

Abstract
Background
The transient receptor potential vanilloid type 1 (TRPV1) is expressed in the cardiovascular system, and increased TRPV1 expression has been associated with cardiac hypertrophy. Nevertheless, the role of TRPV1 in the pathogenesis of cardiac hypertrophy and the underlying molecular mechanisms remain unclear.
Methods and Results
In cultured cardiomyocytes, activation of TRPV1 increased cell size and elevated expression of atrial natriuretic peptide mRNA and intracellular calcium level, which was reversed by TRPV1 antagonist capsazepine. Increased expression of phosphorylated calmodulin‐dependent protein kinase IIδ and mitogen‐activated protein kinases were found in TRPV1 agonist capsaicin‐treated cardiomyocytes. Selective inhibitor of calmodulin‐dependent protein kinase IIδ decreased phosphorylation of extracellular signal–regulated kinases and p38. Capsaicin induced an increase in expression of ornithine decarboxylase protein, which is the key enzyme in polyamine biosynthesis in cardiomyocytes. Nevertheless, there was no obvious change of ornithine decarboxylase expression in TRPV1 knockdown cells after capsaicin treatment, and specific inhibitors of calmodulin‐dependent protein kinase IIδ or p38 downregulated the capsaicin‐induced expression of ornithine decarboxylase. Capsazepine alleviated the increase in cross‐sectional area of cardiomyocytes and the ratio of heart weight to body weight and improved cardiac function, including left ventricular internal end‐diastolic and ‐systolic dimensions and ejection fraction and fractional shortening percentages, in mice treated with transverse aorta constriction. Capsazepine also reduced expression of ornithine decarboxylase and cardiac polyamine levels. Transverse aorta constriction induced increases in phosphorylated calmodulin‐dependent protein kinase IIδ and extracellular signal–regulated kinases, and p38 and Serca2a were attenuated by capsazepine treatment.
Conclusions
This study revealed that the mitogen‐activated protein kinase signaling pathway and intracellular polyamines are essential for TRPV1 activation–induced cardiac hypertrophy.
Keywords: calcium, calcium signaling, capsaicin, cardiac hypertrophy, ornithine decarboxylase, phosphoprotein, polyamine, transient receptor potential vanilloid type 1
Subject Categories: Heart Failure
Introduction
Transient receptor potential vanilloid type 1 (TRPV1) is a 6‐transmembrane tetrameric nonselective cation channel found mainly in the nociceptive neurons of the peripheral nervous system. Activation of TRPV1 leads to a painful sensation.1, 2 TRPV1 is activated by a wide variety of physical and chemical stimuli. Exogenous activators of TRPV1 include capsaicin (CAP) and temperatures >43°C, whereas endogenous activators include endocannabinoids anandamide (ANA) and N‐arachidonoyl dopamine.3, 4, 5
In addition to the peripheral sensory neurons, TRPV1 is found in the cardiovascular system in, for example, cardiomyocytes, cardiac blood vessels, and coronary endothelial cells.6, 7 Studies have implicated the role of endogenous activator ANA in multiple cardiovascular diseases, such as myocardial ischemia reperfusion injury and hypertension.8, 9 Recently, increased TRPV1 expression was associated with cardiac hypertrophy in mice, and functional knockout of TRPV1 protected heart function in a model of cardiac hypertrophy.6, 10 Furthermore, administration of a TRPV1 antagonist can overcome loss of heart function.11
Although these studies imply that TRPV1 antagonism may represent an exploitable therapy for cardiac hypertrophy and heart failure, much less is known about the underlying molecular events associated with cardiac hypertrophy. Considering the function of TRPV1 as a Ca2+‐permeable channel and the pivotal role that Ca2+ plays in regulation of long‐term structural remodeling or degenerative processes in the heart,12 we reasoned that increased TRPV1 expression would contribute to the elevated influx of Ca2+ and downstream signaling pathways that play a role in cardiac hypertrophy.
The aim of this study was to explore intracellular TRPV1 signaling in cultured cardiomyocytes cells in vitro and in cardiac hypertrophy induced by transverse aorta constriction (TAC) in vivo. The findings from this study provide justification for further studies into the role of TRPV1 in cardiac hypertrophy and raise the possibility that TRPV1 antagonists currently used as antihyperalgesics could be repurposed as agents against cardiac hypertrophy.
Materials and Methods
Culture of Neonatal and Embryonic Rat Cardiomyocytes
All animal experiments and procedures were approved by the institutional animal care and use committee at the Fourth Military Medical University. In vitro culture of neonatal rat cardiomyocytes was performed as described previously.13 Briefly, Wister rats aged 2 to 3 days were anesthetized and decapitated. The hearts were minced and dissociated with 0.25% trypsinase (Sigma‐Aldrich) in an incubator with 5% CO2 at 37°C for 15 minutes. Dispersed cells were seeded at 2×105 cells/cm2 with DMEM supplemented with 10% fetal bovine serum (Gibco) in culture dishes and cultured in a 5% CO2 incubator at 37°C. The H9C2 cell is a subclone of the original clonal cell line derived from embryonic rat heart tissue, which was purchased from American Type Culture Collection (ATCC, Rockville, MD) and cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in a humidified incubator containing 95% air and 5% CO2.
Transverse Aorta Constriction Procedures and TRPV1 Antagonist Capsazepine Treatment In Vivo
The TAC or sham surgery described previously was performed.14 Briefly, C57BL/6 male mice (aged 7 weeks; Fourth Military Medical University Medical Laboratory Animal Center, Xi'an, People's Republic of China) were anesthetized with a mixture of ketamine and xylazine. The transverse aorta between the brachiocephalic and left carotid arteries was banded using 6‐0 silk ligature around the vessel and a 26‐gauge needle, after which the needle was withdrawn. Sham surgeries were identical apart from the constriction of the aorta. All animal experiments and care procedures were reviewed and approved by the Animal Resource Center of the Fourth Military Medical University, and the methods were carried out in strict accordance with the approved guidelines. To study the role of TRPV1 blocking in TAC‐induced cardiac hypertrophy mice, the TRPV1 antagonist capsazepine (CPZ; 0.25 and 1.25 mg/kg) was given to the mice intraperitoneally twice daily starting 24 hours after TAC operation through 4 or 8 weeks. Control mice were treated using 10% DMSO in saline, and the final volume was 120 μL.
Downregulation of Trpv1 Gene Expression by Small Interfering RNA
To silence the Trpv1 gene in cardiomyocytes, we performed transfection with small interfering RNA (siRNA) duplex, as described by the manufacturer (Santa Cruz Biotechnology). A pool of 3 different Trpv1 siRNAs was synthesized by Sangon Biotech (Shanghai, People's Republic of China): siRNA 1, sense GAAGACCUGUCUGCUGAAAtt, antisense UUUCAGCAGACAGGUCUUCtt; siRNA 2, sense CGAGCAUGUACAAUGAGAUtt, antisense AUCUCAUUGUACAUGCUCGtt; siRNA 3, sense CGCAUCUUCUACUUCAACUtt, antisense AGUUGAAGUAGAAGAUGCGtt. A nonrelated scrambled siRNA was used as a control. All measurements were performed 72 hours after transfection, at which time TRPV1 levels were decreased by 60% to 65%.
