

Abstract
It is of significance to discover genes related to fiber quality and yield traits and tightly linked markers for marker-assisted selection (MAS) in cotton breeding. In this study, 188 F8 recombinant inbred lines (RILs), derived from a intraspecific cross between HS46 and MARCABUCAG8US-1-88 were genotyped by the cotton 63K single nucleotide polymorphism (SNP) assay. Field trials were conducted in Sanya, Hainan Province, during the 2014–2015 cropping seasons under standard conditions. Results revealed significant differences (P < 0.05) among RILs, environments and replications for fiber quality and yield traits. Broad-sense heritabilities of all traits including fiber length, fiber uniformity, micronaire, fiber elongation, fiber strength, boll weight, and lint percentage ranged from 0.26 to 0.66. A 1784.28 cM (centimorgans) linkage map, harboring 2618 polymorphic SNP markers, was constructed, which had 0.68 cM per marker density. Seventy-one quantitative trait locus (QTLs) for fiber quality and yield traits were detected on 21 chromosomes, explaining 4.70∼32.28% phenotypic variance, in which 16 were identified as stable QTLs across two environments. Meanwhile, 12 certain regions were investigated to be involved in the control of one (hotspot) or more (cluster) traits, mainly focused on Chr05, Chr09, Chr10, Chr14, Chr19, and Chr20. Nineteen pairs of epistatic QTLs (e-QTLs) were identified, of which two pairs involved in two additive QTLs. These additive QTLs, e-QTLs, and QTL clusters were tightly linked to SNP markers, which may serve as target regions for map-based cloning, gene discovery, and MAS in cotton breeding.
Keywords: upland cotton, cotton 63K SNP array, linkage analysis, molecular marker, QTLs
Introduction
The Gossypium genus is the most important source of natural textile fiber. It consists of 50 species approximately, including four cultivated species, G. arboreum, G. herbaceum, G. hirsutum, and G. barbadense. G. hirsutum, accounting for 95% of overall cotton production, is characterized by high yield, moderate fiber quality, and wide adaptability (Cai et al., 2014). In the past few years, area under cotton cultivation has declined worldwide, mainly due to high production costs and strong market competition with other crops (Mei et al., 2013). Thus, developing new cotton cultivars with superior fiber quality and high yields are immense to meet the demand of textile industry demand and to maintain the profitability of cotton for its growers.
Fiber quality and yield traits are complex traits, controlled by a multitude of quantitative trait locus (QTLs; Said et al., 2013). There is a complicated genetic correlation between fiber quality and yield due to different population types and parental lines (Qin et al., 2008; Yu et al., 2013; Zhang et al., 2014; Cao et al., 2015; Wang et al., 2015a). Therefore, improving yield and fiber quality, simultaneously, is a long-term challenge for cotton breeders. Traditional breeding procedures are increasingly difficult because of long duration and low selective efficiency (Shen et al., 2005). Marker-assisted selection (MAS) could be one of the more efficient approaches for breeding elite upland cotton cultivars. To date, majority of cotton genetic maps have been developed based on interspecific populations (Reinisch et al., 1994; Lacape et al., 2003; Nguyen et al., 2004; Rong et al., 2004; Guo et al., 2007; Yu et al., 2011b, 2013), which had little usage in upland cotton breeding programs (Ulloa et al., 2002). The intraspecific genetic maps of upland cotton have been constructed to detect QTLs for fiber quality and yield traits (Zhang et al., 2003, 2009; Shen et al., 2006; Wang et al., 2006, 2015b; Qin et al., 2008; Wu et al., 2008; Sun et al., 2011; Liang et al., 2013; Tan et al., 2014; Islam et al., 2015; Zhang Z. et al., 2015). However, due to low levels of intraspecific DNA marker polymorphisms in upland cotton, most intraspecific maps had a relatively low density and could not satisfy for MAS and map-based cloning. Therefore, it is necessary to develop new type of markers which can enable mapping upland cotton populations to obtain high polymorphism.
In comparison with other molecular markers, single nucleotide polymorphisms (SNPs) marker which provides the most abundant form of genetic variations, and is characterized by lower mutation rates, higher numbers, and higher accuracy (Ball et al., 2010; Yu et al., 2011a). Previous research has showed that SNPs plays role in phenotypic changes and act as functional marker for traits in MAS, when located in a gene or promoter region (Beales et al., 2005; Konishi et al., 2006). It led to the discovery of superior high-density SNP gene-chip technology was then developed as a superior method for linkage mapping and QTL detection. Now, it is being used extensively to detect QTL in bi-parental populations of many crop species (McNally et al., 2009; Gao et al., 2015). But only a few studies had reported its use in intraspecific populations of upland cotton, either with a limited number (Byers et al., 2012; Yu et al., 2012; Gore et al., 2014; Zhu et al., 2014) or with lower density (Hulse-Kemp et al., 2015; Wang Y. et al., 2015). So, there is a huge knowledge gap to be filled by a comprehensive study.
Herein, a 63 K Illumina SNP assay was used to screen 188 recombinant inbred lines (RILs) derived from the cross of HS46/MARCABUCAG8US-1-88, and a final map with 2618 loci and 0.68 cM high density map was constructed. The aims of this study were to identifying stable QTLs for fiber quality and yield traits and their tightly linked SNP markers for MAS in upland cotton breeding.
Materials and Methods
Plant Materials
A RIL population of 188 individual lines was developed following a modified single-hill procedure (bulked progeny row; Wu et al., 2008) by crossing two upland cotton cultivars, HS46 and MARCABUCAG8US-1-88. The former is a commercial cultivar with good fiber qualities and higher yield, and the later, a male parent was a germplasm with good resistance. The RILs and their parents were kindly provided by USDA-ARS, Starkville, MS, USA in 1999 (Liu et al., 2012).
In September 2014, the seeds of 188 RILs and two parents were sown in Yacheng and Baogang (two different environments were hereinafter referred to as Yc and Bg, respectively) in Sanya, Hainan Province, China. A completely randomized block design with two replications was applied in each location. Plot size was one row with 7.0 m long and 0.8 m wide. Standard cultivation, weed and insect control practices were followed throughout the growing season.
Phenotypic Measurement and Analysis
A total of 20 normally open bolls were hand-harvested from each line. Approximately 20 g of fiber from each sample was measured by HVI 1000 (Uster®Hvispectrum, Spinlab, USA) under controlled environmental conditions (20°C and 65% RH) in the Cotton Quality Supervision, Inspection and Testing Center, Ministry of Agriculture, Anyang, Henan province, China. The fiber quality traits include fiber length (FL, mm), fiber length uniformity (FU, %), micronaire (MIC), fiber elongation (FE, %), and fiber strength (FS, cN.tex-1). Yield traits consist of boll weight (BW, g) and lint percent (LP, %).
The basic statistics for the phenotypic data of the RILs, the significance of differences for each trait between the two parents, and the correlation among different traits were calculated by SPSS20.0. The variance components for fiber quality and yield traits were estimated by QTModel1.
SNP Maker Analysis and Genotyping
Genomic DNA was extracted from young leaves of the 188 RILs and two parents using modified CTAB method (Paterson et al., 1993).
The 188 RILs and their parents were genotyped with cotton 63K SNP array (Hulse-Kemp et al., 2015) from Emei Tongde Technology Development, Co. Ltd (EMTD; Beijing, China2). The array, consisted of 63,058 SNPs, were derived from published literatures (Van Deynze et al., 2009; Byers et al., 2012; Lacape et al., 2012; Rai et al., 2013). Candidate SNPs suitable for further analysis were identified as follows: (1) SNPs were filtered by excluding those with monomorphic markers or with poor quality data; (2) SNPs which the parental genotypes were inconsistent with progeny genotypic ratios or parental genotypes data had missing information were removed from the dataset; (3) SNPs of 188 RILs with missing values more than 40% were removed.
Candidate SNPs, obtained from the array, were further aligned to the tetraploid upland cotton (TM-1) reference genome (Zhang T. et al., 2015), using BWA software (Li and Durbin, 2009). Only SNPs with less than two mismatches based on a high quality sequence and a mapping Q-Value > 20 were used. The retained SNPs were sent to samtools (Li et al., 2009) and then screened for polymorphism between the mapping parents. Polymorphic SNPs were classified based on Illumina GenTrain score and call frequencies across samples (Hulse-Kemp et al., 2015). Minor allele frequencies of polymorphic markers were only used to genotype 188 RILs.
Map Construction
Linkage maps were constructed by JoinMap 4.0 Version Software (Van Ooijen, 2006), using a regression approach with the log of odds (LODs) score of 3∼10 and the jump threshold of 0.5. Converting recombination frequencies into map distances were calculated using Kosambi’s mapping function (Kosambi, 1944).
The chi-square analysis was performed to test segregating markers which deviated from 1:1 expected segregation ratio. A segregation distorted region (SDR) was defined as region with at least three adjacent loci showing significant segregation distortion (P < 0.05; Yu et al., 2011b).
Based on the results of SNPs aligned to the G. hirsutum reference genome by BWA (Li and Durbin, 2009), software CIRCOS 0.69 was used to compare the collinearity of SNPs based on their genetic positions and physical positions.
