

Abstract
Background
Bioavailability of nitric oxide (NO) and hydrogen sulfide (H2S) is reduced in heart failure (HF). Recent studies suggest cross‐talk between NO and H2S signaling. We previously reported that sodium nitrite (NaNO 2) ameliorates myocardial ischemia‐reperfusion injury and HF. Nuclear factor‐erythroid‐2‐related factor 2 (Nrf2) regulates the antioxidant proteins expression and is upregulated by H2S. We examined the NaNO 2 effects on endogenous H2S bioavailability and Nrf2 activation in mice subjected to ischemia‐induced chronic heart failure (CHF).
Methods and Results
Mice underwent 60 minutes of left coronary artery occlusion and 4 weeks of reperfusion. NaNO 2 (165 μg/kgic) or vehicle was administered at reperfusion and then in drinking water (100 mg/L) for 4 weeks. Left ventricular (LV), ejection fraction (EF), LV end diastolic (LVEDD) and systolic dimensions (LVESD) were determined at baseline and at 4 weeks of reperfusion. Myocardial tissue was analyzed for oxidative stress and respective gene/protein‐related assays. We found that NaNO 2 therapy preserved LVEF, LVEDD and LVSD at 4 weeks during ischemia‐induced HF. Myocardial malondialdehyde and protein carbonyl content were significantly reduced in NaNO 2‐treated mice as compared to vehicle, suggesting a reduction in oxidative stress. NaNO 2 therapy markedly increased expression of Cu,Zn‐superoxide dismutase, catalase, and glutathione peroxidase during 4 weeks of reperfusion. Furthermore, NaNO 2 upregulated the activity of Nrf2, as well as H2S‐producing enzymes, and ultimately increased H2S bioavailability in ischemia‐induced CHF in mice as compared with vehicle.
Conclusions
Our results demonstrate that NaNO 2 therapy significantly improves LV function via increasing H2S bioavailability, Nrf2 activation, and antioxidant defenses.
Keywords: antioxidant, H2S, heart failure, nitric oxide, Nrf2, reactive oxygen species
Subject Categories: Heart Failure, Oxidant Stress, Ischemia
Introduction
Nitric oxide (NO) is a gaseous signaling molecule that plays a pivotal role in controlling cardiovascular homeostasis.1, 2 NO is synthesized endogenously via 3 isoforms of NO synthase (NOS) as well as by nonenzymatic reduction of nitrate () and nitrite (). The anion forms as a consequence of NO oxidation and it is present at a concentration of 0.3 to 1.0 μmol/L in plasma and 1 to 20 μmol/L in tissue.3, 4, 5 This nitrite is physiologically recycled in the blood and tissues on demand,2 acting as precursor of NO during hypoxia and acidosis.6, 7 In the setting of heart failure (HF), endothelial nitric oxide synthase (eNOS) activity is reduced, leading to decreased bioavailability of NO, an increase in reactive oxygen species (ROS) levels, and decrease of antioxidant defense.8 Ischemia and reperfusion (I/R) is characterized by the formation of ROS upon reintroduction of molecular oxygen and metabolic substrates in ischemic tissue, resulting in widespread lipid and protein oxidative modifications, tissue apoptosis, and necrosis.9 Several studies have shown that eNOS‐derived NO protects against myocardial ischemia reperfusion (MI/R) injury, attenuates the severity of HF,10 and decreases apoptosis.11 NO, NO donors, NO synthase activation, and/or transgenic overexpression have been shown to protect against I/R injury in a number of animal models.12, 13, 14 Indeed, evidence that eNOS/NO provides protection against HF comes from animal studies in which overexpression of eNOS protects against,12 whereas genetic deficiency of eNOS enhances the development of HF.15 In addition, experimental studies have shown that enhancing NO levels through genetic manipulation leads to improved survival,12 decreased remodeling,16 and improved cardiac function following ischemia‐induced HF in mice. Among NO donors, sodium nitrite (NaNO2) represents a promising therapeutic agent for the treatment of HF,17 being an attractive candidate for restoring physiological NO signaling in states of chronic NO insufficiency such as myocardial ischemia. NaNO2 is rapidly absorbed from the circulation by peripheral tissues and stored in cells until conversion to NO is needed.18, 19, 20 Indeed, NaNO2 administration has been demonstrated to protect against I/R injury21 in multiple tissues and organs, such as skeletal muscle,22 renal,23, 24 and liver.25, 26, 27 In addition, eNOS transgenic mice have higher levels of in the plasma and they are protected against I/R injury.28
Hydrogen sulfide (H2S) is a critical cell‐signaling molecule required for cardiovascular homeostasis, much like NO.29, 30, 31 The production of H2S in mammalian systems has been attributed to 3 endogenous enzymes: cystathionine β‐synthase (CBS), cystathionine γ‐lyase (CSE), and 3‐mercaptopyruvate sulfur transferase (3‐MST).32 Although the precise mechanisms by which H2S protects the cardiovascular system are still under investigation, both endogenous and exogenous H2S elicit a wide range of protective actions including vasodilation, anti‐inflammatory, antioxidant, anti‐apoptotic, and modulation of cellular metabolism.33 Both NO and H2S are known to increase heme oxygenase 1 (HO‐1) levels, an enzyme that produces carbon monoxide.34 This suggests that the activation of 1 of the endogenously produced gases can lead to the activation of the other 2 gases. Under these conditions, the 3 gases have the ability to synergize their anti‐apoptotic, anti‐inflammatory, and antihypertrophic effects, which lead to cardioprotection. Although H2S and NO are thought to modulate independent pathways, there is evidence of cross‐talk between these 2 signaling molecules.35, 36 Our laboratory previously demonstrated that treatment with exogenous H2S or modulation of the endogenous production of H2S through the cardiac‐specific overexpression of the H2S‐generating enzyme CSE protects against acute MI/R injury and HF by attenuating oxidative stress, inhibiting apoptosis, and reducing inflammation.34, 37 Furthermore, H2S therapy improves survival after cardiac arrest and cardiopulmonary resuscitation in an eNOS‐dependent38 manner and provides cardioprotection against MI/R injury by activating eNOS/NO.