Real‐Time Polymerase Chain Reaction for TRPV1, Ornithine Decarboxylase, and Atrial Natriuretic Peptide
Total RNA was extracted using TRIzol (Life Technologies Corporation). Using the RT‐PCR Access Kit (Life Technologies Corporation), 1 µg total RNA was reverse transcribed into cDNA; 1 μL cDNA was used for polymerase chain reaction. TRPV1, ornithine decarboxylase (ODC), atrial natriuretic peptide, and β‐actin mRNAs were amplified by polymerase chain reaction using the primers shown in Table 1. The polymerase chain reaction mixture was incubated in a DNA Thermal Cycler (Stratagene). Reactions were prepared in triplicate and heated to 95°C for 5 minutes, followed by 40 cycles at 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds. The resulting relative increase in reporter fluorescent dye (SYBR green) emission was monitored with the use of the ABI PRISM7700 sequence detector (Applied Biosystems). Relative quantification was measured using the comparative threshold method (ΔΔCT), as described previously.15 After determining the comparative threshold values for target and reference genes, fold changes in target mRNA expression level were calculated after normalization to reference gene β‐actin.
Table 1.
Sequences of Primers for Real‐Time Polymerase Chain Reaction
| mRNA | Sense Primer | Antisense Primer |
|---|---|---|
| TRPV1 | 5′‐CCTACAGGAACTTCAACCAATGC ‐3′ | 5′‐TCCTTATCAGTAAAACGGGGACA ‐3′ |
| ODC | 5′‐GCCAGG GTG CTC ACT ATG‐3′ | 5′‐GCAGCT TTA CTA GGAAGAGT‐3′ |
| ANP | 5′‐AGGATTGGAGCCCAGAGTGGACTAGG‐3′ | 5′‐TGATAGATGAAGGCAGGAAGCCGC‐3′ |
| β‐actin | 5′‐TTGTAACCAACTGGGACGATATGG‐3′ | 5′‐GATCTTGATCTTCATGGTGCTAGG‐3′ |
ANP indicates atrial natriuretic peptide; TRPV1, transient receptor potential vanilloid type 1; ODC, ornithine decarboxylase.
Western Blot Measurement for Calmodulin‐Dependent Protein Kinase IIδ, Extracellular Signal–Regulated Kinases, c‐Jun N‐Terminal Kinase, p38, TRPV1, TRPV2, TRPV4, TRPM6, ODC, and β‐Actin
Cells or tissue samples were harvested in lysis buffer (50 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 1% NP40, 0.5% sodium deoxycholate, 1 mmol/L EDTA, 0.1% SDS) containing protease inhibitor cocktail (Sigma‐Aldrich), and protein concentrations were determined using the DC Protein Assay (Bio‐Rad Laboratories). Protein was electrophoresed on 7.5% SDS‐PAGE, transferred to nitrocellulose membranes (Amersham Bioscience), and incubated with specific primary antibodies at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase–conjugated secondary antibodies for 1 hour and visualized with enhanced chemiluminescent substrate. Membranes were also incubated with 1:2000 antibody of β‐actin to validate the technique.
Measurement of Intracellular Calcium in Cardiomyocytes
Intracellular Ca2+ transient measurements and analyses were performed as described below, and myocytes were loaded with 0.67 μmol/L Fura‐2‐acetoxymethyl ester for 15 minutes at 37°C and then transferred to the recording chamber mounted on an inverted microscope (Nikon) in a solution consisting of 145 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L glucose, and 10 mmol/L HEPES with 0 or 2.5 mmol/L CaCl2 at pH 7.2. Background‐corrected fluorescent images were acquired at 1 Hz with a slow scan CCD camera system (Princeton Instruments). Emitted light (510 nm) was acquired with equal exposure time (200 ms) for the excitation wavelengths of 340 and 380 nm.
Echocardiography Measurement of Left Ventricular Hypertrophy and Systolic Function
Echocardiography was performed using an Accuson Sequoia instrument paired with a 14‐MHz transducer to study left ventricular (LV) hypertrophy and systolic function. After light anesthesia (100 mg/kg ketamine and 10 mg/kg xylazine), the animal was shaved, placed in a supine position, and warmed using an isothermal heating pad. LV internal end‐diastole and ‐systole dimensions were measured from M‐mode tracings as well as from interventricular septum and posterior wall thickness. LV fractional shortening was calculated using the following equation: (LV internal end‐diastole dimension−LV internal end‐systole dimension/LV internal end‐diastole dimension)×100. Ejection fraction was calculated as follows: (LV end‐diastole volume−LV end‐systole volume/LV end‐diastole volume)×100.
High‐Performance Liquid Chromatography Analysis of Polyamines
According to our previously reported method,16 the samples were homogenized in 700 μL of 0.9% sodium chloride mixed with 10 μL (200 nmol/mL) internal standard (1, 7‐diaminoheptane). To precipitate proteins, 300 μL of trichloroacetic acid (0.5 mol/L) was added to the homogenate. After 30 minutes of incubation in ice, the homogenate was centrifuged for 30 minutes at 14 500g at 4°C. The derivatization reaction was carried out with 9‐fluorenylmethyl chloroformate, and the fluorescent‐polyamine derivatives were performed using C18 high‐performance liquid chromatography columns (150×4.6 mm, 5 μm) with a fluorescence detector (Jasco 821‐FP) filled with 3 g reverse‐phase material from Chrompack Nederland (chloroquine phosphate microspheres). The excitation and emission wavelengths of the detector were set at 264 and 310 nm, respectively. The solvent flow was 2 mL/min (acetonitrile:acetate 60/40 vol/vol) and was followed by a linear increase of acetonitrile concentration to 95% in 30 minutes. The samples were dissolved in 50 mmol/L sodium acetonitrile:acetate 50/50 (vol/vol). The injection volume was 20 μL.
Materials
CAP, CPZ, putrescine, spermidine, spermine, and KN‐93 were purchased from Sigma‐Aldrich. ANA was purchased from Tocris. BIRB‐796 (doramapimod) was purchased from Selleckchem. Antibodies for calmodulin‐dependent protein kinase IIδ (CaMKIIδ), phosphorylated CaMKIIδ, extracellular signal–regulated kinases (ERKs), phosphorylated ERKs, c‐Jun N‐terminal kinase (JNK), phosphorylated JNK, p38, phosphorylated p38, TRPV1, TRPV4, TRPM6, and ODC were purchased from Abcam; TRPV2 antibody was purchased from Abnova; and phospholamban (PLN), PLN–phosphorylated threonine 17, sarcoplasmic reticulum Ca2+‐ATPase 2a (Serca2a), and β‐actin antibody were purchased from Santa Cruz Biotechnology.
Statistical Analyses
Values are shown as mean±SEM. Comparisons between the groups were conducted with ANOVA and Student t tests for unpaired and paired samples (t test). A post hoc comparison for ANOVA was done with the Fisher protected least squares difference test, and differences were considered significant at P<0.05.
Results
TRPV1 Activation Induced Cardiac Hypertrophy In Vitro
To examine the role of TRPV1 in cardiac hypertrophy, we treated isolated rat neonatal cardiomyocytes and the H9C2 cells with CAP and ANA, respectively. We found that 0.5 or 2 μmol/L CAP significantly increased the cell size in H9C2 cells, and 2 μmol/L CPZ reversed the increased cell size; however, only 2 μmol/L ANA induced a significant increase in size of H9C2 cells, whereas 2 μmol/L CPZ reversed this effect (Figure 1A). In cultured rat neonatal cardiomyocytes, cell size was increased by 2 μmol/L CAP or ANA, and that effect was ameliorated by 2 μmol/L CPZ treatment (Figure 1B). Next, atrial natriuretic peptide transcript expression, a marker of the hypertrophic response, was analyzed in H9C2 cells after CAP or ANA treatment, and atrial natriuretic peptide expression was increased significantly by CAP or ANA; 2 μmol/L CPZ treatment attenuated the increased atrial natriuretic peptide expression level induced by TRPV1 agonist CAP or ANA (Figure 1C).