QTL Analysis
Fiber quality and yield traits related QTLs detection were performed by WinQTLCart2.5 software (Wang et al., 2001), using composite interval mapping (CIM) approach. The LOD threshold of significant QTL was calculated by 1,000 permutation tests with a significance level of P < 0.05, a mapping step of 1.0 cM, and five control markers. LOD score values between 2.5 and permutation test LOD threshold were used to declare suggestive QTL. E-QTLs were detected by IciMapping ver. 4.0 software (Li et al., 2007) using multi-environment trials (METs) function and the inclusive composite interval mapping (ICIM) method. The e-QTLs identification was done with pre-adjusted IciMapping parameters: Scan = 5 cM, LOD = 5.0, and PIN = 0.0001. A graphical representation of the linkage groups and QTLs was created by Map Chart 2.2 (Voorrips, 2002).
QTLs were named as following: q + trait abbreviation+ chromosome number+ QTL number.
Results
Phenotypic Evaluation of RIL Populations
Descriptive statistics for the fiber quality and yield traits of the RIL population, as well as their parents across two environments were presented in Table 1. FL, FU, BW, FS, and MIC of HS46 showed significantly higher than those of MARCABUCAG8US-1-88 in one or both locations; however, there were no significant differences were found for FE and LP between parental lines. In the RIL population, all analyzed traits presented continuous variation and transgressive segregation, and accorded with normal distributions.
Table 1.
Phenotypic variation of five fiber quality traits and two yield traits for the upland cotton RILs and their parents.
| Traitsa | Environmentb | Parents |
RILs |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HS46(P1) | MAR(P2) | P1–P2 | Mean | SD | Skewness | Kurtosis | Minimum | Maximum | ||
| FL | Yc | 31.00 | 29.84 | 1.17∗∗ | 30.49 | 1.15 | 0.18 | 0.35 | 27.26 | 34.12 |
| Bg | 30.47 | 29.17 | 1.31∗∗ | 30.21 | 1.02 | 0.06 | 0.16 | 27.20 | 33.39 | |
| FU | Yc | 86.35 | 85.27 | 1.08∗∗ | 85.57 | 0.83 | 0.28 | 0.17 | 83.60 | 88.50 |
| Bg | 85.92 | 84.90 | 1.02∗ | 85.39 | 0.86 | -0.30 | -0.38 | 83.10 | 87.30 | |
| MIC | Yc | 4.12 | 3.77 | 0.36∗ | 3.83 | 0.32 | 0.20 | 0.14 | 3.06 | 4.76 |
| Bg | 3.88 | 3.75 | 0.13 | 3.50 | 0.39 | 0.21 | -0.29 | 2.68 | 4.54 | |
| FE | Yc | 6.43 | 6.52 | -0.08 | 6.38 | 0.70 | 0.08 | -0.40 | 4.90 | 8.20 |
| Bg | 5.98 | 6.07 | -0.09 | 5.76 | 0.66 | 0.33 | 0.14 | 4.10 | 8.10 | |
| FS | Yc | 30.09 | 28.09 | 2.01∗∗ | 29.92 | 1.66 | 0.48 | 0.09 | 26.61 | 35.67 |
| Bg | 29.45 | 28.67 | 0.78 | 29.67 | 1.59 | 0.05 | 0.28 | 25.13 | 34.11 | |
| BW | Yc | 5.43 | 4.91 | 0.51∗∗ | 5.37 | 0.56 | 0.47 | 0.61 | 3.96 | 7.09 |
| Bg | 5.81 | 5.37 | 0.44∗ | 5.60 | 0.65 | -0.18 | 0.01 | 3.92 | 7.41 | |
| LP | Yc | 37.07 | 38.74 | -1.67 | 37.73 | 1.94 | 0.22 | 0.63 | 31.63 | 43.87 |
| Bg | 39.07 | 39.87 | -0.81 | 38.16 | 1.75 | -0.47 | 0.71 | 31.69 | 42.15 | |
∗,∗∗Significant at P = 0.05 and P = 0.01, respectively. aFL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage. bYc, Yacheng of Hainan Province; Bg, Baogang of Hainan Province.
Results showed that the FL had the highest broad-sense heritability among all traits, indicating that it could be mainly controlled by genotype, while remaining six traits had lower broad-sense heritability with values of 0.26 for FU, 0.27 for MIC, 0.37 for FE, 0.28 for FS, 0.31 for BW, and 0.38 for LP, suggesting the environmental effects were important for the performance of these traits (Table 2).
Table 2.
Analysis of variance (ANOVA) for five fiber quality traits and two yield traits in the upland cotton RIL population across two environments.
| Factorb | DF | Sum of squares |
||||||
|---|---|---|---|---|---|---|---|---|
| FLa | FUa | MICa | FEa | FSa | BWa | LPa | ||
| Gen | 187 | 803.03ˆ** | 373.93ˆ** | 77.42ˆ** | 288.82ˆ** | 1437.08ˆ** | 199.49ˆ** | 1953.4ˆ** |
| Env | 1 | 14.82ˆ** | 6.22ˆ** | 20.48ˆ** | 70.59ˆ** | 11.45 | 12.34ˆ** | 38.72ˆ** |
| Gen∗Env | 187 | 82.57 | 157.72 | 17.31 | 58.16 | 539.28 | 62.06 | 522.32 |
| Rep | 1 | 0.27 | 0.87 | 8.8ˆ** | 4.57ˆ** | 0.36 | 5.64ˆ** | 5.88 |
| Residual | 375 | 172.19 | 304.43 | 54.00 | 126.38 | 1140.54 | 139.02 | 1108.54 |
| Broad-sense heritability | 0.66 | 0.26 | 0.27 | 0.37 | 0.28 | 0.31 | 0.38 | |
∗,∗∗Significant at P = 0.05 and P = 0.01, respectively. aFL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage. bGen, genotype; Env, environment; rep, replication.
The correlation analysis for fiber and yield traits based on RILs data over two environments (Table 3). Results revealed significant negative correlation of LP with FS and FU, and significant positive correlation with MIC and FE. Similarly, BW was significant positively correlated with MIC and FS. Among the five fiber traits, all trait pairs presented significantly correlation except for FU – FE, FU – MIC, and FS – FE.
Table 3.
Correlation coefficients among all traits involved in fiber quality and yield in the upland cotton RIL population across two environments.
| Traitsa | FL | FU | MIC | FE | FS | BW | LP |
|---|---|---|---|---|---|---|---|
| FL | 1 | ||||||
| FU | 0.56∗∗ | 1 | |||||
| MIC | -0.30∗∗ | -0.04 | 1 | ||||
| FE | -0.26∗∗ | 0.00 | 0.41∗∗ | 1 | |||
| FS | 0.17∗∗ | 0.24∗∗ | 0.15∗∗ | 0.01 | 1 | ||
| BW | -0.02 | 0.05 | 0.38∗∗ | 0.03 | 0.17∗∗ | 1 | |
| LP | -0.18 | -0.14∗∗ | 0.09∗∗ | 0.14∗∗ | -0.18∗∗ | -0.07 | 1 |
∗,∗∗Significant at P = 0.05 and P = 0.01, respectively. aFL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage.
Map Construction
Among 63,058 SNPs used for screening RIL population, 3120 SNP markers (4.9%) were polymorphic between the two parents. Of those, 2618 were mapped on 26 chromosomes of upland cotton. The total length of this map was 1784.28 cM with average marker density of 0.68 cM (Table 4; Supplementary Table S1; and Figures 1–5). There were 101 SNPs on each chromosome averagely, with 1198 SNPs on At subgenome and 1420 SNPs on Dt subgenome, respectively. Uneven distribution of SNP markers on cotton chromosomes was observed. Chr14 had the highest number of SNPs (274 loci), while Chr12 had the lowest SNPs (15 loci). The average chromosome length was 68.63 cM, and the longest chromosome was Chr18 with 119.97 cM and the shortest one was Chr23 with 30.93 cM. The total lengths of At and Dt subgenomes were 888.61 and 902.67 cM, respectively. There were more loci on Dt subgenome than At subgenome. Twenty-two gaps (marker interval > 10 cM) were found on this genetic map, in which, 10 on At subgenome and 12 on Dt subgenome were observed.
Table 4.