In addition, recently we and others have demonstrated that 1 mechanism by which H2S exerts cytoprotective actions is via upregulation of cellular antioxidants in a nuclear factor‐erythroid‐2‐related factor 2 (Nrf2)‐dependent manner.34 H2S has recently been shown to sulfhydrate Keap1, which results in the release and translocation of Nrf2 to the nucleus.39 Nrf2 regulates the gene expression of a number of enzymes that serve to detoxify pro‐oxidative stressors,40 such as glutathione peroxidase (GPX) and HO‐1 by binding to the antioxidant response element found in the gene promoter region.34 In the same regard, there is also evidence that NO possesses antioxidant effects and can activate Nrf2 and HO‐1.41, 42 However, the mechanism by which NO reduces oxidative stress following MI/R remains unknown. Furthermore, the role of NO is being re‐evaluated with the appreciation of H2S that also serves many regulatory roles in physiological systems. To date, no evidence exists as to whether increasing NO bioavailability promotes H2S signaling activation, leading to protection of cardiac function.
In our study, we demonstrate that long term administration of NaNO2 during ischemia‐induced HF results in improved left ventricular (LV) function through increased H2S bioavailability, Nrf2 activation, elevation of antioxidant proteins, and consequently suppression of myocardial oxidative stress.
Materials and Methods
Mice
Male C57BL/6J mice 8 to 10 weeks of age were purchased from the Jackson Laboratory (Bar Harbor, ME). All experimental protocols were approved by the Institute for Animal Care and Use Committee at Louisiana State University Health Sciences Center and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication NO. 86‐23, revised 1996), and with Federal and state regulation.
MI/R Injury
Male C57BL/6J mice underwent 60 minutes of MI induced by transient occlusion of the left coronary artery followed by 4 weeks of reperfusion. Saline (vehicle [VEH]) or NaNO2 was administered at a dose of 165 μg/kg by intracardiac injection at reperfusion, and then 50 or 100 mg/L was administered in the drinking water for 4 weeks. Two‐dimensional echocardiography was performed at baseline before MI/R injury and at 4 weeks post‐MI to assess left ventricular ejection fraction, left ventricular end diastolic dimension, and left ventricular end systolic dimension. After 4 weeks of reperfusion, mice were euthanized and myocardial and blood samples were saved to perform further analysis.
Measurement of NO Metabolites
Nitrite concentrations in plasma and myocardial tissues of VEH and nitrite (100 mg/L)‐treated mice were quantified by ion chromatography (ENO20 Analyzer; Eicom, Kyoto, Japan).
Immunoblot Analysis
Protein samples obtained from heart tissues of VEH and nitrite‐treated mice were analyzed by immunoblotting43 using specific antibodies to superoxide dismutase 1 (SOD1; Santa Cruz Biotechnology, Santa Cruz, CA), GPX (Santa Cruz Biotechnology, Santa Cruz, CA), eNOS (BD Bioscience, San Jose, CA), eNOS‐phospho‐Ser1177 (abcam, Cambridge, UK), eNOS‐phospho‐Thr495 (Santa Cruz Biotechnology, Santa Cruz, CA), catalase (Santa Cruz Biotechnology, Santa Cruz, CA), Nrf2 (Santa Cruz Biotechnology, Santa Cruz, CA), CBS (Santa Cruz Biotechnology, Santa Cruz, CA), CSE (Abnova, Walnut, CA), 3‐MST (Novus, Littleton, CO), fibrillarin (Cell Signaling, Danvers, MA), and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA).
RNA Isolation and Reverse Transcriptase Real‐Time Quantitative Polymerase Chain Reaction
RNA was isolated from the heart tissue of VEH and nitrite (100 mg/L)–treated mice at 4 weeks of reperfusion. One microgram of RNA was transcribed using an I‐script cDNA synthesis kit from Bio‐Rad. TaqMan primers for CBS, CSE, 3‐MST, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), myosin heavy chain beta (Myh7), SOD1, GPX, catalase, beclin‐1, autophagy‐related gene 5 (ATG‐5) and 7 (ATG‐7) from Life Technology (Carlsbad, CA) were used to amplify quantitative polymerase chain reaction and 18s was used as a housekeeping gene. 2^‐delta‐delta cycle threshold (Ct) was used for the data analysis of quantitative polymerase chain reaction.43
Measurement of Total Antioxidant Capacity
Total antioxidant capacity for plasma and heart tissues obtained from VEH and nitrite (100 mg/L)–treated CHF mice were measured by the Trolox equivalent antioxidant capacity assay kit (abcam).
Determination of Protein Carbonyl Contents
Protein carbonyl contents in heart tissues of VEH and nitrite (100 mg/L)–treated chronic heart failure (CHF) mice were measured as described previously.44
Measurement of Malondialdehyde Levels
Malondialdehyde (MDA) levels in heart tissues of VEH and nitrite (100 mg/L)–treated CHF mice were assayed as described previously.45
Measurement of H2S
H2S levels were measured in plasma and protein extracts from heart tissue of VEH and mice treated with nitrite (100 mg/L) at 4 weeks reperfusion by gas chromatography chemiluminescence.46
Statistical Analysis
All data were expressed as the mean±SEM. Statistical significance of multiple treatments was determined by 1‐way or 2‐way ANOVA Bonferroni multiple comparison test. Multiple comparison adjustment was also performed. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). A P value of <0.05 was considered statistically significant.
Results
To investigate the effects of nitrite therapy in ischemia‐induced CHF mice, we followed the protocol as mentioned in the Materials and Methods section. At baseline and 4 weeks of reperfusion after 60 minutes of ischemia, mice were subjected to cardiac function and structure evaluation. We tested doses of 50 and 100 mg/L of NaNO2 administered in drinking water for 4 weeks. Significant improvements in LV performance (LV ejection fraction) and remodeling (LV end diastolic dimension and LV end systolic dimension) were observed when CHF mice were treated with the dosage of 100 mg/L of NaNO2 (Figure 1A through 1C) as compared with VEH. Therefore, mice that received a dose of NaNO2 100 mg/L were selected for further experiments.
Figure 1.