Figure 1.
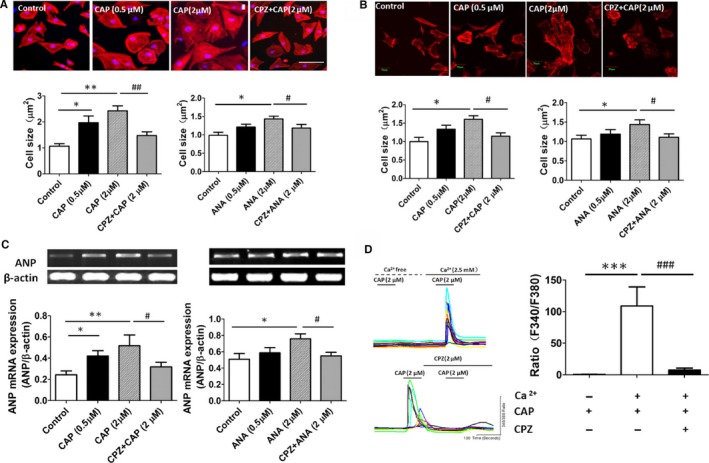
Activation of TRPV1 induced a cardiohypertrophic response and elevated intracellular calcium level in cultured cardiomyocytes. A, Histological staining of H9C2 cells treated with vehicle, CAP, and CPZ plus CAP for 48 hours is shown; cardiomyocyte cross‐sectional area was measured after treatment with TRPV1 agonist CAP or ANA (6 independent experiments per group, 20 cells counted per experiment). *P<0.05, **P<0.01 versus control, # P<0.05 versus 2 μmol/L ANA, ## P<0.01 versus 2 μmol/L CAP. B, Morphologies of isolated rat neonatal cardiomyocytes were examined after CAP or CPZ plus CAP treatment for 48 hours (5 independent experiments per group, 20 cells counted per experiment), and cardiomyocyte cross‐sectional area was measured after CAP or ANA treatment. *P<0.05 versus control, # P<0.05 versus 2 μmol/L CAP or 2 μmol/L ANA. C, Representative mRNA products of atrial natriuretic peptide in the CAP‐ or ANA‐treated H9C2 cells (6 independent experiments per group) and expression of ANP relative to β‐actin mRNA were assayed. *P<0.05, **P<0.01 versus control, # P<0.05 versus 2 μmol/L CAP or 2 μmol/L ANA group. D, Representative recording of change in intracellular Ca2+ induced by 2 μmol/L CAP with 0 or 2.5 mmol/L calcium in bath solution was reversed by 2 μmol/L CPZ, and the F340/F380 Fura‐2 fluorescence ratio was measured (8 independent experiments per group, 50 cells counted per experiment).***P<0.001 versus calcium‐free bath solution group, ### P<0.001 versus CAP with calcium‐treated group. ANA indicates anandamide; CAP, capsaicin; CPZ, capsazepine.
CaMKIIδ and the Mitogen‐Activated Protein Kinase Signaling Pathway Were Involved in TRPV1 Activation–Induced Cardiac Hypertrophy
Given that TRPV1 functions as a Ca2+‐permeable channel, and Ca2+ plays a pivotal role in cardiac hypertrophy, intracellular Ca2+ was measured in CAP‐treated H9C2 cells. Results showed that 2 μmol/L CAP evoked an increase in intracellular Ca2+ when the extracellular solution contained 2.5 mmol/L Ca2+, and this increase was blocked by CPZ treatment. Furthermore, in the absence of extracellular calcium, CAP‐induced intracellular Ca2+ increase was not observed (Figure 1D). Ca2+/CaMKII is activated by increased intracellular Ca2+ concentration, and the CaMKIIδ isoform found predominantly in the heart has been shown to play an essential role in cardiac hypertrophy.17 We explored whether CaMKIIδ activity and phosphorylation were altered in TRPV1 activation–induced cardiac hypertrophy. Western blot analysis showed that CAP treatment (0.5 or 2 μmol/L) for 24 or 48 hours induced an increase in expression of phosphorylated CaMKIIδ in cardiac myocytes (Figure 2A and 2B) that was attenuated by CPZ treatment.
Figure 2.
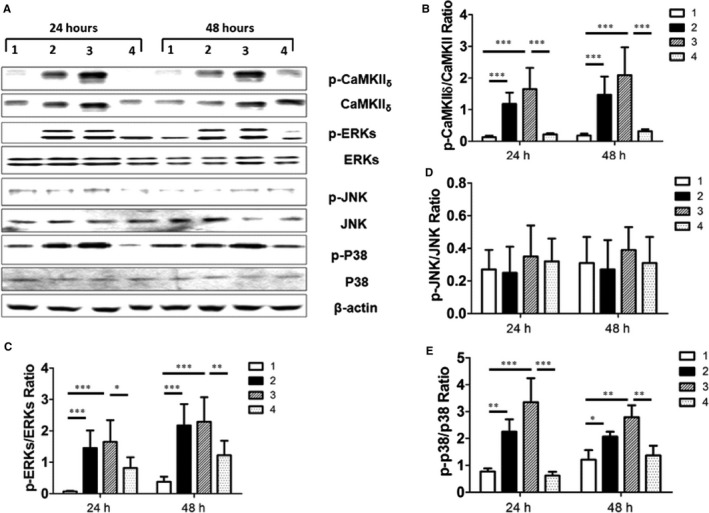
CAP induced cardiac hypertrophy in H9C2 cells through CaMKII δ and the mitogen‐activated protein kinase signaling pathway. A, Representative Western blotting products are shown from H9C2 cells treated with vehicle control (panel 1), 0.5 μmol/L CAP (panel 2), 2 μmol/L CAP (panel 3), and 2 μmol/L CPZ plus CAP (panel 4) for 24 or 48 hours; β‐actin was used as internal control. Quantitative assay of relative expression of p‐CAMKII δ to CaMKII δ (B) and relative expression ratios of p‐ERKs to ERKs (C), p‐JNK to JNK (D), and p‐P38 to P38 in (E) cells treated by vehicle control (panel 1), 0.5 μmol/L CAP (panel 2), 2 μmol/L CAP (panel 3) and 2 μmol/L CPZ plus CAP (panel 4) for 24 hour or 48 hours (6 independent experiments per group) are shown. *P<0.05, **P<0.01, ***P<0.001. CaMKII indicates calmodulin‐dependent protein kinase II; CAP, capsaicin; CPZ, capsazepine; ERK, extracellular signal–regulated kinase; JNK, c‐Jun N‐terminal kinase; p, phosphorylated.
Next, we observed the possible change in mitogen‐activated protein kinases (MAPKs) including ERKs, JNK, and p38 in CAP‐treated cardiac myocytes. The expression of phosphorylated ERKs, p38, and JNK proteins was assessed in H9C2 cells cultured with CAP for 24 or 48 hours. We observed increased expression of phosphorylated ERKs and p38 but not JNK in CAP‐cultured H9C2 cells (Figure 2C–2E), and those increases were largely attenuated by CPZ treatment.