Summary of the high-density SNP map based on upland cotton RIL population.
| Chromosome | Size (cM) | Mean distance (cM) | No SNP | Gap > 10 cM | Distortion ratio (%)a | SDRb |
|---|---|---|---|---|---|---|
| Chr01 | 52.90 | 0.40 | 133 | 0 | 15.79 | 2 |
| Chr02 | 95.22 | 0.84 | 114 | 1 | 11.40 | 1 |
| Chr03 | 116.96 | 1.72 | 68 | 3 | 27.94 | 3 |
| Chr04 | 34.26 | 1.14 | 30 | 0 | 3.33 | 0 |
| Chr05 | 69.80 | 0.52 | 135 | 0 | 7.41 | 0 |
| Chr06 | 56.76 | 1.77 | 32 | 0 | 15.63 | 1 |
| Chr07 | 67.03 | 0.96 | 70 | 0 | 15.71 | 1 |
| Chr08 | 56.77 | 0.52 | 109 | 1 | 18.35 | 3 |
| Chr09 | 99.90 | 0.62 | 162 | 1 | 9.26 | 1 |
| Chr10 | 69.07 | 1.82 | 38 | 2 | 21.05 | 1 |
| Chr11 | 41.46 | 0.86 | 48 | 0 | 18.75 | 1 |
| Chr12 | 52.71 | 3.51 | 15 | 2 | 33.33 | 1 |
| Chr13 | 68.76 | 0.28 | 244 | 0 | 12.70 | 3 |
| At subgenome | 881.61 | 0.74 | 1198 | 10 | 14.02 | 18 |
| Chr14 | 86.01 | 0.31 | 274 | 0 | 1.46 | 0 |
| Chr15 | 47.63 | 0.68 | 70 | 0 | 0.00 | 0 |
| Chr16 | 74.37 | 0.43 | 172 | 1 | 10.47 | 2 |
| Chr17 | 59.04 | 1.00 | 59 | 0 | 32.20 | 2 |
| Chr18 | 119.97 | 1.02 | 118 | 2 | 23.73 | 2 |
| Chr19 | 62.99 | 0.52 | 121 | 0 | 9.92 | 0 |
| Chr20 | 80.10 | 0.90 | 89 | 2 | 21.35 | 3 |
| Chr21 | 72.88 | 1.46 | 50 | 4 | 12.00 | 0 |
| Chr22 | 50.24 | 0.72 | 70 | 0 | 8.57 | 1 |
| Chr23 | 30.93 | 1.00 | 31 | 1 | 19.35 | 1 |
| Chr24 | 73.43 | 0.34 | 219 | 0 | 7.76 | 2 |
| Chr25 | 79.85 | 0.94 | 85 | 1 | 41.18 | 3 |
| Chr26 | 65.21 | 1.05 | 62 | 1 | 16.13 | 1 |
| Dt subgenome | 902.67 | 0.64 | 1420 | 12 | 12.68 | 17 |
| Total | 1784.28 | 0.68 | 2618 | 22 | 13.29 | 35 |
aThe percentage of distortion loci account for the total loci each chromosome. bSDR, segregation distorted region.
FIGURE 1.

Genetic maps of Chr01/Chr15, Chr02/Chr14, and Chr03/Chr17 homoeologous chromosomes and QTL detection for fiber quality and yield traits in RIL population. Map distances were given in centimorgans (cM). Markers showing segregation distortion are underlined and indicated by red color and asterisks (∗P < 0.05; ∗∗P < 0.01). Number sign (#) markers represent skewing toward the HS46 allele, ampersand (&) markers represent skewing toward the MARCABUCAG8US-1-88 allele. Segregation distortion regions (SDRs) are named as ‘Chromosome + No. SDR,’ for example, SDR01-1 refers to the first SDR on Chr01. Solid bars with different colors represent different traits and the legend is given at the end of figure. FL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage.
FIGURE 5.

Genetic maps of Chr13/Chr18 homoeologous chromosomes and QTL detection for fiber quality and yield traits in RIL population. All legends are same as described for Figure 1.
FIGURE 2.
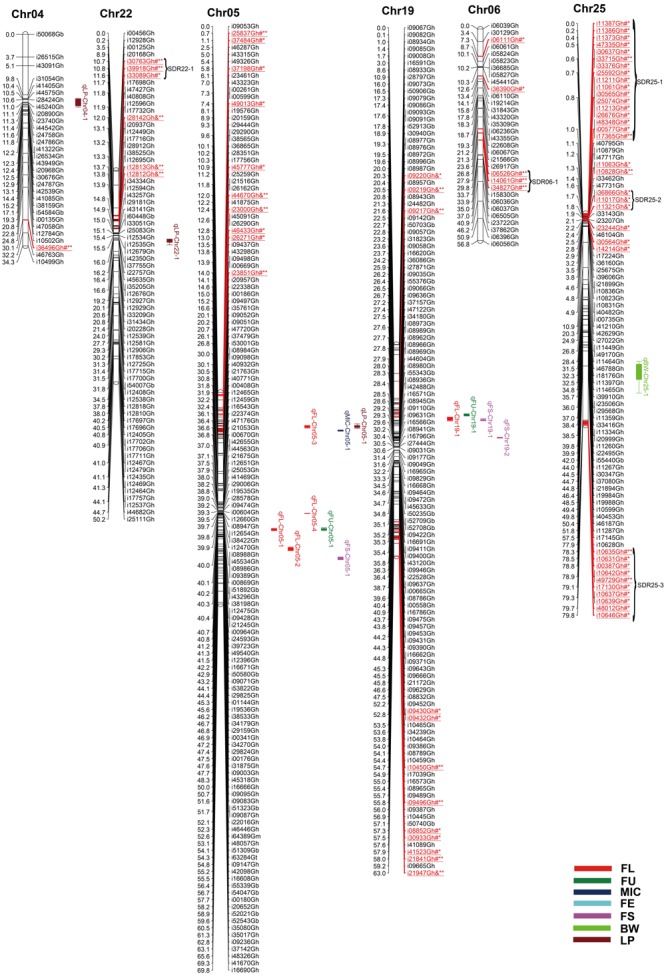
Genetic maps of Chr04/Chr22, Chr05/Chr19 and Chr06/Chr25 homoeologous chromosomes and QTL detection for fiber quality and yield traits in RIL population. All legends are same as described for Figure 1.
FIGURE 3.
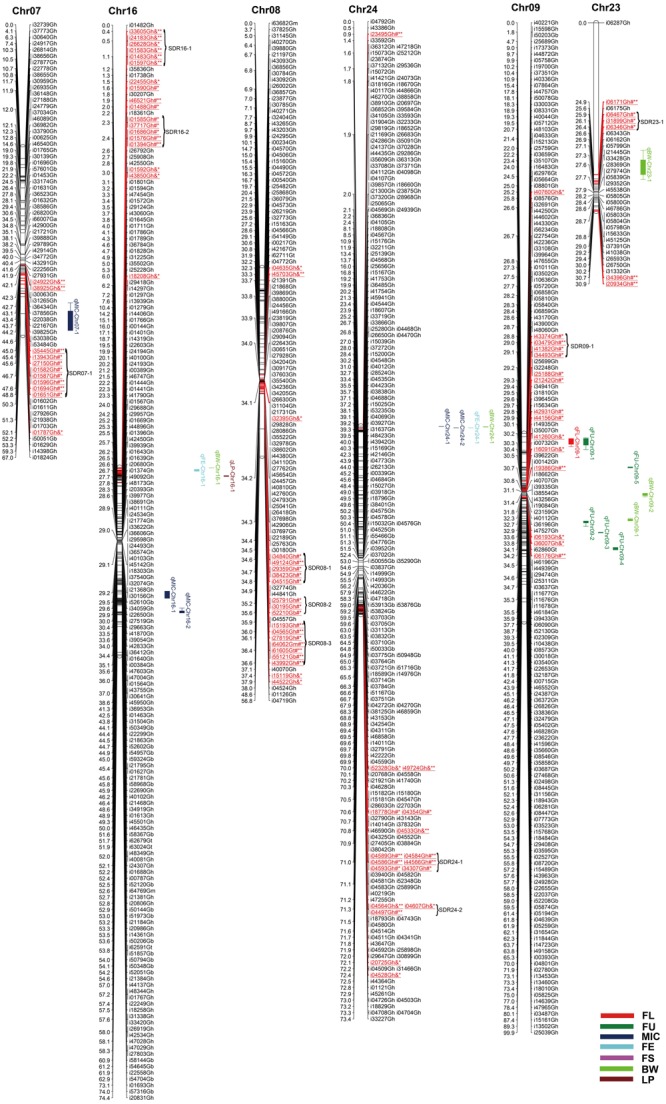
Genetic maps of Chr07/Chr16, Chr08/Chr24, and Chr09/Chr23 homoeologous chromosomes and QTL detection for fiber quality and yield traits in RIL population. All legends are same as described for Figure 1.
FIGURE 4.

Genetic maps of Chr10/Chr20, Chr11/Chr21, and Chr12/Chr26 homoeologous chromosomes and QTL detection for fiber quality and yield traits in RIL population. All legends are same as described for Figure 1.
Segregation Distortion
Among the 2618 mapped SNPs, 13.29% (348) showed segregation distortion and most loci (71.26%) showed a higher allelic frequency from the female parent (Table 4; Figures 1–5). These SNPs were unevenly distributed on the 26 cotton chromosomes and ranged from 0 to 35 loci on each chromosome (Table 4). As previous report (Yu et al., 2011b), more distorted loci were located on the Dt subgenome than on the At subgenome (180 versus 168). Segregation distortion was non-random across the linkage map. Three chromosomesChr12, Chr17, and Chr25, showed serious segregation distortions of 33.33, 32.20, and 41.18%, respectively. Furthermore, a total of 35 SDRs were found on 20 chromosomes with 18 SDRs on the At subgenome and 17 SDRs on the Dt subgenome (Table 4; Figures 1–5). Interestingly, the distorted loci in some of the SDRs (SDR03-2, SDR06-1, SDR20-1, and SDR25-2) skewed toward the same allele and showed similar degree of segregation(Figures 1–5).
Collinearity Analysis
All the mapped 2618 SNPs were aligned to the G. hirsutum reference genome to validate the genetic map. Alignments indicated that the genetic map constructed in the present study had good collinearity with the physical map (Figure 6), suggesting the high quality of the RIL map. However, several deviation on Chr04, Chr05, and Chr10 in the At subgenome and Chr16, Chr17, Chr18, Chr21, Chr23, and Chr25 in the Dt subgenome were detected between the genetic map and the physical map. The Dt subgenome showed good coverage of the physical map, representing 96.81% of the genome assembly length, while the At subgenome showed a lower coverage of 88.33% (Supplementary Table S2; Figure 6).
FIGURE 6.
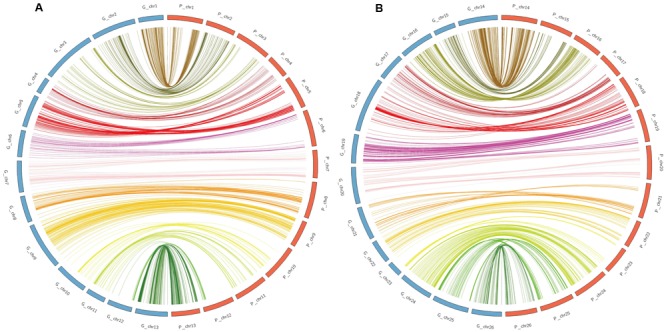
Collinearity between the genetic map (blue) and the physical map (orange). (A) Collinearity of the At subgenome between the genetic map and the physical map. (B) Collinearity of the Dt subgenome between the genetic map and the physical map.
QTL Analysis of Fiber and Yield Traits
A total of 71 QTLs for fiber quality and yield traits with 4.70∼32.28% of the total explained phenotypic variance (PV) were identified by CIM analysis (Supplementary Table S3; Figures 1–5). In these QTLs, 35 were positive additive indicating HS46 contributed alleles leading to an increase in relevant traits, while 36 were negative additive which meant MARCABUCAG8US-1-88 contributed alleles to increase fiber quality and yield traits. These QTLs were detected on 21 chromosomes, except Chr02, Chr06, Chr08, Chr13, and Chr26. Sixteen QTLs explaining 5.35∼32.28% of the PV were detected in both locations (Table 5). Among them, three were three QTLs each for FL, FU, MIC, FE, and LP, and one for BW.
Table 5.
Stable quantitative trait locus (QTLs) for fiber quality and yield identified in the upland cotton RIL population.
| Traitsa | QTL | Environmentb | Flanking markers | Positionc | LODd | Additivee | R2(%)f |
|---|---|---|---|---|---|---|---|
| FL | qFL-Chr10-1 | Yc | i11502Gh and i33011Gh | 44.51 | 2.65 | 0.29 | 6.30 |
| Bg | i11502Gh and i33011Gh | 44.51 | 2.68 | 0.26 | 6.27 | ||
| qFL-Chr14-3 | Yc | i15340Gh and i34657Gh | 23.31 | 7.71 | -0.46 | 15.05 | |
| Bg | i34657Gh and i40518Gh | 25.71 | 2.50 | -0.27 | 6.21 | ||
| qFL-Chr15-1 | Yc | i02955Gh and i02314Gh | 13.11 | 3.69 | -0.36 | 8.38 | |
| Bg | i02955Gh and i02315Gh | 12.31 | 5.21 | -0.38 | 11.12 | ||
| FU | qFU-Chr09-1 | Yc | i50203Gb and i17373Gh | 3.81 | 3.33 | -0.24 | 7.73 |
| Bg | i50203Gb and i17373Gh | 3.81 | 4.17 | -0.29 | 10.94 | ||
| qFU-Chr09-2 | Yc | i41596Gh and i26826Gh | 47.11 | 3.12 | -0.24 | 5.97 | |
| Bg | i41596Gh and i26826Gh | 47.11 | 2.68 | -0.24 | 5.58 | ||
| qFU-Chr09-3 | Yc | i06281Gh and i07773Gh | 52.61 | 3.63 | 0.30 | 7.48 | |
| Bg | i18943Gh and i07773Gh | 52.61 | 2.76 | 0.29 | 6.31 | ||
| MIC | qMIC-Chr14-1 | Yc | i15340Gh and i34657Gh | 23.31 | 2.51 | 0.08 | 5.52 |
| Bg | i15345Gh and i00465Gh | 20.91 | 2.52 | 0.11 | 5.64 | ||
| qMIC-Chr16-1 | Yc | i46435Gh and i62679Gt | 51.01 | 2.89 | 0.08 | 6.53 | |
| Bg | i01613Gh and i58367Gb | 49.31 | 2.54 | 0.09 | 5.77 | ||
| qMIC-Chr16-2 | Yc | i21384Gh and i22249Gh | 57.01 | 3.36 | 0.09 | 7.35 | |
| Bg | i44137Gh and i18258Gh | 57.41 | 4.23 | 0.15 | 9.20 | ||
| FE | qFE-Chr14-1 | Yc | i15343Gh and i21369Gh | 16.81 | 7.80 | 0.30 | 17.53 |
| Bg | i15343Gh and i21369Gh | 15.71 | 7.42 | 0.26 | 15.15 | ||
| qFE-Chr20-1 | Yc | i47006Gh and i17500Gh | 41.51 | 3.28 | 0.20 | 7.40 | |
| Bg | i17500Gh and i47439Gh | 47.11 | 6.19 | 0.44 | 32.28 | ||
| qFE-Chr24-1 | Yc | i04503Gh and i04704Gh | 73.31 | 2.62 | 0.75 | 5.78 | |
| Bg | i04503Gh and i04704Gh | 73.31 | 2.49 | 0.62 | 5.35 | ||
| BW | qBW-Chr10-1 | Yc | i25267Gh and i30274Gh | 32.21 | 2.82 | 0.61 | 27.96 |
| Bg | i25267Gh and i30274Gh | 32.21 | 2.69 | 0.60 | 24.73 | ||
| LP | qLP-Chr04-1 | Yc | i20890Gh and i24786Gh | 11.41 | 3.50 | 0.57 | 8.20 |
| Bg | i44575Gh and i24758Gh | 11.11 | 3.28 | 0.56 | 7.49 | ||
| qLP-Chr10-1 | Yc | i38146Gh and i22401Gh | 35.11 | 4.20 | -2.32 | 8.86 | |
| Bg | i25267Gh and i30274Gh | 33.21 | 2.68 | -2.27 | 20.71 | ||
| qLP-Chr12-1 | Yc | i40974Gh and i48211Gh | 4.01 | 3.38 | 0.66 | 10.42 | |
| Bg | i40974Gh and i48211Gh | 4.01 | 2.81 | 0.64 | 11.79 |
aFL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage. bYc, Yacheng of Hainan Province; Bg, Baogang of Hainan Province. cPosition of QTL located on chromosome: as cM distance from the top of each chromosome. dA LOD threshold of 2.5 was used for declaration of QTL, based on 1000 permutations at a significance level of 0.01. ePositive “additive effect” indicates an increasing effect from HS46; negative “additive effect” indicates an increasing effect fromMARCABUCAG8US-1-88. fPhenotypic variance explained by QTL.
Fiber Length
Twelve QTLs were detected, explaining 5.28∼17.98% of the PV, and located on Chr05, Chr09, Chr10, Chr12, Chr14, Chr15, and Chr19 (Supplementary Table S3; Figures 1–5). Alleles that increased FL at 10 loci were derived from MARCABUCAG8US-1-88, whereas two positive alleles were contributed by HS46. Across both environments, qFL-Chr10-1, qFL-Chr14-3, and qFL-Chr15-1 were stably identified at marker intervals of i11502Gh-i33011Gh, i15340Gh-i40518Gh, and i02955Gh-i02315Gh, explaining 6.27∼6.30, 6.21∼15.05, and 8.38∼11.12% of the PV, respectively (Table 5).
Fiber Uniformity
A total of eight QTLs were mapped on Chr01, Chr05, Chr09, and Chr19 (Supplementary Table S3; Figures 1–5), which explained 5.28∼17.98% of the PV. Notably, all of the loci showed negative effects originating from MARCABUCAG8US-1-88. The details of three stable QTLs detected in all environments were as follows: qFU-Chr09-1, flanked by markers i50203Gb and i17373Gh, and explained 7.73∼10.94% of PV; qFU-Chr09-2, between markers i41596Gh and i26827Gh, accounting for 5.58∼5.97% of the FU variance; qFU-Chr09-3 located in the intervals between markers i06281Gh and i07773Gh, explaining 6.31∼7.48% of the PV. Interestingly, all these three stable QTLs were on Chr09 (Table 5).
Micronaire
Ten QTLs were identified and located on 8 chromosomes (Chr01, Chr05, Chr07, Chr10, Chr14, Chr16, Chr17, and Chr24), explaining 5.52∼18.24% of the PV (Supplementary Table S3; Figures 1–5). Alleles for increasing MIC at nine loci were contributed by HS46, and one loci was contributed by MARCABUCAG8US-1-88. qMIC-Chr14-1, qMIC-Chr16-1, and qMIC-Chr16-2, detected in both environments, were located in the intervals between i15345Gh and i34657Gh, i46435Gh and i62679Gt, and i21384Gh and i22249Gh, respectively. They explained 5.52∼5.64, 5.77∼6.53, and 7.35∼9.20% of the PV, accordingly (Table 5).
Fiber Elongation
Nine QTLs were mapped on Chr11, Chr14, Chr16, Chr17, Chr18, Chr20, and Chr24, explaining 5.65∼32.28% of the PV (Supplementary Table S3; Figures 1–5). All favorable QTL effects were contributed by HS46. Stable detection of qFE-Chr14-1, qFE-Chr20-1, and qFE-Chr24-1 flanked by SNP markers i15343Gh and i21369Gh, i47006Gh and i47439Gh, i04503Gh and i04704Gh were in both environments, explained15.15∼17.53, 7.40∼32.28, and 5.35∼5.78% of the FE variance, respectively (Table 5). Three major QTLs, qFE-Chr14-1, qFE-Chr14-3, and qFE-Chr20-1, explained more than 10% of variation.
Fiber Strength
Eight QTLs, explaining 5.14∼9.96% of the total PV, were detected on Chr05, Chr14, Chr19, and Chr20 (Supplementary Table S3; Figures 1–5). Alleles for increasing FS on Chr05 and Chr19 were contributed by HS46, and positive alleles on Chr14 and Chr20 came from MARCABUCAG8US-1-88. All of these eight QTLs were detected in single environment, indicating that the environmental effects were important for the performance of FS.
Boll Weight
For BW, among eight identified QTLs, seven were located on six chromosomes (Chr10, Chr16, Chr18, Chr23, Chr24, and Chr25) and the remaining two on Chr09 (Supplementary Table S3; Figures 1–5). Alleles for increasing BW at the loci on Chr09, Chr10, Chr18, Chr23, and Chr24, were contributed by HS46, and on Chr16 was from MARCABUCAG8US-1-88. All other seven QTLs were identified only in one environment (Table 5), with the exception of qBW-Chr10-1 which was identified in both environments. Four major QTLs, qBW-Chr09-2, qBW-Chr10-1, qBW-Chr18-1, and qBW-Chr23-1, explaining 10.62%, 24.73%/27.96% (detected in two environments), 12.28, and 23.55% of the PV, respectively, were important in the improvement of BW.
Lint Percent
Fifteen QTLs were detected on Chr03, Chr04, Chr05, Chr10, Chr12, Chr14, Chr16, Chr17, Chr20, Chr21, and Chr22 (Supplementary Table S3; Figures 1–5). Among the 15 non-over lapping QTLs, qLP-Chr04-1, qLP-Chr10-1, and qLP-Chr12-1 were detected in both environments. Four major QTLs, qLP-Chr10-1, qLP-Chr12-1, qLP-Chr14-1, and qLP-Chr17-1, explained more than 10% of variation. Among them, alleles increasing LP at Chr10, Chr14, and Chr17 came from MARCABUCAG8US-1-88, whereas the one on Chr12 was derived from HS46.
QTL Clusters and Hotspots
Quantitative trait locus were not randomly distributed across chromosomes and chromosomal regions. Some QTLs were identified as “cluster” and “hotspot,” where clusters and hotspots were defined to contain multiple QTLs within 20 cM regions, approximately, for different and same traits, respectively (Guo et al., 2007; Rong et al., 2007; Said et al., 2013).
In the current study, there were two QTL clusters on Chr05 which contained three and five QTLs, respectively (Table 6). The Chr05-cluster-1, which possessed three QTLs, was found at 11∼13 cM for FL, MIC, and LP and Chr05-cluster-2 with five QTLs was located at 40∼55 cM for FL, FU, and FS. The FL hotspot, Chr05-hotspot-1, carrying three QTLs, was located at 40∼53 cM. It should be noted that the position of the hotspot coincided with the second identified cluster.
Table 6.
Quantitative trait locus Cluster/Hotspot for fiber quality and yield in the upland cotton RIL population across two environments.
| Cluster/Hotspota | Locationb | QTL |
|---|---|---|
| Chr05-cluster-1 | 11–13 cM | qFL-Chr05-3, qLP-Chr05-1, qMIC-Chr05-1 |
| Chr05-cluster-2 | 40–55 cM | qFL-Chr05-4, qFL-Chr05-1, qFU-Chr05-1, qFL-Chr05-2, qFS-Chr05-1 |
| Chr05-hotspot-1 | 40–53 cM | qFL-Chr05-4, qFL-Chr05-1, qFL-Chr05-2 |
| Chr09-cluster-1 | 46–62 cM | qBW-Chr09-1, qFU-Chr09-2, qFU-Chr09-3, qFU-Chr09-4 |
| Chr09-hotspot-1 | 47–62 cM | qFU-Chr09-2, qFU-Chr09-3, qFU-Chr09-4 |
| Chr10-cluster-1 | 32–45 cM | qBW-Chr10-1, qLP-Chr10-1, qLP-Chr10-2, qFL-Chr10-1 |
| Chr14-cluster-1 | 6–26 cM | qFS-Chr14-1, qFL-Chr14-1, qFS-Chr14-2, qFE-Chr14-1, qFL-Chr14-2, qFS-Chr14-3, qFL-Chr14-3, qMIC-Chr14-1 |
| Chr14-hotspot-1 | 14–26 cM | qFL-Chr14-1, qFL-Chr14-2, qFL-Chr14-3 |
| Chr14-hotspot-2 | 6–22 cM | qFS-Chr14-1, qFS-Chr14-2, qFS-Chr14-3 |
| Chr14-hotspot-3 | 1–17 cM | qFE-Chr14-2, qFE-Chr14-3, qFE-Chr14-1 |
| Chr19-cluster-1 | 20–28 cM | qFU-Chr19-1, qFL-Chr19-1, qFS-Chr19-1, qFS-Chr19-2 |
| Chr20-cluster-1 | 41–60 cM | qFE-Chr20-1, qFS-Chr20-1, qFS-Chr20-2 |
aCluster, a number of QTLs for different traits within approximately 20 cM regions; Hotspot, a multiple QTLs for the same traits within a 20 cM region. bPosition of QTL Cluster/Hotspot located on chromosome: as cM distance from the top of each chromosome.
Chr09 contained one cluster and one hotspot (Table 6). The Chr09-cluster-1, identified at 46∼62 cM, carried four QTLs for FU and BW while Chr09-hotspot-1, identified at 47∼62 cM, carried three QTLs of FU and overlapped the cluster Chr09-cluster-1.
Chr10 contained one cluster (Chr10-cluster-1) pertaining to FL, BW, and LP QTLs (Table 6). This cluster was located at 32∼45 cM, containing four QTLs.
Chr14 contained one cluster and three hotspots (Table 6). The Chr14-cluster-1 was located at 6∼26 cM and carried eight QTLs for FL, FS, MIC, and FE. The two hotspots, Chr14-hotspot-1 and Chr14-hotspot-2, identified fell into the range of the Chr14-cluster-1, and were FL and FS related hotspots, respectively. Furthermore, the three hotspots Chr14-hotspot-1, Chr14-hotspot-2, and Chr14-hotspot-3 for FE, FS, and FL overlapped at 1∼17, 6∼22, and 14∼26 cM, respectively, indicating a possible fiber quality hotspot cluster in the region.
Chr19 contained one cluster, Chr19-cluster-1, which ranged from 20∼28 cM, and carried four QTLs for FL, FU, and FS (Table 6).
Chr20 had one cluster Chr20-cluster-1, which was identified at 41∼60 cM and carried three QTLs related to FE and FS(Table 6).
Identification of the Epistatic and QTL × Environment Interactions Loci
Nineteen e-QTLs were identified by the MET analysis of the multi-environment phenotypic values (Supplementary Table S4; Figure 7). Fifteen of them had both an epistatic main effect and minor epistasis × environment interaction effect while remaining four had the former effect only. The explained PV by each QTL ranged from 3.89 to 6.22%, while that by each QTL × environment interaction only ranged from 0.0 to 0.67%. Among them, the highest number of e-QTLs (9) was detected for FL, three for MIC, two for FE, two for FS, one for BW, and two for LP. No e-QTL was detected for FU. For FL, there were two loci i38186Gh-i11502Gh and i15345Gh-i18849Gh overlapped with two additive QTLs, qFL-Chr10-1 and qFL-Chr14-2, indicating both additive and epistatic value of these two loci had played important role in FL. One pair of interacting marker intervals, i08786Gh-i00558Gh and i09371Gh-i09643Gh, was detected for FL on the same chromosome of Chr19, whereas other interacted loci were located on different chromosomes. In addition, some marker intervals had interactions with other multiple marker intervals to control same or different traits. The marker interval, i22642Gh-i41613Gh on Chr21, had interaction with two marker intervals, i35903Gh-i20966Gh on Chr03 and i44474Gh-i03341Gh on Chr17, for FL. The marker interval i16566Gh-i08941Gh on Chr19 interacted with two marker intervals, i14920Gh-i51624Gb on Chr17 and i08832Gh-i09452Gh on Chr19, for MIC. The marker interval i10502Gh-i36496Gh on Chr04 had interactions with two marker intervals, i08933Gh-i28797Gh on Chr19 and i05035Gh-i22015Gh on Chr14, to control two traits, FL and FS, respectively. The marker interval i32883Gh-i13851Gh on Chr18 had interactions with two marker intervals including i38186Gh-i11502Gh on Chr10 and i02298Gh-i42430Gh on Chr01 pertaining to FL and FE, respectively.
FIGURE 7.
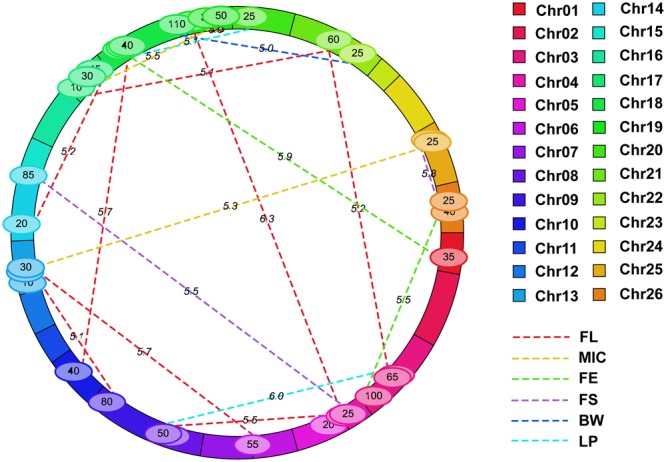
Epistatic and QE interaction loci for fiber quality and yield of RILs across two environments by IciMapping. The solid bars with different colors represent different chromosomes and the dotted line with different colors indicates different chromosomes, for which the legend is given at the side of figure. FL, fiber length; FU, fiber uniformity; MIC, micronaire; FE, fiber elongation; FS, fiber strength; BW, boll weight; LP, lint percentage.
Discussion
SNP Discovery and map Construction
With the development of theoretical and applied genetic breeding research, high-density genetic maps are becoming more and more important. SNPs were proved to be the most abundant form of genetic variation, providing a rich source of DNA markers (Agarwal et al., 2008). Recently, SNP arrays for many crops had been developed and utilized in MAS breeding (Fang et al., 2013; Gao et al., 2015; Wang X. et al., 2015). In present work, 2618 polymorphic SNP markers from a 63K SNP assay (Hulse-Kemp et al., 2015) were used to construct a relative high-density genetic map for 188 RILs derived from the combination of HS46/MARCABUCAG8US-1-88.
The total length of the linkage map was 1784.28 cM, far longer than previously map lengths reported with the same cross (Shappley et al., 1998; Wu et al., 2008). The interval of the map was 0.68 cM/marker, increasing the density of the previous linkage map to the thickest linkage map in upland cotton and representing a considerable advance over previously map researches based on RFLP, SSR, AFLP, ITISJ, SRAPetc (Shappley et al., 1998; Ulloa et al., 2002; Shen et al., 2005, 2006; Zhang et al., 2005, 2009; Wu et al., 2008; Tan et al., 2014; Liu et al., 2015). The collinearity analysis showed that the constructed map had good collinearity with the G. hirsutum reference genome, indicating the high quality and accuracy of the map. One main reason for the lower coverage of At subgenome than Dt subgenome was that two linkage groups, Chr12 and Chr10, only represent 10.91 and 70.34% of the corresponding chromosomes, respectively.
Consistent with previous studies (Tan et al., 2014; Cao et al., 2015; Hulse-Kemp et al., 2015), more markers were found on Dt subgenome (54.24%) than At subgenome (45.76%) in the present map, which was attributed to the lower level of polymorphism in At subgenome of upland cotton. The Dt subgenome was longer than At subgenome in this map, duo to Dt subgenome with more loci experienced a higher frequency of recombination (Guo et al., 2007). Although the average density was high, there were still some gaps in several chromosomes. Moreover, like previous maps (Shen et al., 2006; Tan et al., 2014; Cao et al., 2015), although the markers of our map distributed evenly on the entire genome, there were still some chromosomes anchored more markers than others, which might be attributed to the non-random distribution of markers and the lack of marker polymorphism between mapping parents on some chromosomes because of relative narrow range of genetic diversity in upland cotton.
Segregation distortion, regarded as the important source of plant evolution, widely exists in plant populations. The species, population types, crosses, and marker types of plants will lead to significant variance in segregation distortion with different origin, genetic effects, and degree (Xu et al., 1997). The reports from intraspecific population of upland cotton indicated that the ratio of segregation distortion enhanced with the increase of divergence level of the parents (Shappley et al., 1998; Ulloa et al., 2002; Shen et al., 2005, 2006), and the ratio of segregation distortion in the RIL population was higher than that in the F2 population, which might mainly result from genetic drift (Shen et al., 2006). In present study, 13.29% of the total loci showed segregation distortion (P < 0.05), which was similar to previous maps of upland cotton (16.6∼36.7%; Tan et al., 2014; Cao et al., 2015; Liu et al., 2015). Most distorted loci skewed to HS46 alleles, which might be the result of chromosomal elements markers diverged via female drive in the meiosis (Fishman and Willis, 2005). Furthermore, most segregation distortion loci occurred in the clusters, like previously reported for interspecific and intraspecific populations (Guo et al., 2007; Zhang et al., 2009; Yu et al., 2011b; Tan et al., 2014; Liu et al., 2015). The loci clustering on the same chromosome or within the same SDR skewed to the same allele suggested that genetic hitchhiking effects existed in upland cotton (Yu et al., 2011b).
Significance and Potential Application of QTL Mapping
The genotypic value for HS46 was greater (P < 0.05) than that for MARCABUCAG8US-1-88 with respect to FL, FU, BW, FS, and MIC. Similar to the same cross used by Wu et al. (2008), no significant difference between the two parents was detected for FE and LP. Even though the two parents were phenotypically similar regarding these two traits, due to genetic dissimilarities between the two parents, significant differences in this RIL population existed. In the present research, FS had a low heritability and easily affected by environment. Similar results could be found in previous literatures about a four-way cross population in upland cotton (Qin et al., 2008). The obvious difference indicated FS was highly influenced by the experimental environment and difficult in genetic improvement. Wang et al. (2015a) suggested FS was a moderate heritability trait and that all the fiber quality and yield component traits presented significant environmental effects. Moreover, in the research based on same population, FS did not show a higher heritability in all the measured traits but a significant VGE/VP instead (Wu et al., 2008). Therefore, the heritability of FS was not stable and affected by different population and environment.
Similarly, BW was highly influenced by environments. Furthermore, BW was significantly and positively correlated with MIC and FS. Consequently, it is feasible to improve BW by selecting these correlated traits, having more accurate repeatability across environments in breeding. Stable QTLs such as qFL-Chr14-3, qFL-Chr15-1, qFU-Chr09-1, qFE-Chr14-1 qFE-Chr20-1, qBW-Chr10-1, qLP-Chr10-1, and qLP-Chr12-1 could be used in breeding. It was more likely that these QTLs could be used to identify candidate genes for these related traits because of the availability of high-density SNP markers. In such case, they will be the potential candidates for fine mapping and ultimate candidate gene discovery.
Although there were many differences in parental lines, mapping populations, and markers type, our results were comparable with earlier identified QTLs. Searching for QTLs of upland cotton in a CottonQTLdb database3 developed by Said et al. (2015) and statistical data on cotton QTL previously, there were seven QTLs for FL, two QTLs for FU, two QTLs for MIC, four QTLs for FE, two QTLs for FS, and five QTLs for LP in present work, sharing same genetic position (spacing distance < 5 cM) and physical position with earlier reports (Wu et al., 2008; Tan et al., 2014; Liu et al., 2015; Zhang Z. et al., 2015). In addition, five QTLs for FL, six QTLs for FU, eight QTLs for MIC, four QTLs for FE, seven QTLs for FS, six QTLs for BW, and seven QTLs for LP had been mapped on the same chromosomes but not on the same position as reported in the previous researches. These inconsistencies might be due to different genetic backgrounds and DNA markers (Shappley et al., 1998; Wu et al., 2008; Said et al., 2015). Remaining nine additive QTLs (qFE-Chr17-1, qBW-Chr09-1, qBW-Chr09-2, qBW-Chr23-1, qBW-Chr24-1, qBW-Chr25-1, qLP-Chr20-1, qLP-Chr21-1, and qLP-Chr21-2) might be novel loci, due to unavailability of any reports for these traits on those chromosomes. As the number of identified QTLs for fiber quality and yield traits increased, the genetic control of fiber quality and yield will be better understood.
Molecular Mechanism of Trait Correlation and Linkage Drag
Co-localization of QTLs on chromosomes, referred to as “QTL cluster/hotspot,” has previously been reported in cotton (Shappley et al., 1998; Qin et al., 2008; Said et al., 2013) and many other species (Gonzalez et al., 2015; Li et al., 2015). In the present study, 12 certain genomic regions, especially, Chr05, Chr09, Chr10, Chr14, Chr19, and Chr20, were investigated for their involvement in controlling one (hotspot) or more (clusters) fiber quality or yield traits, the similar result was also reported in the publications (Qin et al., 2008; Sun et al., 2011; Said et al., 2013; Yu et al., 2013). The existence of QTL clusters explained why so many traits were highly interrelated.
Based on the comprehensive analysis of clusters and hotspots in this study, breeding programs targeting fiber quality or yield traits can focus on hotspot clustering regions and select around the region. Notably, almost all the hotspots overlapped QTL clusters. The presence of QTL clusters and hotspots proved that genes related to certain traits were more heavily concentrated in certain regions of genome than others (Said et al., 2013). The discovery of cluster and hotspot may be useful in MAS breeding program and may help breeders to select the traits of interests and find novel QTLs once the markers have anchored these regions.
Interaction between Loci within and Across Chromosomes
It was successful to identify e-QTLs with both additive and epistatic effects, e-QTL pairs and epistasis × environment interactions in the present work, which were often neglected in some complex trait studies. Generally, if the proportion of PV explained by the identified additive QTL is close to broad-sense heritability, epistasis is less important (Li et al., 2007). However, for FL, the total PV explained by additive QTLs were much lower than the broad-sense heritability (66%, Table 2), indicating that there were epistatic interactions in these loci. Finally, nine pairs of e-QTLs for FL were detected in our work, of which there were two loci, i38186Gh-i11502Gh on Chr10 and i15345Gh-i18849Gh on Chr14, locating on the same position with two additive QTLs (qFL-Chr10-1 and qFL-Chr14-2), suggesting that both addictive and epistatic effects had played important roles in genetic control of FL. In fact, interactions among loci or QTL × environmental factors contributed a substantial effect to complex trait phenotypic variation (Carlborg and Haley, 2004). Several novel QTLs and specific trait relationships between loci, within and across chromosomes, could be considered as the interactions between loci (Grosse-Brinkhaus et al., 2010).
Conclusion
A high-density linkage map was constructed in the upland cotton RIL population using the 63K cotton SNP array. Nine novel QTLs, seven pleiotropic QTL clusters, five hotspots, and 19 e-QTLs for fiber quality and yield traits were identified with tightly linked SNP markers. These QTLs could serve as target regions for map-based gene cloning and MAS in cotton breeding.
Author Contributions
CoL and SZ designed and conducted the experiments, analyzed data and wrote the manuscript. CoL and YD performed phenotyping and data analysis, CoL, TZ, and LL participated in field trials. ChL, EY, LM, QH, and MD prepared and reviewed the manuscript. SZ and JC designed and supervised the experiments. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. XU (Zhejiang University, China), Li Chen (Zhejiang University, China), and Zhen Zhang (Chinese Academy of Agricultural Sciences, China) for technical assistance. The work was funded by the National High Technology Research and Development Program of China (2013AA102601), China Agriculture Research System (CARS-18-25), and Jiangsu Collaborative Innovation Center for Modern Crop Production.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01356
References
- Agarwal M., Shrivastava N., Padh H. (2008). Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 27 617–631. 10.1007/s00299-008-0507-z [DOI] [PubMed] [Google Scholar]
- Ball A. D., Stapley J., Dawson D. A., Birkhead T. R., Burke T., Slate J. (2010). A comparison of SNPs and microsatellites as linkage mapping markers: lessons from the zebra finch (Taeniopygia guttata). BMC Genomics 11:218 10.1186/1471-2164-11-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales J., Laurie D. A., Devos K. M. (2005). Allelic variation at the linked AP1 and PhyC loci in hexaploid wheat is associated but not perfectly correlated with vernalization response. Theor. Appl. Genet. 110 1099–1107. 10.1007/s00122-005-1938-3 [DOI] [PubMed] [Google Scholar]
- Byers R. L., Harker D. B., Yourstone S. M., Maughan P. J., Udall J. A. (2012). Development and mapping of SNP assays in allotetraploid cotton. Theor. Appl. Genet. 124 1201–1214. 10.1007/s00122-011-1780-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C., Ye W., Zhang T., Guo W. (2014). Association analysis of fiber quality traits and exploration of elite alleles in upland cotton cultivars/accessions (Gossypium hirsutum L.). J. Integr. Plant Biol. 56 51–62. 10.1111/jipb.12124 [DOI] [PubMed] [Google Scholar]
- Cao Z., Zhu X., Chen H., Zhang T. (2015). Fine mapping of clustered quantitative trait loci for fiber quality on chromosome 7 using a Gossypium barbadense introgressed line. Mol. Breed. 35 1–13. 10.1007/s11032-015-0393-3 [DOI] [Google Scholar]
- Carlborg O., Haley C. S. (2004). Epistasis: too often neglected in complex trait studies? Nat. Rev. Genet. 5 618–625. 10.1038/nrg1407 [DOI] [PubMed] [Google Scholar]
- Fang H., Zhou H., Sanogo S., Lipka A. E., Fang D. D., Percy R. G., et al. (2013). Quantitative trait locus analysis of Verticillium wilt resistance in an introgressed recombinant inbred population of Upland cotton. Mol. Breed. 33 709–720. 10.1007/s11032-013-9987-9 [DOI] [Google Scholar]
- Fishman L., Willis J. H. (2005). A novel meiotic drive locus almost completely distorts segregation in mimulus (monkey flower) hybrids. Genetics 169 347–353. 10.1534/genetics.104.032789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Wen W., Liu J., Rasheed A., Yin G., Xia X., et al. (2015). Genome-wide linkage mapping of QTL for yield components, plant eight and yield-related physiological traits in the Chinese wheat cross zhou 8425B/Chinese spring. Front. Plant Sci. 6:1099 10.3389/fpls.2015.01099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A. M., Yuste-Lisbona F. J., Rodino A. P., De Ron A. M., Capel C., Garcia-Alcazar M., et al. (2015). Uncovering the genetic architecture of Colletotrichum lindemuthianum resistance through QTL mapping and epistatic interaction analysis in common bean. Front. Plant Sci. 6:141 10.3389/fpls.2015.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore M. A., Fang D. D., Poland J. A., Zhang J., Percy R. G., Cantrell R. G., et al. (2014). Linkage pap construction and quantitative trait locus analysis of agronomic and fiber quality traits in cotton. Plant Genome 7 1–10. 10.3835/plantgenome2013.07.0023 [DOI] [Google Scholar]
- Grosse-Brinkhaus C., Jonas E., Buschbell H., Phatsara C., Tesfaye D., Jungst H., et al. (2010). Epistatic QTL pairs associated with meat quality and carcass composition traits in a porcine Duroc x Pietrain population. Genet. Sel. Evol. 42:39 10.1186/1297-9686-42-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Cai C., Wang C., Han Z., Song X., Wang K., et al. (2007). A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics 176 527–541. 10.1534/genetics.107.070375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse-Kemp A. M., Lemm J., Plieske J., Ashrafi H., Buyyarapu R., Fang D. D., et al. (2015). Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 (Bethesda) 5 1187–1209. 10.1534/g3.115.018416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. S., Thyssen G. N., Jenkins J. N., Fang D. D. (2015). Detection, validation, and application of genotyping-by-sequencing based single nucleotide polymorphisms in upland cotton. Plant Genome 8 10.3835/plantgenome2014.07.0034 [DOI] [PubMed] [Google Scholar]
- Konishi S., Izawa T., Lin S. Y., Ebana K., Fukuta Y., Sasaki T., et al. (2006). An SNP caused loss of seed shattering during rice domestication. Science 312 1392–1396. 10.1126/science.1126410 [DOI] [PubMed] [Google Scholar]
- Kosambi D. D. (1944). The estimation of map distance from recombination values. Ann. Eugen. 12 172–175. 10.1111/j.1469-1809.1943.tb02321.x [DOI] [Google Scholar]
- Lacape J. M., Claverie M., Vidal R. O., Carazzolle M. F., Guimaraes Pereira G. A., Ruiz M., et al. (2012). Deep sequencing reveals differences in the transcriptional landscapes of fibers from two cultivated species of cotton. PLoS ONE 7:e48855 10.1371/journal.pone.0048855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacape J. M., Nguyen T. B., Thibivilliers S., Bojinov B., Courtois B., Cantrell R. G., et al. (2003). A combined RFLP-SSR-AFLP map of tetraploid cotton based on a Gossypium hirsutum x Gossypium barbadense backcross population. Genome 46 612–626. 10.1139/g03-050 [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Ye G., Wang J. (2007). A modified algorithm for the improvement of composite interval mapping. Genetics 175 361–374. 10.1534/genetics.106.066811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang W., Wang Z., Li K., Lim Y. P., Piao Z. (2015). Construction of chromosome segment substitution lines enables QTL mapping for flowering and morphological traits in Brassica rapa. Front. Plant Sci. 6:432 10.3389/fpls.2015.00432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q., Hu C., Hua H., Li Z., Hua J. (2013). Construction of a linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Chin. Sci. Bull. 58 3233–3243. 10.1007/s11434-013-5807-1 [DOI] [Google Scholar]
- Liu D., Liu F., Shan X., Zhang J., Tang S., Fang X., et al. (2015). Construction of a high-density genetic map and lint percentage and cottonseed nutrient trait QTL identification in upland cotton (Gossypium hirsutum L.). Mol. Genet. Genomics 290 1683–1700. 10.1007/s00438-015-1027-5 [DOI] [PubMed] [Google Scholar]
- Liu H. Y., Quampah A., Chen J. H., Li J. R., Huang Z. R., He Q. L., et al. (2012). QTL analysis for gossypol and protein contents in upland cottonseeds with two different genetic systems across environments. Euphytica 188 453–463. 10.1007/s10681-012-0733-x [DOI] [Google Scholar]
- McNally K. L., Childs K. L., Bohnert R., Davidson R. M., Zhao K., Ulat V. J., et al. (2009). Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. U.S.A. 106 12273–12278. 10.1073/pnas.0900992106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H., Zhu X., Zhang T. (2013). Favorable QTL alleles for yield and its components identified by association mapping in Chinese upland cotton cultivars. PLoS ONE 8:e82193 10.1371/journal.pone.0082193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. B., Giband M., Brottier P., Risterucci A. M., Lacape J. M. (2004). Wide coverage of the tetraploid cotton genome using newly developed microsatellite markers. Theor. Appl. Genet. 109 167–175. 10.1007/s00122-004-1612-1 [DOI] [PubMed] [Google Scholar]
- Paterson A. H., Brubaker C. L., Wendel J. F. (1993). A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Rep. 11 122–127. 10.1007/BF02670470 [DOI] [Google Scholar]
- Qin H., Guo W., Zhang Y. M., Zhang T. (2008). QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor. Appl. Genet. 117 883–894. 10.1007/s00122-008-0828-x [DOI] [PubMed] [Google Scholar]
- Rai K. M., Singh S. K., Bhardwaj A., Kumar V., Lakhwani D., Srivastava A., et al. (2013). Large-scale resource development in Gossypium hirsutum L. by 454 sequencing of genic-enriched libraries from six diverse genotypes. Plant Biotechnol. J. 11 953–963. 10.1111/pbi.12088 [DOI] [PubMed] [Google Scholar]
- Reinisch A. J., Dong J. M., Brubaker C. L., Stelly D. M., Wendel J. F., Paterson A. H. (1994). A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138 829–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong J., Abbey C., Bowers J. E., Brubaker C. L., Chang C., Chee P. W., et al. (2004). A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166 389–417. 10.1534/genetics.166.1.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong J., Feltus F. A., Waghmare V. N., Pierce G. J., Chee P. W., Draye X., et al. (2007). Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176 2577–2588. 10.1534/genetics.107.074518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said J. I., Knapka J. A., Song M., Zhang J. (2015). Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol. Genet. Genomics 290 1615–1625. 10.1007/s00438-015-1021-y [DOI] [PubMed] [Google Scholar]
- Said J. I., Lin Z., Zhang X., Song M., Zhang J. (2013). A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genomics 14:776 10.1186/1471-2164-14-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shappley Z. W., Jenkins J. N., Meredith W. R., McCarty J. C. (1998). An RFLP linkage map of upland cotton, Gossypium hirsutum L. Theor. Appl. Genet. 97 756–761. 10.1007/s001220050952 [DOI] [Google Scholar]
- Shen X., Guo W., Lu Q., Zhu X., Yuan Y., Zhang T. (2006). Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in upland cotton. Euphytica 155 371–380. 10.1007/s10681-006-9338-6 [DOI] [Google Scholar]
- Shen X., Guo W., Zhu X., Yuan Y., Yu J. Z., Kohel R. J., et al. (2005). Molecular mapping of QTLs for fiber qualities in three diverse lines in upland cotton using SSR markers. Mol. Breed. 15 169–181. 10.1007/s11032-004-4731-0 [DOI] [Google Scholar]
- Sun F. D., Zhang J. H., Wang S. F., Gong W. K., Shi Y. Z., Liu A. Y., et al. (2011). QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 30 569–582. 10.1007/s11032-011-9645-z [DOI] [Google Scholar]
- Tan Z., Fang X., Tang S., Zhang J., Liu D., Teng Z., et al. (2014). Genetic map and QTL controlling fiber quality traits in upland cotton (Gossypium hirsutum L.). Euphytica 203 615–628. 10.1007/s10681-014-1288-9 [DOI] [Google Scholar]
- Ulloa M., Meredith W. R., Jr., Shappley Z. W., Kahler A. L. (2002). RFLP genetic linkage maps from four F(2.3) populations and a joinmap of Gossypium hirsutum L. Theor. Appl. Genet. 104 200–208. 10.1007/s001220100739 [DOI] [PubMed] [Google Scholar]
- Van Deynze A., Stoffel K., Lee M., Wilkins T. A., Kozik A., Cantrell R. G., et al. (2009). Sampling nucleotide diversity in cotton. BMC Plant Biol. 9:125 10.1186/1471-2229-9-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ooijen J. W. (2006). JoinMap 4.0 Software for the Calculation of Genetic Linkage Maps in Experimental Populations. Wageningen: Kyazma B.V. [Google Scholar]
- Voorrips R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93 77–78. 10.1093/jhered/93.1.77 [DOI] [PubMed] [Google Scholar]
- Wang B., Guo W., Zhu X., Wu Y., Huang N., Zhang T. (2006). QTL mapping of fiber quality in an elite hybrid derived-RIL population of upland cotton. Euphytica 152 367–378. 10.1007/s10681-006-9224-2 [DOI] [Google Scholar]
- Wang H., Huang C., Guo H., Li X., Zhao W., Dai B. (2015a). QTL mapping for fiber and yield traits in upland cotton under multiple environments. PLoS ONE 10:e0130742 10.1371/journal.pone.0130742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Jin X., Zhang B., Shen C., Lin Z. (2015b). Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. 22 147–160. 10.1093/dnares/dsu047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Basten C. J., Zeng Z. B. (2001). Windows QTL Cartographer 2.5. Department of Statistics, North Carolina.State University, Raleigh, NC. Available at: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm [Accessed February 1, 2013]. [Google Scholar]
- Wang X., Yu K., Li H., Peng Q., Chen F., Zhang W., et al. (2015). High-density SNP map construction and QTL identification for the apetalous character in Brassica napus L. Front. Plant Sci. 6:1164 10.3389/fpls.2015.01164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ning Z., Hu Y., Chen J., Zhao R., Chen H., et al. (2015). Molecular mapping of restriction-site associated DNA markers in allotetraploid upland cotton. PLoS ONE 10:e0124781 10.1371/journal.pone.0124781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Gutierrez O. A., Jenkins J. N., McCarty J. C., Zhu J. (2008). Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165 231–245. 10.1007/s10681-008-9748-8 [DOI] [Google Scholar]
- Xu Y., Zhu L., Xiao J., Huang N., McCouch S. R. (1997). Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid, and recombinant inbred populations in rice (Oryza sativa L.). Mol. Gen. Genet. 253 535–545. 10.1007/s004380050355 [DOI] [PubMed] [Google Scholar]
- Yu H., Xie W., Wang J., Xing Y., Xu C., Li X., et al. (2011a). Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 6:e17595 10.1371/journal.pone.0017595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zhang K., Li S., Yu S., Zhai H., Wu M., et al. (2013). Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 126 275–287. 10.1007/s00122-012-1980-x [DOI] [PubMed] [Google Scholar]
- Yu J. Z., Kohel R. J., Fang D. D., Cho J., Van Deynze A., Ulloa M., et al. (2012). A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 (Bethesda) 2 43–58. 10.1534/g3.111.001552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Yuan D., Liang S., Li X., Wang X., Lin Z., et al. (2011b). Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genomics 12:15 10.1186/1471-2164-12-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lu Y., Yu S. (2005). Cleaved AFLP (cAFLP), a modified amplified fragment length polymorphism analysis for cotton. Theor. Appl. Genet. 111 1385–1395. 10.1007/s00122-005-0070-8 [DOI] [PubMed] [Google Scholar]
- Zhang S., Wang T., Liu Q., Gao X., Zhu X., Zhang T., et al. (2014). Quantitative trait locus analysis of boll-related traits in an intraspecific population of Gossypium hirsutum. Euphytica 203 121–144. 10.1007/s10681-014-1281-3 [DOI] [Google Scholar]
- Zhang T., Hu Y., Jiang W., Fang L., Guan X., Chen J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33 531–537. 10.1038/nbt.3207 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Li J., Muhammad J., Cai J., Jia F., Shi Y., et al. (2015). High resolution consensus mapping of quantitative trait loci for fiber strength, length and micronaire on chromosome 25 of the upland cotton (Gossypium hirsutum L.). PLoS ONE 10:e0135430 10.1371/journal.pone.0135430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Yuan Y., Yu J., Guo W., Kohel R. J. (2003). Molecular tagging of a major QTL for fiber strength in upland cotton and its marker-assisted selection. Theor. Appl. Genet. 106 262–268. 10.1007/s00122-002-1101-3 [DOI] [PubMed] [Google Scholar]
- Zhang Z. S., Hu M. C., Zhang J., Liu D. J., Zheng J., Zhang K., et al. (2009). Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol. Breed. 24 49–61. 10.1007/s11032-009-9271-1 [DOI] [Google Scholar]
- Zhu Q. H., Spriggs A., Taylor J. M., Llewellyn D., Wilson I. (2014). Transcriptome and complexity-reduced, DNA-based identification of intraspecies single-nucleotide polymorphisms in the polyploid Gossypium hirsutum L. G3 (Bethesda) 4 1893–1905. 10.1534/g3.114.012542 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.