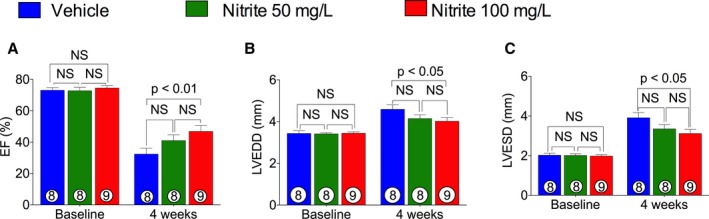
Effects of nitrite doses on ischemia‐induced CHF. After 60 minutes of ischemia, mice were given 50 and 100 mg/L of NaNO 2 in drinking water daily during 4 weeks of reperfusion. Left ventricular (LV) ejection fraction (EF) (A), LV end diastolic dimension (LVEDD) (B), and LV end systolic dimension (LVESD) (C) were measured at baseline and at 4 weeks of reperfusion. The number inside the bar denotes the number of animals used per group. Statistical significance of multiple treatments was determined by 2‐way ANOVA Bonferroni multiple comparison test. Multiple comparison adjustment was also performed. CHF indicates chronic heart failure; NS, not significant.
Since there is considerable evidence showing reduction of NO bioavailability during HF,8 we determined whether oral nitrite therapy restores or ameliorates the levels of circulatory and tissue NO. Therefore, samples obtained from sham, VEH, and nitrite‐treated mice were analyzed for nitrite levels. As can be seen in Figure 2A and 2B, the induction of myocardial ischemia significantly reduced NO levels in both myocardium and plasma of VEH as compared to sham animals. Interestingly, nitrite administration significantly increased both myocardial and circulating nitrite levels in ischemia‐induced CHF mice as compared with VEH. These results indicate that long term nitrite therapy restores physiological circulating levels of NO and augments its bioavailability within myocardial tissue, suggesting a significant role in protection of cardiac function and structure during CHF.
Figure 2.
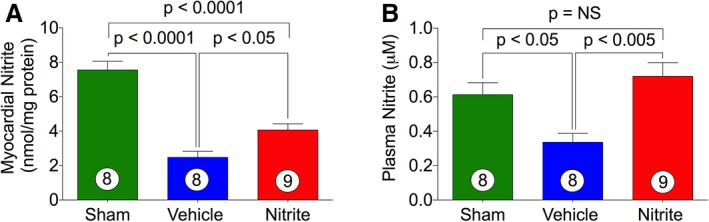
Effects of nitrite therapy on myocardial and circulatory nitrite levels. Heart (A) and plasma (B) nitrite levels in sham and CHF mice. Nitrite (100 mg/L) was given in the drinking water during 4 weeks of reperfusion period. The number inside the bar denotes the number of animals used per experiment. Statistical significance of multiple treatments was determined by 1‐way Bonferroni multiple comparison test. Multiple comparison adjustment was also performed. CHF indicates chronic heart failure; NS, not significant.
We then measured the nitrite effects on the expression of hypertrophic genes ANP, BNP, and Myh7 (myosin heavy chain β), which are reported to be upregulated during HF. Figure 3A through 3C show that ANP, BNP, and Myh7 expression were significantly decreased by nitrite treatment in ischemia‐induced CHF mice as compared with VEH. These data suggest that nitrite administration preserves cardiac function in part via suppressing hypertrophic genes.
Figure 3.
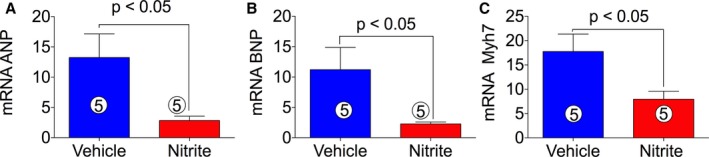
Effects of nitrite therapy on hypertrophic gene expression in ischemia‐induced CHF mice. Nitrite (100 mg/L) was given in the drinking water during reperfusion. cDNA was prepared from RNA obtained from mouse heart tissues followed by analysis of mRNA of ANP (A), BNP (B), and Myh7 (C) using TaqMan PCR assay system. The number inside the bar denotes the number of animals used per experiment. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). ANP indicates atrial natriuretic peptide; BNP, brain natriuretic peptide; Myh7, myosin heavy chain beta; PCR, polymerase chain reaction.
We studied the effect of oral nitrite therapy on total antioxidant capacity and oxidative damage during ischemia‐induced CHF in mice. Total antioxidant capacity was estimated by Trolox equivalent capacity assay in both heart tissues and plasma (Figure 4A and 4B) while oxidative modifications were determined by measuring MDA and protein carbonyl contents in myocardial tissue samples (Figure 4C and 4D). Figure 4A and 4B show that nitrite treatment increased the total antioxidant capacity in both myocardium and plasma obtained from ischemia‐induced CHF mice. Additionally, Figure 4C and 4D show that levels of both MDA and protein carbonyl contents were significantly decreased in nitrite‐treated CHF mice as compared with VEH. For further confirmation of the antioxidant effects of oral nitrite therapy, we also measured the antioxidant proteins levels. Figure 5A through 5G shows that nitrite treatment increased both myocardial mRNA and protein levels of SOD1, catalase, and GPX in ischemia‐induced CHF mice as compared with VEH.
Figure 4.
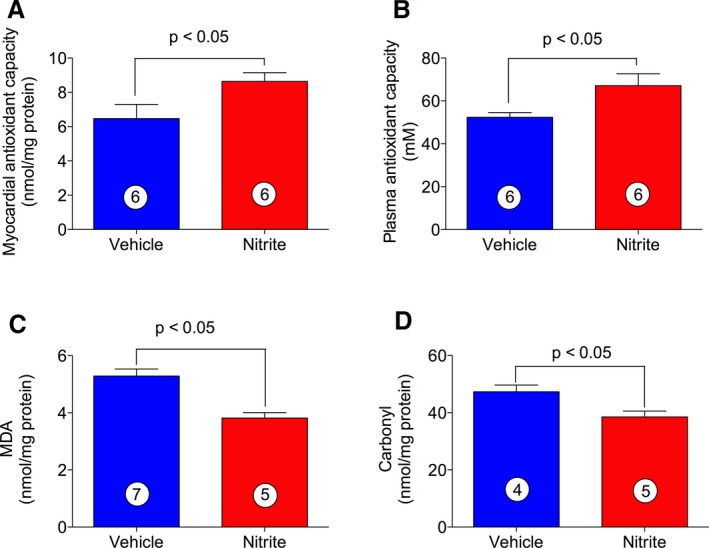
Effects of nitrite on oxidative stress during ischemia‐induced CHF. Total antioxidant capacity in both heart tissues (A) and plasma (B) were measured by the Trolox equivalent capacity assay. Malondialdehyde (C) and carbonyl contents (D) were measured in total protein extracts obtained from mouse heart tissues. The number inside the bar denotes the number of animals per group. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). CHF indicates chronic heart failure.