Downregulation of CaMKIIδ‐Dependent MAPKs in Trpv1‐Silencing Cardiomyocytes
To confirm whether phosphorylated CaMKIIδ was induced by TRPV1 activation and whether ERKs or p38 is downstream of phosphorylated CaMKIIδ, we knocked down expression of TRPV1 in H9C2 cells using Trpv1 siRNA and KN‐93, the selective inhibitor of CaMKIIδ. Compared with the scrambled siRNA–treated cardiac myocytes, CAP (0.5 and 2 μmol/L) was not able to induce an increase in expression of phosphorylated CaMKIIδ, ERKs, and p38 in TRPV1 siRNA–treated cells. Moreover, in the presence of 2 μmol/L KN‐93, CAP‐induced expression of phosphorylated ERKs or p38 protein was significantly attenuated (Figure 3).
Figure 3.
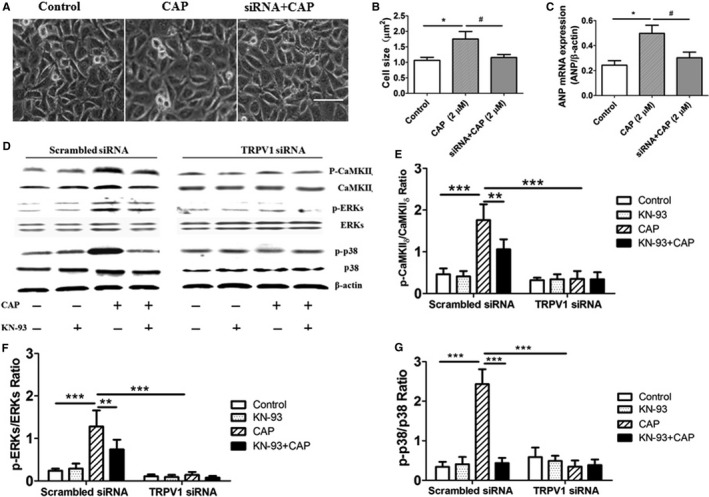
Expression of CaMKII δ‐dependent mitogen‐activated protein kinase signaling in TRPV1 siRNA–treated H9C2 cells. A, Morphologies of H9C2 cells were examined after CAP treatment on control or TRPV1 siRNA–treated groups for 48 hours. Scale bar=25 μm. B, Cardiomyocyte cross‐sectional area was measured after CAP treatment. C, Representative mRNA products of ANP in the CAP‐treated H9C2 cells (5 independent experiments per group) are shown. *P<0.05 versus control, # P<0.05 versus 2 μmol/L CAP. D, In scrambled siRNA– and TRPV1 siRNA–treated cells, 1 μmol/L CaMKII δ inhibitor KN‐93, 2 μmol/L CAP, or 1 μmol/L KN‐93 plus 2 μmol/L CAP were added in culture media for 48 hours, and then cell extracts were analyzed by Western blotting for CaMKII δ, ERKs, and p38 protein; β‐actin was used as internal control. Relative expression ratios of p‐CaMKII δ to CaMKII δ (E), p‐ERKs to ERKs (F), and p‐p38 to p38 (G) in cells with different treatment (5 independent experiments per group) are shown. **P<0.01, ***P<0.001. ANP indicates atrial natriuretic peptide; CaMKII, calmodulin‐dependent protein kinase II; CAP, capsaicin; ERK, extracellular signal–regulated kinase; p, phosphorylated; siRNA, small interfering RNA.
We evaluated whether MAPKs were able to induce expression of ODC, the first and rate‐limiting enzyme in polyamine biosynthesis.18, 19 We found that CAP (2 μmol/L) was able to induce a significant increase in protein expression of ODC; however, in the presence of KN‐93 or the p38‐selective inhibitor BIRB‐796, the expression of ODC was greatly downregulated. Interestingly, in the Trpv1 siRNA–treated cardiomyocytes, CAP was not able to induce a significant increase in ODC protein expression (Figure 4). This indicated that TRPV1 activation–induced ODC expression was dependent on the CaMKIIδ–MAPK pathway.
Figure 4.
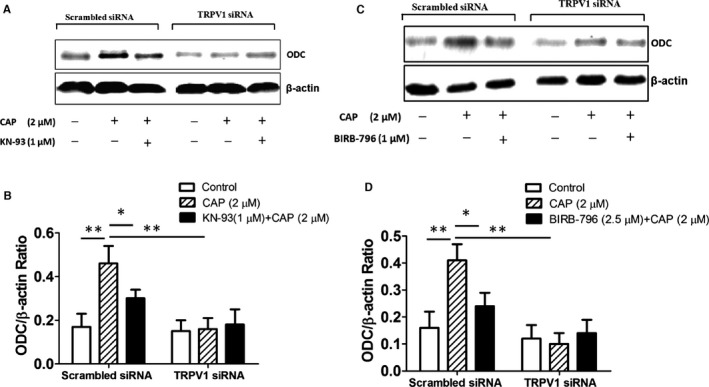
TRPV1 activation–induced ODC expression was mediated by calmodulin‐dependent protein kinase IIδ and p38. A, In scrambled siRNA– and TRPV1 siRNA–treated H9C2 cells, 2 μmol/L CAP with or without 1 μmol/L KN‐93 were added in culture media for 48 hours, and then cell extracts were analyzed by Western blotting for ODC protein expression; β‐actin was used as internal control. B, Relative expression ratio of ODC to β‐actin in the cells with above treatment (5 independent experiments per group) is shown. *P<0.05, **P<0.01. C, In above cells, 2 μmol/L CAP or 2.5 μmol/L p38 inhibitor BIRB‐796 plus 2 μmol/L CAP were added in culture media for 48 hours, and then cell extracts were analyzed by Western blotting for ODC protein expression. D, Relative expression ratio of ODC to β‐actin in cells from above groups (5 independent experiments per group) is shown. *P<0.05, **P<0.01. CAP indicates capsaicin; ODC, ornithine decarboxylase; siRNA, small interfering RNA.
TRPV1 Antagonist Treatment Produced Beneficial Effects in TAC‐Induced Cardiac Hypertrophy
Because the presented data showed that the TRPV1 antagonist CPZ alleviated CAP‐ or ANA‐induced cardiac hypertrophy in vitro, we next evaluated the effect of CPZ in TAC‐induced cardiac hypertrophy in vivo. Histological analysis showed that CPZ‐ or vehicle‐treated mice that underwent sham surgery showed no significant difference in cell size, whereas vehicle‐treated mice that underwent TAC had a significantly larger cross‐sectional area of cardiomyocytes compared with CPZ‐treated mice (0.5 and 2.5 mg/kg) that underwent TAC (Figure 5A and 5B). In addition, heart weight/body weight ratios in mice treated with 0.5 and 2.5 mg/kg CPZ and TAC were significantly lower compared with vehicle‐treated mice that underwent TAC (Figure 5C).
Figure 5.
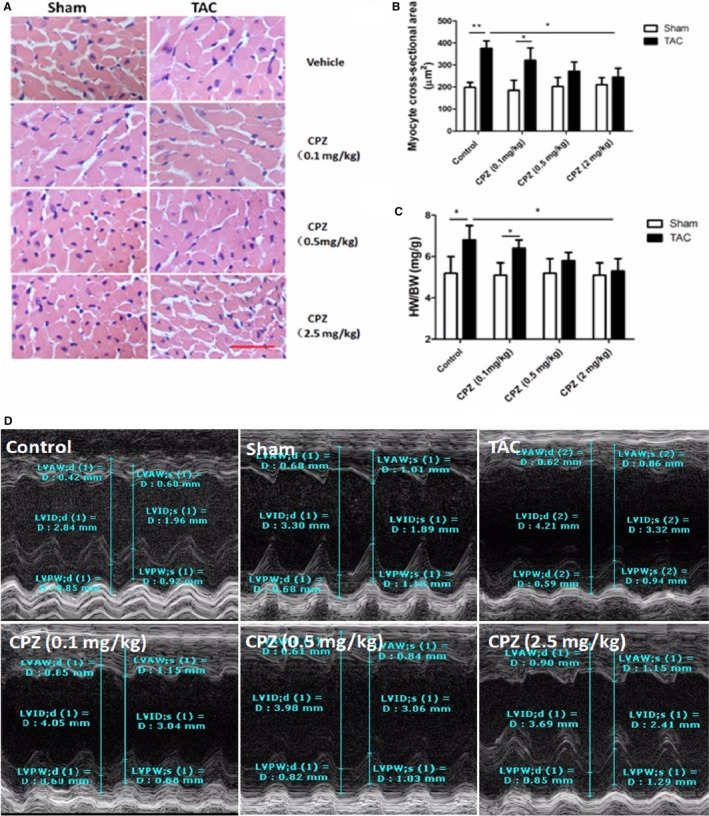
Antihypertrophy effect of TRPV1 antagonist in TAC‐treated mouse. A, Representative histological staining of cardiomyocytes in mice treated with vehicle control or CPZ (0.1, 0.5, or 2.5 mg/kg per day) at 8 weeks after TAC or sham surgery. Scale bar=25 μm. Analysis of cardiomyocyte cross‐sectional area (B) and ratio of heart weight normalized to body weight (C) was measured in the above groups (n=5 mice per group). *P<0.05, **P<0.01. D, Representative serial M‐model echocardiographic tracings in control, sham, vehicle, and CPZ (0.1, 0.5, or 2.5 mg/kg per day) treatment after TAC show LVIDs and LVIDd. CPZ indicates capsazepine; d, end‐diastole; D, diameter; HW/BW, heart weight/body weight ratio; LVAW, left ventricular anterior wall thickness; LVID, left ventricular internal dimension; LVPW, left ventricular posterior wall thickness; s, end‐systole; TAC, transverse aorta constriction.
Next we measured the effect of CPZ on cardiac function in sham‐ or TAC‐treated mice. There was no significant difference between the sham‐ and TAC‐treated mice in heart rate, but measurements of LV internal end‐diastole and ‐systole diameters revealed that vehicle‐treated mice had significantly more dilated ventricles at 4 and 8 weeks after TAC than mice treated with 0.5 and 2.5 mg/kg CPZ. Most notably, the ejection fraction percentage was significantly better at 36 days after TAC in mice treated with 0.5 and 2.5 mg/kg CPZ than in vehicle‐treated mice. This fractional shortening percentage indicates significantly better heart function (Figure 5D, Table 2). There is a linear trend between the CPZ concentration and progress in cardiac function.
Table 2.
Effect of TRPV1 Antagonist on Cardiac Function in Mice (n=6 mice per group)
| Control | Sham | TAC | ||||
|---|---|---|---|---|---|---|
| Vehicle | CPZ (0.1 mg/kg) | CPZ (0.5 mg/kg) | CPZ (2.5 mg/kg) | |||
| HR, bpm in s | 512±38 | 523±65 | 517±63 | 509±45 | 518±38 | 507±32 |
| BW, g | 28.6±3.7 | 27.9±4.3 | 29.8±4.4 | 28.5±3.6 | 28.7±2.9 | 28.3±3.1 |
| HW, mg | 105±15 | 106±13 | 137±19b | 126±21 | 118±16 | 110±14 |
| LW, mg | 171±24 | 173±23 | 192±28 | 181±26 | 183±27 | 185±26 |
| TL, mm | 16.7±4.3 | 17.1±5.4 | 17.1±4.3 | 16.9±4.4 | 17.5±3.6 | 17.2±4.5 |
| HW/TL, mg/mm | 7.6±1.9 | 7.5±2.4 | 9.2±2.6a | 9.0±3.1 | 8.9±2.5 | 8.2±2.2c |
| AW/TL, mg/mm | 0.83±0.06 | 0.82±0.08 | 0.93±0.06a | 0.90±0.05 | 0.88±0.06 | 0.85±0.09c |
| AW/LW, mg/g | 67.4±14.2 | 66.7±15.5 | 93.6±22.5b | 85.6±17.4 | 83.6±16.2c | 76.5±18.5d |
| LVIDd, mm | 3.32±0.36 | 3.42±0.31 | 4.18±0.35b | 3.92±0.68 | 3.61±0.22c | 3.52±0.38c |
| LVIDs, mm | 2.21±0.53 | 2.17±0.32 | 3.34±0.63a | 3.24±0.49 | 2.43±0.31c | 2.37±0.52c |
| EF, % | 65.82±6.37 | 66.84±8.81 | 38.05±10.63b | 39.81±6.77 | 56.83±6.10c | 57.11±6.14c |
| FS, % | 35.58±4.97 | 36.46±6.51 | 18.29±5.34b | 19.57±5.61 | 29.65±3.52d | 30.57±3.41d |
AW indicates atria weight; BW, body weight; CPZ, capsazepine; EF, ejection fraction; FS, fractional shortening; HR, heart rate; HW, heart weight; LVIDd, left ventricular interior end‐diastole dimension; LVIDs, left ventricular interior end‐systole dimension; LW, lung weight; TL, tibia length; TRPV1, transient receptor potential vanilloid type 1.
P<0.05.
P<0.01 versus sham group.
P<0.05.
P<0.01 versus transverse aorta constriction vehicle group.
Increased Expression of TRPV1 in Pressure Overload–Induced Cardiac Hypertrophy In Vivo
It has been reported that TRPV1, TRPV4, and TRPM6 were the TRP family members expressed primarily in the cardiovascular system; therefore, we tested whether expression of these TRP family members was altered during cardiac hypertrophy and CPZ treatment in vivo. The results showed that TAC induced a significant increase in expression of TRPV1 protein (Figure 6A) but not TRPV4 or TRPM6 (Figure 6B and 6C). After CPZ treatment of 2.5 mg/kg per day for 8 weeks, expression of TRPV1 protein was decreased, and there was no obvious change in TRPV4 or TRPM6 protein expression after CPZ treatment (Figure 6). These results revealed that TRPV1 played a pivotal role in the TRPV family for cardiac pathophysiology, and CPZ treatment attenuated the TAC‐induced expression of TRPV1.
Figure 6.
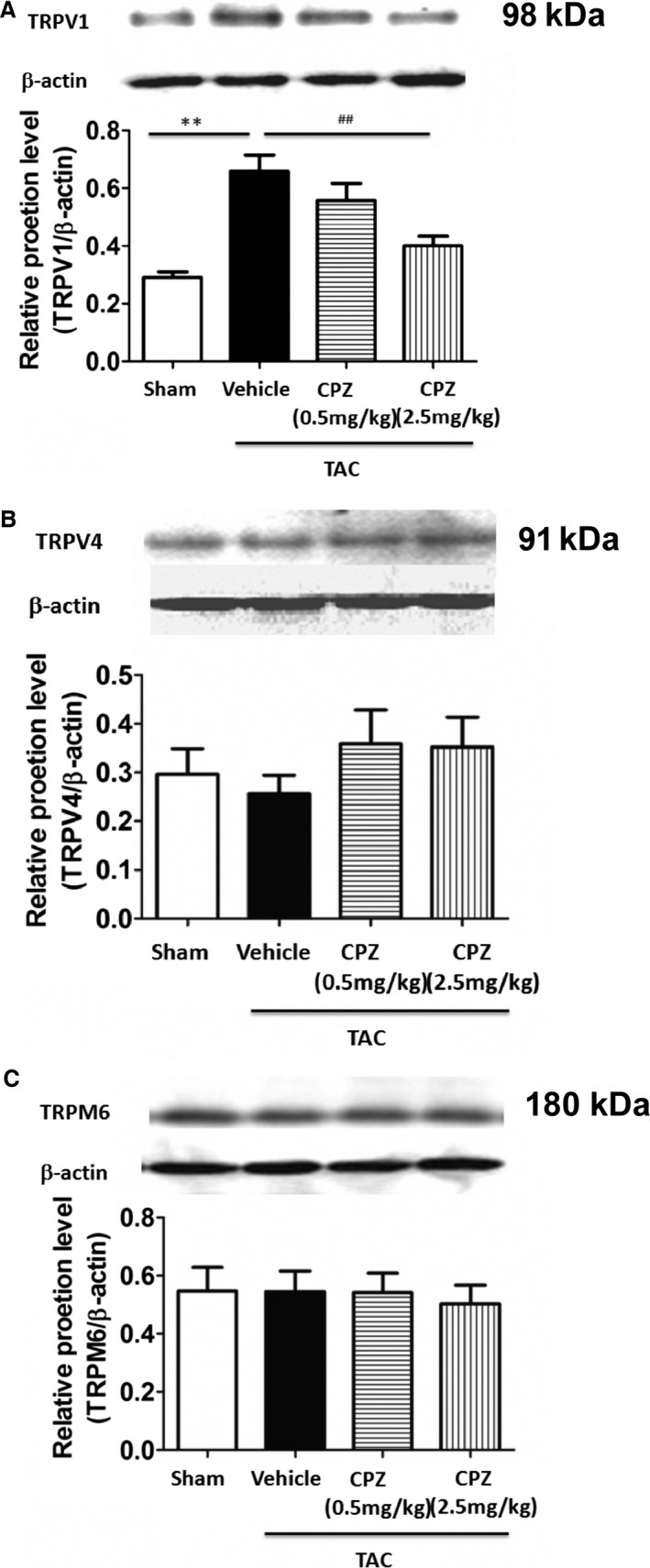
Expression of transient receptor potential family proteins in the left ventricle of TAC‐operated mice after CPZ treatment. A, Western blot analyses of TRPV1 protein expression in the left ventricle of mice show levels were increased significantly after TAC treatment, and CPZ (2.5 mg/kg per day) reversed the effect. **P<0.01 versus sham, ## P<0.01 versus vehicle. Western blotting analyses of TRPV4 (B) and TRPM6 (C) protein expression in the left ventricle of mice show no significant difference within the above groups (n=5 mice per group). CPZ indicates capsazepine; TAC, transverse aorta constriction.
TRPV1 Antagonist Treatment Decreased Expression of Cardiac ODC and Polyamine Level in TAC‐Treated Mice
Because our results showed that TRPV1 activation induced an increase in ODC expression through CaMKII and the MAPK signaling pathway in vitro, we further evaluated the effects of TAC treatment on the expression of ODC and polyamine level and whether CPZ was able to reverse the effect in vivo. Real‐time polymerase chain reaction and Western blot analysis showed that ODC expression increased significantly for 4 or 8 weeks after TAC treatment compared with sham surgery, whereas 2.5 mg/kg per day CPZ treatment reversed the increase in ODC expression (Figure 7A–7D). High‐performance liquid chromatography data showed that TAC‐treated mice had higher concentrations of cardiac polyamines, including putrescine, spermidine, and spermine, and CPZ caused a reduction in cardiac polyamine level following TAC treatment (Figure 7E and 7F). Because polyamines contribute to several physiological and pathological processes, including cardiac hypertrophy,14 modifications in polyamine homeostasis by CPZ treatment might benefit cardiac function.
Figure 7.
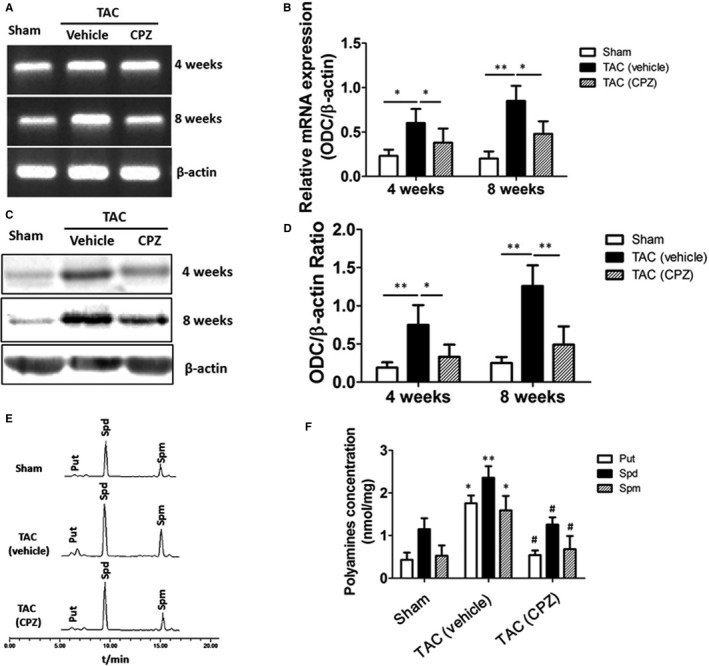
Expression of ODC and polyamine levels were downregulated in CPZ‐treated TAC‐operated mice. A, Expression of ODC mRNA in left ventricle tissue collected from vehicle control‐ and CPZ‐treated mice at 4 or 8 weeks after sham or TAC operation is shown. B, Analyses of cardiac ODC mRNA levels in the above groups (n=6 mice per group) are shown. *P<0.05, **P<0.01. C, Western blotting results from left ventricle tissue of the vehicle control‐ and CPZ‐treated mice at 4 or 8 weeks after sham or TAC operation are shown. D, Analyses of ODC protein in the above groups (n=6 mice per group) are shown. *P<0.05, **P<0.01. E, Representative figure shows the chromatographic separation of polyamines including putrescine, spermidine, and spermine in left ventricle tissue of the sham‐operated and vehicle control‐ and CPZ‐treated TAC‐operated mice. F, Measurement of polyamine level in the above group (n=6 mice per group) is shown. *P<0.05, **P<0.01 versus sham group, # P<0.05 versus TAC vehicle control group. CPZ indicates capsazepine; ODC, ornithine decarboxylase; Put, putrescine; Spd, spermidine; Spm, spermine; TAC, transverse aorta constriction.
CPZ Reverses TAC‐Induced Cardiac Hypertrophy Through CaMKIIδ and the MAPK Signaling Pathway In Vivo
Our results showed that activation of TRPV1 induced an increase in intracellular calcium and phosphorylated CaMKIIδ–dependent MAPK signaling and that expression of TRPV1 was upregulated in TAC‐induced hypertrophic hearts in vivo; therefore, we assayed the expression of CaMKIIδ and MAPK proteins in TAC‐induced cardiac hypertrophy in vivo. Western blot analysis showed that TAC treatment for 4 or 8 weeks also induced an increase in phosphorylated CaMKIIδ in cardiac myocytes (Figure 8A and 8B) that was attenuated by CPZ treatment of 2.5 mg/kg per day in vivo. To observe the change of MAPK signaling components after CPZ treatment in TAC‐induced cardiac hypertrophy in vivo, we further measured the expression of phosphorylated ERKs, p38, and JNK proteins in the sham‐ and TAC‐operated mice and analyzed the effect of CPZ on expression of these MAPKs. The results showed increased expression of phosphorylated ERKs and p38 but not phosphorylated JNK in TAC vehicle groups, and this effect was reversed by CPZ treatment of 2.5 mg/kg per day (Figure 8A, 8C–8E).
Figure 8.
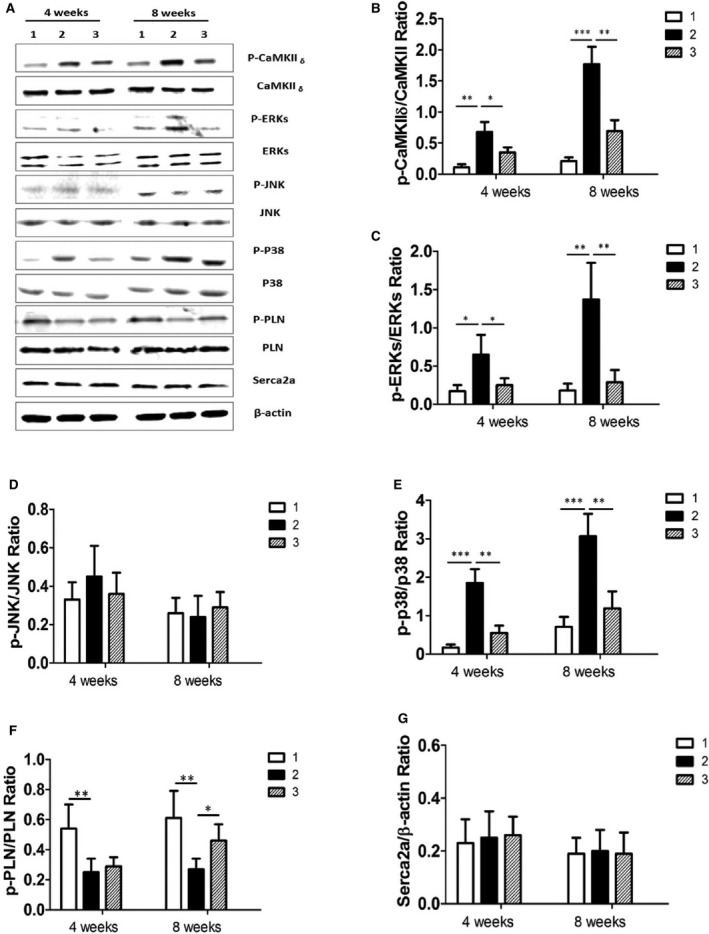
Therapeutic effect of CPZ in TAC‐induced cardiac hypertrophy by inhibiting the CaMKII δ–MAPK signaling pathway in vivo. A, Expression of proteins were analyzed by Western blotting for CaMKII δ, ERKs, p38, JNK, PLN, and Serca2a in the sham‐operated group (panel 1), the TAC group (panel 2), and the group treated with CPZ 2.5 mg/kg per day (panel 3). In the TAC group at 4 or 8 weeks after TAC operation, β‐actin was used as an internal control. Relative expression ratios of p‐CaMKII δ to CaMKII δ (B), p‐ERKs to ERKs(C), p‐JNK to JNK(D), p‐p38 to p38 (E), p‐PLN to PLN (F), and Serac2a to β‐actin (G) in left ventricle tissue from the above groups (6 independent experiments per group) are shown. *P<0.05, **P<0.01, ***P<0.001. CaMKII indicates calmodulin‐dependent protein kinase II; CPZ, capsazepine; ERK, extracellular signal–regulated kinase; JNK, c‐Jun N‐terminal kinase; p, phosphorylated; p, phosphorylated; PLN, phospholamban; TAC, transverse aorta constriction.
Sarcoplasmic reticulum Ca2+ handling plays an important role in chronic cardiac hypertrophy and remodeling, and the coupling of workload is partly dependent on mitochondrial Ca2+ uniporter activity.15 To address this issue, we observed the expression of cardiac muscle Serca2a, which is the key regulator of cardiac diastolic function, and PLN, a 52–amino acid integral membrane protein that regulates the function of Serca2a. The results showed that expression of phosphorylated PLN was significantly decreased in the hearts of TAC‐treated mice for 8 weeks, and this decrease was reversed by CPZ treatment; however, there was no significant difference in expression of Serca2a between sham and TAC groups (Figure 8F and 8G).
Discussion
TRPV1 receptors act as transducers and molecular integrators of nociceptive stimuli in the periphery.20, 21 They are also found in multiple target tissues and cell types in the heart, and a number of research articles show that TRPV1 regulation can affect cardiopathologies.6, 22, 23 Previous results showed that mice lacking functional TRPV1 or mice treated with TRPV1 antagonist had improved heart function and reduced hypertrophic, fibrotic, and apoptotic markers using TAC to model pressure overload cardiac hypertrophy.10, 24 The present study reveals that increased expression of TRPV1 in TAC‐treated mice and activation of TRPV1 is able to induce cardiohypertrophic response in vitro. Although TRPV1 was initially identified as the receptor for CAP—the pungent ingredient in hot chili peppers—it has other functions, and multiple signals and inputs, such as pH, endogenous anandamide, protein kinase C phosphorylation, mechanosensitivity, and baroreflex, that activate TRPV1 are also associated with cardiohypertrophic conditions.25, 26, 27, 28, 29 This indicates that TRPV1 plays an important role in cardiac physiology and in the progression of cardiac hypertrophy.
Cardiac hypertrophy is characterized by a dysregulation in intracellular Ca2+ homeostasis within cardiomyocytes, and it has been confirmed that this Ca2+ influx is induced by a persistent increase of intracellular calcium concentration through l‐type Ca2+ channels and the activation of β‐adrenergic receptors in failing hearts.30, 31 Whether Ca2+ influx through TRPV1 plays a role in this process remains unclear. In this study, Ca2+ influx in cardiomyocytes subjected to TRPV1 activation was significantly increased. To investigate the mechanisms underlying TRPV1‐induced Ca2+ influx in the development of cardiac hypertrophy, we modulated calcium‐dependent kinase activity by compound KN‐93 for inhibition of CaMKII, which is widely used for this purpose.32 CAP‐induced increase in cardiac hypertrophy was reduced by KN‐93, and this implies that Ca2+ CaMKII is involved in TRPV1 activation–induced cardiac hypertrophy.
MAPKs are well characterized and have been reported to be important for the induction of hypertrophic responses and signaling in pressure overload–induced cardiac hypertrophy and remodeling.33, 34 Although the link between TRPV1‐mediated increases in intracellular calcium level and MAPK signaling is not fully understood, CAP‐induced phosphorylation of ERKs and p38 was found in load‐induced skeletal muscle hypertrophy,35 whereas p38 was also required for TRPV1‐mediated CAP‐induced apoptosis of glioma cells.36 The present study confirmed that CAP could induce phosphorylation of ERKs and p38 but not JNK in cardiomyocytes. Inhibition of CaMKII decreased the phosphorylation of ERKs and p38 induced by CAP. Our data continued to highlight the importance of TRPV1‐mediated calcium signaling in intracellular signaling pathways, especially in the heart.
The present results also demonstrated that TRPV1 activation markedly increased the concentrations of polyamines in the heart, including putrescine, spermidine, and spermine, and expression of ODC mRNA and protein. Because ODC and polyamines are required for cell growth,37 the common belief is that the induction of cardiac ODC could be an obligatory step in heart hypertrophy induced by catecholamines.38 Nevertheless, experiments using transgenic mice overexpressing ODC in the heart demonstrated that the robust expression of ODC is not sufficient by itself to promote heart hypertrophy but indicated that ODC may cooperate with other factors inducing cardiac growth.39 Cardiac ODC induction by β‐adrenoreceptor agonist was associated with the phosphorylation of ERKs, and the blockade of ODC induction by l‐type calcium channel antagonists also prevented the phosphorylation of ERKs. This finding strongly suggests that the activation of the MAPK cascade appears to be important for the induction of cardiac ODC by catecholamines but requires critical concentration of free intracellular calcium.40 Our results showed that TRPV1 activation–induced cardiac ODC was dependent on phosphorylation of p38 because the p38‐selective inhibitor BIRB‐796 could downregulate the expression of cardiac ODC induced by CAP. Moreover, CAP also induced an increase in intracellular calcium level, possibly requiring the activation of the p38 signaling pathway and ODC induction at the same time.
Interestingly, our results also revealed that phosphorylation of PLN was decreased in TAC‐induced cardiac hypertrophy. It has been reported that PLN inhibits Serca2a activity by decreasing the apparent affinity of the Serca2a for Ca2+, and this inhibition is relieved by phosphorylation of PLN at serine 16 or threonine 17.41 This suggests that the activity of Serca2a in TAC‐induced cardiac hypertrophy is inhibited because of decreased phosphorylation of PLN, resulting in a decrease in the rate of relaxation of cardiac muscle and a positive inotropic effect. In skeletal muscle sarcoplasmic reticulum, it was found that the Serca2a activity was inhibited by a variety of polyamines including spermine and spermidine,42 so the activity of Serca2a in TAC‐induced cardiac hypertrophy is inhibited either indirectly by a decrease in PLN phosphorylation or directly by elevated intracellular polyamine levels. Because the potential nonspecific nature of using TRPV1 agonists or antagonists in vitro may be magnified in the in vivo experiments, in which their effects likely affect all tissue types including nervous system and result in off‐target effects, it will be interesting to observe the possible structural and functional change in heart‐specific TRPV1 knockout mice after TAC treatment.
The TRPV1 receptor is of great interest in the pharmaceutical industry as the molecular target for CAP, and CAP has been used as a treatment for pain because of its ability to desensitize TRPV1 channels in neurons.43 As the first reported antagonist of TRPV1, CPZ has also been used in pharmacological studies.44 Although early TRPV1 antagonists in the clinic showed worrisome adverse effects including hyperthermia and impaired noxious heat sensation, it is reported that at least 7 orally active TRPV1 antagonist substances have progressed into clinical development, and several more are in preclinical development. The ligand GRC 6211 (Eli Lilly and Company–Glenmark) is the most advanced and is currently in phase IIb clinical trials.45
Taken together, upregulation of TRPV1 involved in cardiac hypertrophy was shown in the present study. Activation of TRPV1 induced increases in intracellular calcium levels and CAMKII/MAPK signaling. Increased cardiac ODC and intracellular polyamine levels as a consequence of phosphorylation of p38 also contributed to the pathogenesis of cardiac hypertrophy. In line with our results, Buckley et al10 demonstrated that mice lacking functional TRPV1 or mice treated with TRPV1 antagonist have improved heart function and reduced hypertrophic, fibrotic, and apoptotic markers in a model of pressure overload. This suggests that TRPV1 plays a role in the progression of cardiac hypertrophy through intracellular calcium and CaMKII/MAPK‐dependent polyamine pathways. Conclusively, TRPV1 antagonism provides an exploitable therapeutic advantage for the treatment of cardiac hypertrophy.
Sources of Funding
This work was supported by grants from the Key Science and Technology Innovation Team in Shaanxi Province of China (No. IRT‐14208) and the National Natural Science Foundation in China (No. 81000062 and No. 81070198).
Disclosures
None.
Acknowledgments
The authors are grateful to Dr C. Colin Brinkman, from the Center for Vascular and Inflammatory Diseases, University of Maryland School of Medicine in Baltimore, MD, for the valuable help with the manuscript review and language editing.
(J Am Heart Assoc. 2016;5:e003718 doi: 10.1161/JAHA.116.003718)
References
- 1. Eid SR, Cortright DN. Transient receptor potential channels on sensory nerves. Handb Exp Pharmacol. 2009;194:261–281. [DOI] [PubMed] [Google Scholar]
- 2. Stucky CL, Dubin AE, Jeske NA, Malin SA, McKemy DD, Story GM. Roles of transient receptor potential channels in pain. Brain Res Rev. 2009;60:2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun H, Li DP, Chen SR, Hittelman WN, Pan HL. Sensing of blood pressure increase by transient receptor potential vanilloid 1 receptors on baroreceptors. J Pharmacol Exp Ther. 2009;331:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yin S, Luo J, Qian A, Du J, Yang Q, Zhou S, Yu W, Du G, Clark RB, Walters ET, Carlton SM, Hu H. Retinoids activate the irritant receptor TRPV1 and produce sensory hypersensitivity. J Clin Invest. 2013;123:3941–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santha P, Jenes A, Somogyi C, Nagy I. The endogenous cannabinoid anandamide inhibits transient receptor potential vanilloid type 1 receptor‐mediated currents in rat cultured primary sensory neurons. Acta Physiol Hung. 2010;97:149–158. [DOI] [PubMed] [Google Scholar]
- 6. Zhong B, Wang DH. Protease‐activated receptor 2‐mediated protection of myocardial ischemia‐reperfusion injury: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1681–R1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Wang J, Wang C, Liu J, Shi L, Xu M, Wang C. Functional expression of transient receptor potential vanilloid‐related channels in chronically hypoxic human pulmonary arterial smooth muscle cells. J Membr Biol. 2008a;223:151–159. [DOI] [PubMed] [Google Scholar]
- 8. Smith SA, Leal AK, Williams MA, Murphy MN, Mitchell JH, Garry MG. The TRPv1 receptor is a mediator of the exercise pressor reflex in rats. J Physiol. 2010;588:1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng J, Li YJ. The vanilloid receptor TRPV1: role in cardiovascular and gastrointestinal protection. Eur J Pharmacol. 2010;627:1–7. [DOI] [PubMed] [Google Scholar]
- 10. Buckley C, Stokes A. Mice lacking functional TRPV1 are protected from pressure overload cardiac hypertrophy. Channels. 2011;5:367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horton JS, Buckley CL, Stokes AJ. Successful TRPV1 antagonist treatment for cardiac hypertrophy and heart failure in mice. Channels. 2013;7:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA. Ca2+/calmodulin‐dependent protein kinase II‐dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem. 2008b;283:25524–25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belostotskaya GB, Golovanova TA. