. 433 genes out of the 1,064 genes replicated from efficient origins have consistent Δcorrelations in all three experiments. In other words, the lagging copy has a bigger or smaller maturation index than the leading copy, respectively, in all experiments. Note that lagging and leading copy profiles are composed of a mixture of Watson and Crick strand profiles, depending on the relative position of the gene with respect to its closest efficient origin.
(B) Ten-genes moving window average of Δcorrelations from the 20-min and the 5-min EdU pulse experiments ordered as in (A).
(C) log2 of enrichment (compared to the whole gene set in the heatmap) for genes in which the lagging nascent strand is also the transcription template for each Δcorrelation quartile. p values from the hypergeometric distribution test are shown on the left. Quartiles 1 and 4 are significantly enriched for genes in which the nascent strand is the transcription template or is transcribed, respectively.
(D) Distribution of Δcorrelations from (A) (lagging index − leading index) from different datasets for quartiles 1 and 2 (left; 216 genes) and 3 and 4 (right; 217 genes) of the 433 gene set defined in (A). The difference in nascent chromatin maturation between the lagging and the leading gene copies is reduced upon thiolutin addition, i.e., the Δcorrelation distribution is shifted to the right or left in the left or right panels, respectively. This is consistent with the hypothesis that transcription elongation is higher on the copy with the higher maturation index, and when transcription is inhibited with thiolutin, the differences in chromatin maturation indices on the two copies are eliminated.
(E) Ten-genes moving window average of Δcorrelations from the 5-min EdU pulse experiment without thiolutin, total chromatin input (orange), and nascent chromatin (purple), ordered as in (A) and (B).
What features unite those genes subject to asymmetric maturation processes? The relatively slower-maturing copies in the top and bottom quartile of the heatmap in Figure 5A are enriched for genes in which the newly synthesized strand also serves as the template for transcription (Figure 5C). This observation is consistent with at least two hypotheses: (1) EdU incorporation on the template strand potentially interferes with RNA Pol2 initiation or elongation, thus delaying transcription-coupled chromatin maturation (Figures S10C and S10D) or (2) asymmetric recruitment of chromatin remodeling enzymes and/or TFs to one copy, resulting in the preferential transcription of that gene copy. For example, asymmetric transcription following replication could result from transcription preferentially occurring on the leading strand copy when the newly synthesized copy of a gene’s promoter and the replication fork are oriented in the same direction, potentially as a result of the underassembly of chromatin on the lagging strand immediately behind the fork.
Several observations suggest that asymmetric chromatin maturation results from differential expression of the two gene copies after replication. First, differences between maturation indices of the leading and lagging copies are substantially reduced in the presence of thiolutin, suggesting that differences in chromatin maturation dynamics on the two copies of a gene may be due to differences in transcription rates of the two copies, i.e., when neither gene copy is transcribed, chromatin maturation is equally slow on either copy (Figures 5D and S11). Second, to test whether EdU interferes with transcription when it is incorporated in the template strand, we compared steady-state mRNA levels in mid and late S phase of EdU-treated and untreated synchronized cells using gene expression microarrays in two independent biological replicates (Figure 6). Consistent with recent studies (Voichek et al., 2016), we find that RNA levels of 95% of cell-cycle-independent genes do not change from mid to late S phase, which could be a consequence of either a 2-fold decrease in transcription rates on both copies or the expression of only one gene copy. For this group of genes, EdU also had no effect on RNA levels (Figures 6A and 6B), arguing against the hypothesis that asymmetric chromatin maturation is an artifact of EdU effects on transcription. Moreover, although a small group of 343 genes (whose expression was not buffered after S phase) exhibited EdU-dependent inhibition of transcription (Figures 6B and 6C), these genes are not enriched for genes that show differences in chromatin maturation between the leading and the lagging gene copies. The non-buffered gene set is enriched for ribosomal genes and genes involved in translation (Figure 6D).
Figure 6.

Effect of EdU on Steady-State mRNA Levels
(A) Heatmap of a gene expression two-channel microarray. Each line represents average log2 ratios (two probes per gene) of mRNA from mid (32 min) and late (40 min) S phase versus genomic DNA isolated from G1-arrested cells from two biological replicates with two dye flip technical replicates each. All yeast genes are grouped by cell-cycle expression and ordered by replication timing. Note that, as expected for S phase cells, G1 and mitotic genes are turning off and on, respectively, in late S phase, whereas S and M/G1 genes are on and off, respectively. Cell-cycle expression annotations were taken from the SGD database. Cell-cycle-independent genes were also ordered by the normalized average (from four microarrays shown on the left) difference in mRNA enrichment between late (40 min) and mid (32 min) S phase time points from cells not treated with EdU (right panel). The average differences were normalized by subtracting the mean difference for all cell-cycle-independent genes from the difference for each gene.
(B) Fifty (top left) and ten (bottom right) genes moving window averages of the average difference in mRNA levels between mid and late S phase in EdU-treated (green) or non-treated (red) cells, all cell-cycle-independent genes (top); genes with an average RNA level difference between late and mid S phase of 0.5 and higher (non-buffered genes) in non-treated cells (343 genes; bottom). Genes are ordered by replication timing (black line).
(C) Distribution of average log2 (RNA/DNA) late S (40 min) − log2 (RNA/DNA) mid S (32 min) of all 5,072 cell-cycle-independent genes (left) and of genes from Figure 5 that showed differences in chromatin maturation between the leading and the lagging gene copies (right). All genes from each set are shown in light blue and yellow for EdU-treated and non-treated cells, respectively, and genes with log2 (RNA/DNA) late S (40 min) − log2 (RNA/DNA) mid S (32 min) ≥ 0.5 (non-buffered genes) are shown in dark blue and orange for EdU-treated and non-treated cells, respectively. EdU interference with transcription can only be detected in genes that “escape” buffering and are probably transcribed from both copies like the ∼343 genes from (C).
(D) GO annotations analysis for 343 non-buffered genes (FuncAssociate 2.1b).
We propose that, for the majority of genes, RNA production is buffered after genome replication by the suppression of transcription in one of the two copies (probably the one with the nascent strand as the transcription template), which we detect as a difference in transcription-dependent chromatin maturation between the two gene copies (Figure 5). Future studies will focus on the mechanisms that regulate gene expression levels in the genome after replication, which should clarify whether the two gene copies are differentially transcribed following replication.
Discussion
Chromatin features change throughout the cell cycle, with the biggest perturbations occurring during DNA replication and mitosis. Histone proteins on the maternal genome are removed from the DNA ahead of the replication fork and are recycled on one or the other daughter chromatid, with newly synthesized histones restoring a full complement of nucleosomes to both daughter genomes. Here, we describe a method for following nucleosome positioning dynamics on newly replicated DNA, which we call NChAP. NChAP allows us to isolate nascent chromatin and follow in parallel changes in nucleosome positioning on the leading and lagging strand chromatids shortly after the passage of the replication fork. Whereas other studies have concentrated on proteomic analysis of bulk nascent chromatin (Alabert et al., 2014, Alabert et al., 2015, Sirbu et al., 2011), we provide a genome-wide locus-specific timeline of nucleosome footprint changes after replication.
An earlier study, which mapped Okazaki fragments, reported an enrichment of Okazaki fragment ends at the positions of nucleosome dyad axes (Smith and Whitehouse, 2012). However, it is not clear whether nucleosome assembly on the lagging strand precedes Okazaki fragment ligation, as proposed. Given that we find that nucleosome maturation is not significantly faster on leading or lagging strand genomes (Figure 5), we conclude that any lagging strand-specific nucleosome deposition processes must occur rapidly relative to the ∼10-min time resolution achieved here and that the nucleosome positioning maturation process described here takes place after Okazaki fragment maturation.
We propose that gene expression programs are maintained from one cell generation to the next by the formation of NDRs on daughter chromatids very early after the passage of the replication fork (Figure 7A). Due to the heterogeneous rates of EdU import in the population, we can only conclude that promoter maturation happens within 10 min after replication fork passage, and this process most likely occurs independently of transcriptional elongation, because transcription-dependent nucleosome phasing is detected later on in our time course.
Figure 7.

Models of Chromatin Maturation and Asymmetric TF Distribution
(A) Model for a timeline of nucleosome positioning maturation in the wake of the replication fork. Replication forks from two different cells are shown: cell 1 (black) and cell 2 (blue).
(B and C) Models for asymmetric distribution of TFs. (B) It is less likely that RNA pol2 ahead of the fork (red triangle) and the replication fork (blue triangle) will collide when replication and transcription travel in the same direction. As the fork travels unhindered, Okazaki fragment ligation lags behind the fork, and TFs (star) bound to the promoter (magenta rectangle) ahead of the fork are more likely to rebind to the leading copy after replication of the promoter sequence. (C) A head-on collision of the fork and RNA pol2 traveling toward each other may cause fork stalling or slowing down, and Okazaki fragment ligation can happen almost simultaneously with synthesis, which allows TFs to bind to either the leading or lagging copy of the promoter.
The precise mechanism of NDR formation is probably locus specific. It may involve a DNA sequence that “repels” nucleosomes (such as poly A tracts) and consequently favors the (re)binding of TFs, chromatin remodelers, and other components of the transcription machinery that were probably present at the locus before replication. Or an initially bound nucleosome may be evicted through the action of a chromatin remodeler recruited to the site by a sequence-specific TF, or the bound TF itself may prevent nucleosome binding. These TFs/remodelers presumably then help establish the positions of the +1 and −1 nucleosomes. A similar model was recently proposed for promoter architecture re-establishment after replication in Drosophila cells (Ramachandran and Henikoff, 2016).
In regions without nucleosome-repelling sequences or without sequences for available TFs, nucleosomes are deposited at regular intervals shortly after the passage of the replication fork. At this stage, nucleosomes are slightly delocalized (i.e., not exactly in the same position), both on the two nascent copies within the same cell and in different cells in the population, resulting in average nucleosome profiles with low peak-to-trough ratios. Nucleosomes become better phased after transcription resumes. Remodelers that accompany the elongating RNA Pol2 (CHD1 or ISW1a) reorder nucleosomes in its wake and position them at more-regular intervals. Consequently, nucleosomes on genes with higher RNA Pol2 occupancy will be better phased over the whole population. Concomitantly, HIR reduces the spacing between nucleosomes from on average 20 bp (found in nascent chromatin) to 13 bp (measured in total chromatin). We nevertheless found a significant number of genes with low RNA Pol2 occupancy and high maturation indices, which, as suggested by our results with thiolutin-treated cells, is probably due to a transcription-independent chromatin maturation process. This could be a consequence of the action of transcription-independent chromatin remodelers, although it is not clear why these remodelers only act on a subset of poorly transcribed genes. It is also possible that, in the absence of transcription, nucleosomes are reorganized passively through statistical positioning within discrete domains located between boundaries akin to chromosomal interaction domains (CIDs) recently observed in yeast (Hsieh et al., 2015). Alternatively, a transient burst of transcription right after replication could also be responsible for high maturation indices on these genes. A detailed analysis of RNA Pol2 occupancy kinetics on nascent DNA will be necessary to distinguish between these possibilities.
Because the nascent strand in the slower-maturing copy tends to be the transcription template strand, observed differences in maturation rates between leading and lagging gene copies could be a consequence of gene expression buffering. Whereas RNA Pol2 progression may be impaired when it encounters EdU in the template strand, transcription of both copies of the gene after replication in the absence of EdU is only seen at 343 mostly highly transcribed genes (note that these are not the same genes that exhibit copy-specific maturation behaviors). The reason there is no detectable EdU effect on steady-state RNA levels in most genes could be a consequence of recently reported buffering effects of H3K56 acetylation by Rtt109 (Voichek et al., 2016) that potentially operate via the preferential expression of only one gene copy after replication. We propose that the suppressed copy would generally be the one with the nascent strand as the transcription template strand. Consequently, differences in maturation rates between the two gene copies might be caused by asymmetric H3K56 acetylation and recruitment of chromatin-remodeling enzymes and/or TFs to one of the copies, which would cause differences in transcription rates and nucleosome positioning maturation rates. Interestingly, when the lagging nascent strand is the transcription template, the replication fork and RNA polymerase advance in the same direction. Conversely, replication and transcription travel in opposite directions when the transcription template strand is the leading nascent strand. It is therefore possible that TFs bound to promoters ahead of the fork rebind preferentially to the leading or lagging copy after replication as a consequence of the differential rates of Okazaki fragment ligation and fork speed. As illustrated in Figure 7B, it is less likely that RNA Pol2 (on the yet un-replicated promoter) and the replication fork will collide when replication and transcription travel in the same direction. As the fork travels unhindered, Okazaki fragment ligation may lag behind the fork and TFs bound to the promoter ahead of the fork are more likely to rebind to the leading copy, thus favoring transcription and consequently chromatin maturation of the leading copy as observed. On the other hand, when the two travel in opposite directions, a head-on collision of the fork and RNA Pol2 that is ahead of the fork is more likely. The fork then possibly stalls and slows down, and Okazaki fragment ligation now happens almost simultaneously with synthesis, which allows TFs to bind to the leading or lagging copy of the promoter. It is, however, difficult to imagine why there would be a bias toward the lagging copy as our results predict. Future studies will test whether only one gene copy is transcribed after replication and consequently find out which copy is suppressed.
Taken together, our data demonstrate that chromatin architecture is rapidly established after genomic replication. Future studies should further illuminate the mechanisms responsible for rapid establishment of nucleosome positions and could potentially identify subtle consequences of slow maturation on genome function.
Experimental Procedures
Detailed protocols are available in the Supplemental Experimental Procedures.
Yeast Strains
All experiments (except those in Figure S2C) were done with the strain PV1 (MATa ade2-1 trp1-1 can1-1000 leu2-3,112 his3-11,15 GAL psi+ RAD5+ ura3::URA3/GPD-TK(7x) AUR1c::ADH-hENT1 Δbar1::KanR).
The experiment in Figure S2C was done with the CvY61HO strain (MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 can1-100 Δbar1::hisG TRP1::BrdU–Inc [BrdU = HSV-TK +hENT1] pJH132 [Gal::HO URA3]).
Mutant strains from Figure 4 are as follows:
hir1Δ: strain AC5 (MATa ade2-1 his3-11,15 leu2-3,112 ura3-1 TRP+ can1-100 GAL psi+ RAD5+ URA3::GDP-TK(7x) AUR1c::ADH-hENT1 Δhir1::Nat Δbar1::kanR);
chd1Δ: strain RZ12 (MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 GAL psi+ RAD5+ URA3::GDP-TK(7x) AUR1c::ADH-hENT1 Δchd1::LEU2); and
ioc3Δ: strain RZ15 (MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 GAL psi+ RAD5+ URA3::GDP-TK(7x) AUR1c::ADH-hENT1 Δioc3::KanR).
Yeast Culture
For the synchronization experiment in Figure 1, cells were arrested in G1 with α factor and then transferred into preheated media containing 10 μM EdU and pronase. Aliquots were taken and fixed with 1% formaldehyde right before release and then at regular intervals after release starting at 25 min from media change. In the EdU-thymidine pulse-chase experiments, exponentially growing cells were transferred to media with 10 μM EdU preheated at 30°C. Thymidine was added after 5 or 20 min and incubated for another 5 or 10 min. Cells were then pelleted and transferred into fresh media with thymidine (and thiolutin when indicated), and aliquots were taken at indicated time points and fixed as above.
MNase Digestion
Cells from frozen pellets were spheroplasted by bead beating in the Bullet Blender (Next Advance). Spheroplasts were treated with MNase, which was adjusted to the cell density in each tube in order to obtain 80%–90% mononucleosomal-sized fragments after 20 min at 37°C. After cross-link reversal, DNA was extracted with phenol- chloroform-iso amyl alcohol (PCI), and 150-bp mono-nucleosomal sized fragments were subsequently purified from 2% agarose gels.
Biotin Conjugation to EdU with the Click Reaction
Purified 150-bp fragments were mixed with biotin azide in a CuBr solution. After a 2-hr incubation at 37°C, DNA was precipitated with sodium acetate and ethanol.
Deep-Sequencing Library Construction
Biotinylated DNA was incubated with streptavidin-coated magnetic beads (blocked with salmon sperm DNA). All the subsequent steps were done with DNA attached to the beads. DNA fragments were blunt ended and phosphorylated. Adenosine overhangs were added with exo-Klenow. Following ligation with Illumina Genome sequencing adaptors with in-line barcodes, DNA was subjected to primer (Illumina PE primer 2.0) extension with 2’deoxyuridine 5′-triphosphate (dUTP) to separate the nascent strand from its complement. After degradation of the dUTP-containing strand with USER enzyme, the nascent DNA strand was PCR amplified. Libraries were gel purified and mixed in equimolar amounts. Paired-end sequencing was done on a HiSeq 2000 (Illumina; CNAG) or on a Next Seq sequencer (Illumina) in O.J.R.’s laboratory.
Flow Cytometry Profiling
Cells were fixed with 70% EtOH. 2.5 million cells were used for fluorescent labeling of incorporated EdU with click chemistry. Another 2.5 million cells were stained with Sytox Green for monitoring DNA content. Measurements were made with FACSCalibur (BD Biosciences; FL-1 filter; forward scattered light [FCS] size cutoff: 70).
Gene Expression Microarray Hybridization
PV1 cells were arrested in G1 as above. Genomic DNA was isolated from G1-arrested flash-frozen cell pellets and sonicated with the Bioruptor Pico cup sonicator. Cells were released into S phase in media with or without 10 μM EdU, as above. Fifty-milliliter aliquots were flash frozen in liquid nitrogen 32 and 40 min after release for RNA isolation.
Total RNA was isolated from frozen PV1 cell pellets with Trizol and treated with DNase I. RNA was reverse transcribed using oligodT as primers. The resulting cDNA was dye coupled with Cy5 or Cy3 N-hydroxysuccinimide (NHS) esters and purified as described previously (Liu et al., 2005).
The Cy5- or Cy3-labeled cDNA was mixed with Cy3- or Cy5-labeled genomic DNA, respectively, and hybridized to Agilent 8x15K yeast gene expression arrays. Images were scanned with the InnoScan 710 MicroArray scanner (Innopsys) and processed with the Mapix software. Data were normalized by dividing the Cy5/Cy3 ratio for each probe with the average Cy5/Cy3 ratio for the whole array.
Data Analysis
Sequences were aligned to the S. cerevisiae genome using BLAST-like alignment tool (BLAT). We kept reads that had at least one uniquely aligned 100% match in the paired-end pair. Read count distribution was normalized to one by dividing each base pair count with the genome-wide average base-pair count. Forward and reverse reads were treated separately.
Genes that are replicated from efficient origins were filtered as follows: (1) efficient origins were defined as origins whose read density peak heights were ≥ 0.6 at the 25-min time point in the experiment from Figure 1. Read densities were normalized to the maximum peak height per chromosome. (2) Genes that were within the boundaries of the read density area around efficient origins at the 25-min time point were considered as being replicated from that particular origin in most cells.
Analysis was done using in-house Perl and R scripts (available upon request).
Statistical Analysis
The analysis in Figures S6, S7, and S11 was performed using Perl (Statistics::Ttest) and R scripts as detailed in the Supplemental Experimental Procedures and Figures S6, S7, and S11.
Author Contributions
M.R.-L. developed NChAP, designed the experiments, performed the experiments in Figure S2, developed analysis tools, analyzed the data, and wrote the manuscript. P.V. optimized NChAP and performed all the experiments except Figures S2 and 6. S.T. and P.V. performed the experiments with the deletion mutants (Figure 4). O.J.R. initiated the development of NChAP, advised on experimental design and data analysis, and wrote the manuscript. R.Z. and A.C. constructed the deletion strains and participated in experimental execution. A.C. performed the gene expression microarray experiments (Figure 6).
Acknowledgments
We thank Etienne Schwob, Oscar Aparicio, Manfred Schmid, David Stillman, and Jim Haber for strains and plasmids. We also thank Etienne Schwob and his group for lab meeting discussions and Tom Owen-Hughes and Ross Fennessy for comments and discussions prior to publication. Thank you to Marta Gut and Julie Blanc from CNAG and Tsung-Han Hsieh from O.J.R.’s group for deep-sequencing services. This work was supported by the ATIP-Avenir (CNRS) and ERC-Consolidator (NChIP 647618) grants (to M.R.-L.) and by the NIH (R01GM079205; to O.J.R.).
Published: August 25, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, eleven figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.083.
Accession Numbers
The accession numbers for the sequencing and gene expression microarray data are GEO: GSE74090 and GSE79384, respectively.
Supplemental Information
References
- Alabert C., Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Alabert C., Bukowski-Wills J.C., Lee S.B., Kustatscher G., Nakamura K., de Lima Alves F., Menard P., Mejlvang J., Rappsilber J., Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Barth T.K., Reverón-Gómez N., Sidoli S., Schmidt A., Jensen O.N., Imhof A., Groth A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L., Kopaczynska M., Hodges C., Lubkowska L., Kashlev M., Bustamante C. The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat. Struct. Mol. Biol. 2011;18:1394–1399. doi: 10.1038/nsmb.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodina T., Adjaye J., Sultan M. A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 2011;500:79–98. doi: 10.1016/B978-0-12-385118-5.00005-0. [DOI] [PubMed] [Google Scholar]
- Brogaard K., Xi L., Wang J.P., Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennessy R.T., Owen-Hughes T. Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic Acids Res. 2016:gkw331. doi: 10.1093/nar/gkw331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T., Schofield P., Singh V., Pinskaya M., Mellor J., Smolle M., Workman J.L., Barton G.J., Owen-Hughes T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E.M., Antczak A.J., Bailey A.O., Franco A.A., Wu K.J., Yates J.R., 3rd, Kaufman P.D. Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol. 2005;15:2044–2049. doi: 10.1016/j.cub.2005.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics. 2012;191:33–64. doi: 10.1534/genetics.111.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T.H., Weiner A., Lajoie B., Dekker J., Friedman N., Rando O.J. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 2015;162:108–119. doi: 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A.L., Rando O.J. Mechanisms underlying nucleosome positioning in vivo. Annu. Rev. Biophys. 2014;43:41–63. doi: 10.1146/annurev-biophys-051013-023114. [DOI] [PubMed] [Google Scholar]
- Hughes A.L., Jin Y., Rando O.J., Struhl K. A functional evolutionary approach to identify determinants of nucleosome positioning: a unifying model for establishing the genome-wide pattern. Mol. Cell. 2012;48:5–15. doi: 10.1016/j.molcel.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., LeProust E.M., Hughes T.R., Lieb J.D., Widom J., Segal E. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.S., Liu C.L., Yassour M., Holik J., Friedman N., Buratowski S., Rando O.J. RNA polymerase mapping during stress responses reveals widespread nonproductive transcription in yeast. Genome Biol. 2010;11:R75. doi: 10.1186/gb-2010-11-7-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieleg C., Ketterer P., Nuebler J., Ludwigsen J., Gerland U., Dietz H., Mueller-Planitz F., Korber P. Nucleosome spacing generated by ISWI and CHD1 remodelers is constant regardless of nucleosome density. Mol. Cell. Biol. 2015;35:1588–1605. doi: 10.1128/MCB.01070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.L., Kaplan T., Kim M., Buratowski S., Schreiber S.L., Friedman N., Rando O.J. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J., Holik J., Green E.M., Rando O.J., Kaufman P.D. Overlapping regulation of CenH3 localization and histone H3 turnover by CAF-1 and HIR proteins in Saccharomyces cerevisiae. Genetics. 2011;187:9–19. doi: 10.1534/genetics.110.123117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski C.A., Hiraga S., Ak P., Benham C.J., Donaldson A.D. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007;35:D40–D46. doi: 10.1093/nar/gkl758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell T.J., Huff J.T., Cairns B.R. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008;27:100–110. doi: 10.1038/sj.emboj.7601946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pointner J., Persson J., Prasad P., Norman-Axelsson U., Strålfors A., Khorosjutina O., Krietenstein N., Svensson J.P., Ekwall K., Korber P. CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J. 2012;31:4388–4403. doi: 10.1038/emboj.2012.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman-Livaja M., Rando O.J. Nucleosome positioning: how is it established, and why does it matter? Dev. Biol. 2010;339:258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman-Livaja M., Verzijlbergen K.F., Weiner A., van Welsem T., Friedman N., Rando O.J., van Leeuwen F. Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol. 2011;9:e1001075. doi: 10.1371/journal.pbio.1001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman-Livaja M., Quan T.K., Valenzuela L., Armstrong J.A., van Welsem T., Kim T., Lee L.J., Buratowski S., van Leeuwen F., Rando O.J., Hartzog G.A. A key role for Chd1 in histone H3 dynamics at the 3′ ends of long genes in yeast. PLoS Genet. 2012;8:e1002811. doi: 10.1371/journal.pgen.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S., Henikoff S. Transcriptional regulators compete with nucleosomes post-replication. Cell. 2016;165:580–592. doi: 10.1016/j.cell.2016.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Gallet D., Quivy J.P., Scamps C., Martini E.M., Lipinski M., Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell. 2002;9:1091–1100. doi: 10.1016/s1097-2765(02)00526-9. [DOI] [PubMed] [Google Scholar]
- Sirbu B.M., Couch F.B., Feigerle J.T., Bhaskara S., Hiebert S.W., Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.J., Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–438. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M., Venkatesh S., Gogol M.M., Li H., Zhang Y., Florens L., Washburn M.P., Workman J.L. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 2012;19:884–892. doi: 10.1038/nsmb.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studitsky V.M., Kassavetis G.A., Geiduschek E.P., Felsenfeld G. Mechanism of transcription through the nucleosome by eukaryotic RNA polymerase. Science. 1997;278:1960–1963. doi: 10.1126/science.278.5345.1960. [DOI] [PubMed] [Google Scholar]
- Tsankov A.M., Thompson D.A., Socha A., Regev A., Rando O.J. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 2010;8:e1000414. doi: 10.1371/journal.pbio.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant C., Palmeira L., Chevereau G., Audit B., d’Aubenton-Carafa Y., Thermes C., Arneodo A. A novel strategy of transcription regulation by intragenic nucleosome ordering. Genome Res. 2010;20:59–67. doi: 10.1101/gr.096644.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voichek Y., Bar-Ziv R., Barkai N. Expression homeostasis during DNA replication. Science. 2016;351:1087–1090. doi: 10.1126/science.aad1162. [DOI] [PubMed] [Google Scholar]
- Weiner A., Hughes A., Yassour M., Rando O.J., Friedman N. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20:90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirges C.T., Gramlich P.M.E., Gutsmiedl K., Gierlich J., Burley G.A., Carell T. Pronounced effect of DNA hybridization on click reaction efficiency. QSAR Comb. Sci. 2007;26:1159–1164. [Google Scholar]
- Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki N., Terashima H., Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- Yang S.C., Rhind N., Bechhoefer J. Modeling genome-wide replication kinetics reveals a mechanism for regulation of replication timing. Mol. Syst. Biol. 2010;6:404. doi: 10.1038/msb.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarragudi A., Miyake T., Li R., Morse R.H. Comparison of ABF1 and RAP1 in chromatin opening and transactivator potentiation in the budding yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 2004;24:9152–9164. doi: 10.1128/MCB.24.20.9152-9164.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarragudi A., Parfrey L.W., Morse R.H. Genome-wide analysis of transcriptional dependence and probable target sites for Abf1 and Rap1 in Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:193–202. doi: 10.1093/nar/gkl1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G.C., Liu Y.J., Dion M.F., Slack M.D., Wu L.F., Altschuler S.J., Rando O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Wippo C.J., Wal M., Ward E., Korber P., Pugh B.F. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332:977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.