Figure 5.
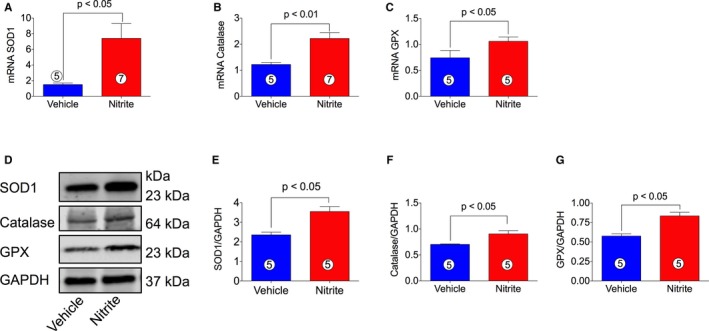
Effects of nitrite on cardiac antioxidant gene and protein levels in CHF mice. cDNA was prepared from RNA obtained from mouse heart tissues followed by analysis of mRNA of SOD1 (A), catalase (B), and GPX (C) using TaqMan PCR assay system. (D) represents proteins levels of SOD1, catalase, and GPX, and (E), (F), and (G) represent the quantitation of the blots in (D). The number in the circle inside the bar denotes the number of animals used. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). CHF indicates chronic heart failure; GPX, glutathione peroxidase; PCR, polymerase chain reaction; SOD1, superoxide dismutase 1.
Nrf2 and its target genes are critical regulators of cardiovascular homeostasis via suppressing oxidative stress/ROS, which is central in the development and progression of HF. Therefore, it was of interest to determine the effects of nitrite therapy on the activation status of myocardial Nrf2 in CHF mice after reperfusion. Figure 6A through 6C shows that nitrite significantly increased both cytosolic and nuclear levels of Nrf2 in myocardial tissue harvested from nitrite‐treated CHF mice as compared with VEH. These data indicate that nitrite therapy increases Nrf2 activation.
Figure 6.
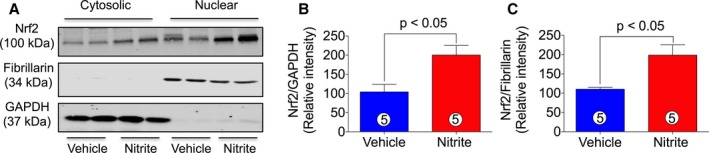
Nitrite treatment promotes Nrf2 activation in ischemia‐induced CHF mice. Cytosolic and nuclear proteins were prepared from mice heart tissues treated with or without nitrite (100 mg/L) during 4 weeks of reperfusion. Nrf2 levels were measured both in cytosolic and nuclear fractions by using immunoblot analysis. (A) represents blots for Nrf2, fibrillarin, and GAPDH. (B and C) represent the quantitation of (A). The number in the circle inside the bar denotes the number of animals used. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). CHF indicates chronic heart failure; Nrf2, nuclear factor‐erythroid‐2‐related factor 2.
It has been reported that Nrf2 increases life span and health span by preventing chronic diseases of oxidative stress through upregulation of the autophagy pathway47 in the cell. Autophagy denotes a ubiquitous cellular pathway that provides nutrients and energy required for cell survival and it is mediated by beclin‐1, and autophagy‐related gene (ATG) family.48 Autophagy has been widely implicated in many pathophysiological processes including cardiovascular diseases and, unsurprisingly, autophagic deficiencies have been associated with a variety of cardiac pathologies.49 Therefore, we were interested in determining the effects of NaNO2 on expression of autophagic genes during CHF. Interestingly, we found upregulation of gene expression of beclin‐1, ATG‐5, and ATG‐7 (Figure 7A through 7C), which contributes to clarifying the cardioprotective effects of NO.
Figure 7.
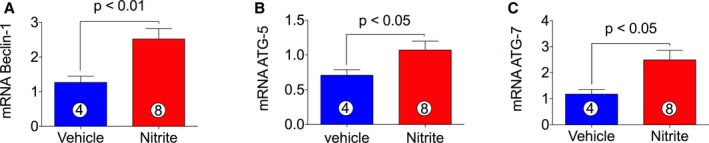
Induction of autophagic genes expression following 4 weeks of nitrite therapy in CHF mice. cDNA was prepared from RNA obtained from mouse heart tissues followed by analysis of mRNA of autophagic markers beclin‐1 (A), ATG‐5 (B), and ATG‐7 (C). The number in the circle inside the bar denotes the number of animals used. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). ATG indicates autophagy related gene; CHF, chronic heart failure.
eNOS is an important enzyme in the cardiovascular system. It catalyzes the production of NO, a key regulator of blood pressure, vascular remodeling, and angiogenesis. Therefore, we examined the effects of nitrite therapy on activation status of eNOS. eNOS activity is regulated by phosphorylation at activation site Ser1177 and the inhibitory site Thr495. As can be seen in Figure 8A and 8D, nitrite therapy significantly increases the phosphorylation of eNOS at Ser1177 (p‐eNOSSer1177). On the other hand, total eNOS (Figure 8A and 8B) and phospho‐eNOS Thr495 (p‐eNOSThr495) (Figure 8A and 8C) were unaltered by nitrite treatment in CHF mice as compared to VEH. These results clearly demonstrate that phosphorylation of eNOS Ser1177 is important for the cardioprotection of NaNO2 in HF.
Figure 8.
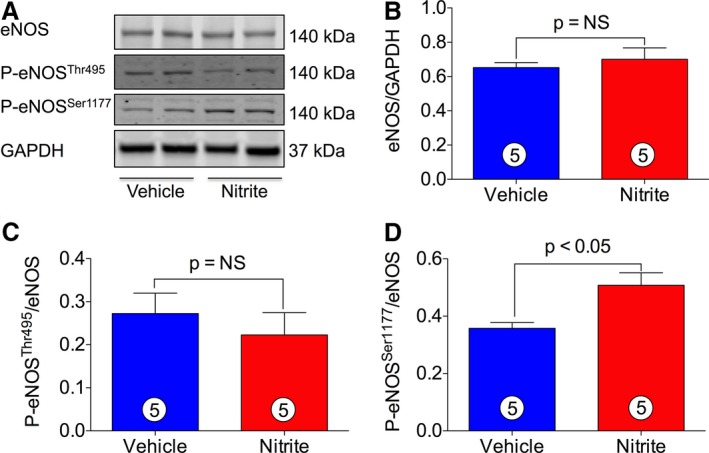
Nitrite therapy induces myocardial eNOS Ser1177 phosphorylation. Total proteins were prepared from mouse heart tissues treated with or without nitrite (100 mg/L) during 4 weeks of reperfusion. Levels of eNOS, including phospho‐eNOS (p‐eNOS), were measured by using immunoblotting. (A) represents blots for eNOS, p‐eNOSThr495, p‐eNOSSer1177, and GAPDH. (B through D) represent the quantitation of blots in (A). The number in the circle inside the bar denotes the number of animals used. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). eNOS indicates endothelial nitric oxide synthase.
Our previous study indicated that H2S therapy increased nitrite levels and phosphorylation of eNOS Ser1177, resulting in cardioprotection in CSE KO mice and suggested significant cross‐talk between the H2S and NO signaling pathways.50 Therefore, we examined the hypothesis that nitrite therapy could also induce H2S production, contributing to the protection of cardiac function. Production of H2S by nitrite therapy has not been studied yet; therefore, we measured H2S levels in blood and myocardial samples of ischemia‐induced CHF mice treated with or without nitrite (Figure 9). Figure 9A and 9B show that H2S levels were significantly increased in both plasma and heart of nitrite‐treated CHF mice as compared with VEH. We then determined the status of mRNA and protein levels of H2S‐producing enzymes, CBS, CSE, and 3‐MST. As can be seen in Figure 9, both mRNA and protein levels of CSE (Figure 9C, 9F, and 9G) and CBS (Figure 9D, 9F, and 9H) significantly increased while mRNA and protein levels of 3‐MST (Figure 9E, 9F, and 9I) were unaltered by nitrite therapy in heart tissue of CHF mice.
Figure 9.
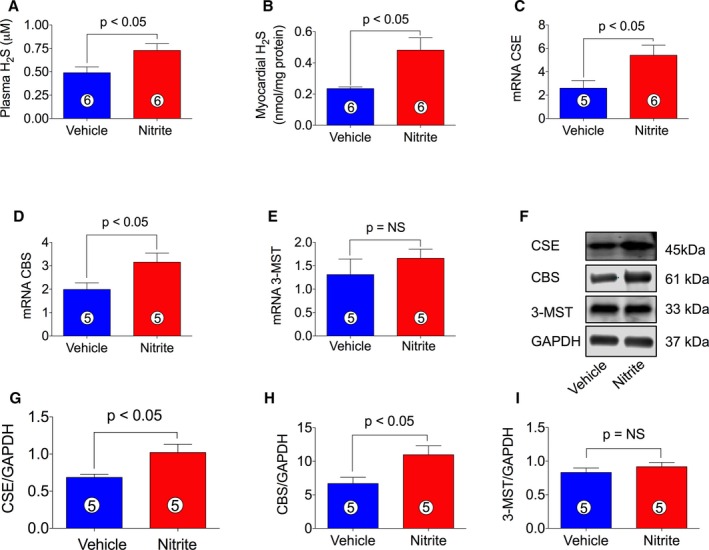
Induction of H2S levels and H2S‐producing enzymes in ischemia‐induced CHF mice by nitrite therapy. Nitrite (100 mg/L) was given to ischemia‐induced CHF mice during 4 weeks of reperfusion. NaNO 2 treatment increases H2S levels both in plasma (A) and heart (B). Induction of H2S‐producing enzymes in the heart of ischemia‐induced CHF mice (C–I). mRNA and protein levels of CSE (C, F, and G), CBS (D, F, and H), and 3‐MST (E, F, and I) following 4 weeks administration of NaNO 2 therapy. The number in the circle inside the bar denotes the number of animals used. Differences in data between the groups were compared using Prism 6 (GraphPad Software, La Jolla, CA) with nonparametric test (Wilcoxon rank sum test). CBS indicates cystathione β‐synthase; CSE, cystationine gamma lyase; 3‐MST, 3‐mercaptopyruvate sulfur transferase; CHF, chronic heart failure.
Discussion
Our recent work showed that H2S therapy activates eNOS and augments NO bioavailability in CSE KO mice.50 Therefore, we investigated whether nitrite therapy induces H2S bioavailability or H2S‐producing enzymes in ischemia‐induced HF in mice. In this study we provide novel insights into the biochemical and molecular mechanisms of NaNO2 therapy. Indeed, we demonstrate that chronic nitrite therapy results in protection against ischemia‐induced HF, activation of H2S‐producing enzymes and increase of H2S bioavailability, Nrf2 activation, and suppression of myocardial oxidative stress.
CHF is characterized by a combination of central and peripheral circulatory dysfunction and is thought to be due, in part, to the reduced NO bioavailability and increased ROS levels8 beyond an increased expression of hypertrophic genes and decreased expression of cardioprotective genes. Biological profiles of H2S and NO are similar, and both molecules are known to protect cells against various injurious states. Previous studies suggest that H2S augments angiogenesis under ischemic conditions both in vitro and in vivo.51, 52 Treatment with exogenous H2S or modulation of the endogenous production of H2S through the cardiac‐specific overexpression of CSE protects against acute myocardial infarction and HF by attenuating oxidative stress, inhibiting apoptosis, and reducing inflammation.34 Although H2S and NO are thought to modulate independent signaling, there is limited evidence of cross‐talk between these 2 molecules,35, 36 indicating a common signaling pathway where NO‐H2S crosstalk mediates their effects on vascular function such as vasodilatation, remodeling, and angiogenesis.51, 53, 54 The interrelation of NO‐H2S and their biochemical interactions are complex and currently unclear. While some studies have shown that NO‐H2S positively affect each other's production and function,35, 51 other studies report contrarian, if not directly opposite findings.55, 56 Studies have shown that H2S has opposing effects on NOS/NO metabolism. Indeed, H2S can downregulate expression or inhibit eNOS activity and subsequent NO production involving altered l‐arginine/BH4, increased heme oxygenase 1/CO, and other unknown mechanisms.55, 57, 58 In addition, recent studies demonstrate H2S‐mediated upregulation of NO and vice‐versa in regulating angiogenesis and attenuation of I/R injury.53 Indeed, H2S upregulates NO production in endothelial cells through the activation of eNOS in an Akt‐dependent manner.59 Likewise, pharmacological NO donors have been shown to enhance the production of H2S from vascular tissues,60 and upregulate substrate bioavailability for and expression of the H2S synthesis enzyme CBS, resulting in H2S production eliciting vasodilatory effects.61, 62 Therefore, it is becoming increasingly clear that a significant ambiguity remains regarding NO‐H2S interactions and related biological effects.
In our current study, we investigated whether nitrite treatment promotes cardioprotection in a chronic model of HF by potentiating H2S signaling. We demonstrated that NaNO2 protects cardiac function in ischemia‐induced CHF, possibly by modulating the expression of hypertrophic and cardioprotective genes expression, inducing activation of H2S‐producing enzymes and reducing oxidative stress. H2S also has been reported to potently regulate cellular redox balance necessary for cytoprotection and inhibition of oxidative stress. In particular, H2S stimulates Nrf2 activation and blunts NOX1 (NADPH oxidase 1) expression and activity,63 leading to increased anti‐oxidant defense responses,34 resulting in the protection of cardiac cells from oxidative injury. Nrf2 and its downstream gene targets play important roles in protecting the heart from ischemic injury via suppressing oxidative stress/ROS, as well as from maladaptive remodeling and cardiac dysfunction.64, 65, 66, 67 For instance, Nrf2 KO mice display exacerbated cardiac injury in response to acute myocardial I/R injury.34 Nrf2 has been reported to be useful in preventing or treating various disease states including chronic diseases of oxidative stress through upregulation of autophagy signaling pathway.47 It is known that the autophagy lysosome pathway is housekeeper in cardiomyocytes under physiological conditions. However, the role of autophagy in HF is controversial. For instance, autophagy may antagonize ventricular hypertrophy by increasing protein degradation and decreasing tissue mass. As a result, autophagy may be an adaptive response to HF. In the mouse heart, autophagy induced by sustained expression of ATG‐7 ameliorates ventricular dysfunction, decreases cardiac hypertrophy, and prolongs survival.68 Another study also indicated that upregulation of autophagy promotes survival during I/R.69 Recent studies have shown that ROS could initiate autophagosome formation and autophagic degradation that act as cellular signaling molecules.70 Autophagy serves to reduce oxidative damage and ROS levels through removal of protein aggregates and damaged organelles such as mitochondria.70 In our study we observed that nitrite therapy induced the activation of signaling pathways of both Nrf2 and autophagy, leading to improvement of cardiac function in CHF mice via inhibiting oxidative stress‐induced damage. These findings suggest that activation of Nrf2 and/or autophagy by nitrite therapy or by NO donors may be a viable therapeutic strategy for the improvement of cardiac function.
In the present study, we found that NaNO2 treatment decreased levels of malondialdehyde and carbonyl protein content, both biomarkers of oxidative stress, during ischemia‐induced CHF as compared with VEH. We also observed that oral NaNO2 therapy increased total antioxidant capacity as well as antioxidant genes and proteins levels of SOD1, GPX, and catalase, and ameliorates myocardial oxidative stress in CHF mice. We may hypothesize that the reduction of tissue and systemic oxidative stress and the enhancement of antioxidant defense occurs via H2S and Nrf2‐dependent mechanisms. However, no evidence currently exists as to whether increasing NO bioavailability through nitrite therapy attenuates ischemia‐induced acute HF or CHF via increasing H2S bioavailability coupled with induction of Nrf2 and inhibition of myocardial oxidative stress. Therefore, we investigated whether NaNO2 treatment induces activation of H2S signaling in CHF mice. We did observe an increase of myocardial mRNA and proteins levels of CBS and CSE with concomitant enhancement of tissue and plasma H2S bioavailability, together with an increase of activation of Nrf2 pathway. Both NO and H2S have antioxidant properties and are capable of decreasing cellular oxidative stress. Therefore, we believe that the beneficial effects of NaNO2 in cardioprotection are further potentiated by the involvement of H2S signaling, which in turn promotes activation of the antioxidant signaling pathway.
In our current study, we investigated whether oral administration of nitrite therapy protects cardiac function in ischemia‐induced CHF mice during reperfusion in an H2S‐dependent manner. Our findings are important because they further corroborate the evidence that there is cross‐talk between the NO and H2S signaling pathways under in vivo pathological conditions, providing strong evidence that NaNO2/NO‐mediated cardioprotection depends at least in part on increased bioavailability of H2S in an in vivo model of cardiovascular disease.
The findings reported in this article argue in favor of cross‐talk between NO and H2S. Despite the plethora of evidence demonstrating that NO and H2S independently act to protect the heart and circulation, additional studies are required to more fully characterize the interdependence and cross‐talk between these 2 gaseous signaling molecules under physiological and pathological conditions. Improved understanding of the importance of H2S and NO cross‐talk will provide new insight into potential therapeutic strategies for heart disease. Therefore, therapeutic intervention aimed at increasing NO and H2S levels may be beneficial for patients affected with cardiovascular disorders.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health; 1R01 HL092141 (DJL), 1R01 HL093579 (DJL), 1U24 HL 094373 (DJL), 1P20 HL113452 (DJL), and COBRE NIGMS 1P30GM106392.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003551 doi: 10.1161/JAHA.116.003551)
References
- 1. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate‐nitrite‐nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. [DOI] [PubMed] [Google Scholar]
- 2. Torregrossa AC, Aranke M, Bryan NS. Nitric oxide and geriatrics: implications in diagnostics and treatment of the elderly. J Geriatr Cardiol. 2011;8:230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gladwin MT. Haldane, hot dogs, halitosis, and hypoxic vasodilation: the emerging biology of the nitrite anion. J Clin Invest. 2004;113:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gladwin MT, Shelhamer JH, Schechter AN, Pease‐Fye ME, Waclawiw MA, Panza JA, Ognibene FP, Cannon RO III. Role of circulating nitrite and s‐nitrosohemoglobin in the regulation of regional blood flow in humans. Proc Natl Acad Sci USA. 2000;97:11482–11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriguez J, Maloney RE, Rassaf T, Bryan NS, Feelisch M. Chemical nature of nitric oxide storage forms in rat vascular tissue. Proc Natl Acad Sci USA. 2003;100:336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim‐Shapiro DB, Schechter AN, Cannon RO III, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. [DOI] [PubMed] [Google Scholar]
- 7. Tiravanti E, Samouilov A, Zweier JL. Nitrosyl‐heme complexes are formed in the ischemic heart: evidence of nitrite‐derived nitric oxide formation, storage, and signaling in post‐ischemic tissues. J Biol Chem. 2004;279:11065–11073. [DOI] [PubMed] [Google Scholar]
- 8. Smith CJ, Sun D, Hoegler C, Roth BS, Zhang X, Zhao G, Xu XB, Kobari Y, Pritchard K Jr, Sessa WC, Hintze TH. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase‐1 in heart failure. Circ Res. 1996;78:58–64. [DOI] [PubMed] [Google Scholar]
- 9. McCord JM, Roy RS, Schaffer SW. Free radicals and myocardial ischemia. The role of xanthine oxidase. Adv Myocardiol. 1985;5:183–189. [PubMed] [Google Scholar]
- 10. Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte‐specific overexpression of NO synthase‐3 protects against myocardial ischemia‐reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:1517–1523. [DOI] [PubMed] [Google Scholar]
- 11. Razavi HM, Hamilton JA, Feng Q. Modulation of apoptosis by nitric oxide: implications in myocardial ischemia and heart failure. Pharmacol Ther. 2005;106:147–162. [DOI] [PubMed] [Google Scholar]
- 12. Jones SP, Greer JJ, van Haperen R, Duncker DJ, de Crom R, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc Natl Acad Sci USA. 2003;100:4891–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lefer DJ. Myocardial protective actions of nitric oxide donors after myocardial ischemia and reperfusion. New Horiz. 1995;3:105–112. [PubMed] [Google Scholar]
- 14. Lefer DJ, Nakanishi K, Johnston WE, Vinten‐Johansen J. Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion of dogs. Circulation. 1993;88:2337–2350. [DOI] [PubMed] [Google Scholar]
- 15. Scherrer‐Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. [DOI] [PubMed] [Google Scholar]
- 16. Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer‐Crosbie M, Picard MH, Szelid Z, Gillijns H, Van de Werf F, Collen D, Bloch KD. Cardiomyocyte‐specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res. 2004;94:1256–1262. [DOI] [PubMed] [Google Scholar]
- 17. Calvert JW, Lefer DJ. Clinical translation of nitrite therapy for cardiovascular diseases. Nitric Oxide. 2010;22:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bryan NS, Fernandez BO, Bauer SM, Garcia‐Saura MF, Milsom AB, Rassaf T, Maloney RE, Bharti A, Rodriguez J, Feelisch M. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005;1:290–297. [DOI] [PubMed] [Google Scholar]
- 19. Feelisch M, Fernandez BO, Bryan NS, Garcia‐Saura MF, Bauer S, Whitlock DR, Ford PC, Janero DR, Rodriguez J, Ashrafian H. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide‐generating and ‐scavenging systems. J Biol Chem. 2008;283:33927–33934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nystrom T, Ortsater H, Huang Z, Zhang F, Larsen FJ, Weitzberg E, Lundberg JO, Sjoholm A. Inorganic nitrite stimulates pancreatic islet blood flow and insulin secretion. Free Radic Biol Med. 2012;53:1017–1023. [DOI] [PubMed] [Google Scholar]
- 21. Bryan NS, Calvert JW, Elrod JW, Gundewar S, Ji SY, Lefer DJ. Dietary nitrite supplementation protects against myocardial ischemia‐reperfusion injury. Proc Natl Acad Sci USA. 2007;104:19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang WZ, Fang XH, Stephenson LL, Zhang X, Williams SJ, Baynosa RC, Khiabani KT, Zamboni WA. Nitrite attenuates ischemia‐reperfusion‐induced microcirculatory alterations and mitochondrial dysfunction in the microvasculature of skeletal muscle. Plast Reconstr Surg. 2011;128:279e–287e. [DOI] [PubMed] [Google Scholar]
- 23. Milsom AB, Patel NS, Mazzon E, Tripatara P, Storey A, Mota‐Filipe H, Sepodes B, Webb AJ, Cuzzocrea S, Hobbs AJ, Thiemermann C, Ahluwalia A. Role for endothelial nitric oxide synthase in nitrite‐induced protection against renal ischemia‐reperfusion injury in mice. Nitric Oxide. 2010;22:141–148. [DOI] [PubMed] [Google Scholar]
- 24. Tripatara P, Patel NS, Webb A, Rathod K, Lecomte FM, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C. Nitrite‐derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J Am Soc Nephrol. 2007;18:570–580. [DOI] [PubMed] [Google Scholar]
- 25. Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia‐reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raat NJ, Noguchi AC, Liu VB, Raghavachari N, Liu D, Xu X, Shiva S, Munson PJ, Gladwin MT. Dietary nitrate and nitrite modulate blood and organ nitrite and the cellular ischemic stress response. Free Radic Biol Med. 2009;47:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elrod JW, Calvert JW, Gundewar S, Bryan NS, Lefer DJ. Nitric oxide promotes distant organ protection: evidence for an endocrine role of nitric oxide. Proc Natl Acad Sci USA. 2008;105:11430–11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. [DOI] [PubMed] [Google Scholar]
- 30. Lowicka E, Beltowski J. Hydrogen sulfide (h2s)—the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- 31. Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. [DOI] [PubMed] [Google Scholar]
- 32. Kimura H. Hydrogen sulfide: its production, release and functions. Amino Acids. 2011;41:113–121. [DOI] [PubMed] [Google Scholar]
- 33. Wang R. Two's company, three's a crowd: can h2s be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. [DOI] [PubMed] [Google Scholar]
- 34. Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through nrf2 signaling. Circ Res. 2009;105:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)s protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Predmore BL, Lefer DJ, Gojon G. Hydrogen sulfide in biochemistry and medicine. Antioxid Redox Signal. 2012;17:119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia‐reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3‐dependent mechanism in mice. Circulation. 2009;120:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Hydrogen sulfide protects against cellular senescence via s‐sulfhydration of keap1 and activation of nrf2. Antioxid Redox Signal. 2013;18:1906–1919. [DOI] [PubMed] [Google Scholar]
- 40. Fisher CD, Augustine LM, Maher JM, Nelson DM, Slitt AL, Klaassen CD, Lehman‐McKeeman LD, Cherrington NJ. Induction of drug‐metabolizing enzymes by garlic and allyl sulfide compounds via activation of constitutive androstane receptor and nuclear factor e2‐related factor 2. Drug Metab Dispos. 2007;35:995–1000. [DOI] [PubMed] [Google Scholar]
- 41. Li Q, Guo Y, Ou Q, Cui C, Wu WJ, Tan W, Zhu X, Lanceta LB, Sanganalmath SK, Dawn B, Shinmura K, Rokosh GD, Wang S, Bolli R. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase‐1 via a nuclear factor‐{kappa}b‐dependent pathway. Circulation. 2009;120:1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates nrf2 through s‐nitrosylation of keap1 in pc12 cells. Nitric Oxide. 2011;25:161–168. [DOI] [PubMed] [Google Scholar]
- 43. Islam KN, Koch WJ. Involvement of nuclear factor kappab (nf‐kappab) signaling pathway in regulation of cardiac g protein‐coupled receptor kinase 5 (grk5) expression. J Biol Chem. 2012;287:12771–12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Islam KN, Kayanoki Y, Kaneto H, Suzuki K, Asahi M, Fujii J, Taniguchi N. Tgf‐beta1 triggers oxidative modifications and enhances apoptosis in hit cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic Biol Med. 1997;22:1007–1017. [DOI] [PubMed] [Google Scholar]
- 45. Bradley JM, Islam KN, Polhemus DJ, Donnarumma E, Brewster LP, Tao YX, Goodchild TT, Lefer DJ. Sustained release nitrite therapy results in myocardial protection in a porcine model of metabolic syndrome with peripheral vascular disease. Am J Physiol. 2015;309:H305–H317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Islam KN, Polhemus DJ, Donnarumma E, Brewster LP, Lefer DJ. Hydrogen sulfide levels and nuclear factor‐erythroid 2‐related factor 2 (nrf2) activity are attenuated in the setting of critical limb ischemia (cli). J Am Heart Assoc. 2015;4 pii:e001986 doi:10.1161/JAHA.115.001986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martin L, Paul SL. Nrf2, a master regulator of detoxification and also antioxidant, anti‐inflammatory and other cytoprotective mechanisms, is raised by health promoting factors. Acta Physiologica Sinica. 2015;67:1–18. [PubMed] [Google Scholar]
- 48. Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo‐Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, Rubinsztein DC, Thumm M, Tooze SA. A comprehensive glossary of autophagy‐related molecules and processes. Autophagy. 2010;6:438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. [DOI] [PubMed] [Google Scholar]
- 50. King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci USA. 2014;111:3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on akt phosphorylation. Cardiovasc Res. 2007;76:29–40. [DOI] [PubMed] [Google Scholar]
- 52. Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC. The hydrogen sulfide donor nahs promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12:1065–1077. [DOI] [PubMed] [Google Scholar]
- 53. Bir SC, Kolluru GK, McCarthy P, Shen X, Pardue S, Pattillo CB, Kevil CG. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia‐inducible factor‐1alpha and vascular endothelial growth factor‐dependent angiogenesis. J Am Heart Assoc. 2012;1:e004093. doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gu Q, Wang B, Zhang XF, Ma YP, Liu JD, Wang XZ. Contribution of hydrogen sulfide and nitric oxide to exercise‐induced attenuation of aortic remodeling and improvement of endothelial function in spontaneously hypertensive rats. Mol Cell Biochem. 2013;375:199–206. [DOI] [PubMed] [Google Scholar]
- 55. Kubo S, Kurokawa Y, Doe I, Masuko T, Sekiguchi F, Kawabata A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology. 2007;241:92–97. [DOI] [PubMed] [Google Scholar]
- 56. Prathapasinghe GA, Siow YL, Xu Z, O K. Inhibition of cystathionine‐beta‐synthase activity during renal ischemia‐reperfusion: role of ph and nitric oxide. Am J Physiol Renal Physiol. 2008;295:F912–F922. [DOI] [PubMed] [Google Scholar]
- 57. Geng B, Cui Y, Zhao J, Yu F, Zhu Y, Xu G, Zhang Z, Tang C, Du J. Hydrogen sulfide downregulates the aortic l‐arginine/nitric oxide pathway in rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1608–R1618. [DOI] [PubMed] [Google Scholar]
- 58. Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, Jeon SB, Jeon WK, Chae HJ, Chung HT. Hydrogen sulfide inhibits nitric oxide production and nuclear factor‐kappab via heme oxygenase‐1 expression in raw264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med. 2006;41:106–119. [DOI] [PubMed] [Google Scholar]
- 59. Predmore BL, Julian D, Cardounel AJ. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt‐dependent mechanism. Front Physiol. 2011;2:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of h(2)s as a novel endogenous gaseous k(atp) channel opener. EMBO J. 2001;20:6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eto K, Kimura H. A novel enhancing mechanism for hydrogen sulfide‐producing activity of cystathionine beta‐synthase. J Biol Chem. 2002;277:42680–42685. [DOI] [PubMed] [Google Scholar]
- 62. Liew HC, Khoo HE, Moore PK, Bhatia M, Lu J, Moochhala SM. Synergism between hydrogen sulfide (h(2)s) and nitric oxide (no) in vasorelaxation induced by stonustoxin (sntx), a lethal and hypotensive protein factor isolated from stonefish synanceja horrida venom. Life Sci. 2007;80:1664–1668. [DOI] [PubMed] [Google Scholar]
- 63. Muzaffar S, Shukla N, Bond M, Newby AC, Angelini GD, Sparatore A, Del Soldato P, Jeremy JY. Exogenous hydrogen sulfide inhibits superoxide formation, nox‐1 expression and rac1 activity in human vascular smooth muscle cells. J Vasc Res. 2008;45:521–528. [DOI] [PubMed] [Google Scholar]
- 64. Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia‐induced heart failure in mice. Circulation. 2010;122:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW. Thioredoxin 1 is essential for sodium sulfide‐mediated cardioprotection in the setting of heart failure. Arterioscler Thromb Vasc Biol. 2013;33:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang W, Li S, Wang H, Li B, Shao L, Lai Y, Horvath G, Wang Q, Yamamoto M, Janicki JS, Wang XL, Tang D, Cui T. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J Mol Cell Cardiol. 2014;72:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, Cui T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. 2009;29:1843–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mei Y, Thompson MD, Cohen RA, Tong X. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. 2015;1852:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li L, Tan J, Miao Y, Lei P, Zhang Q. Ros and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]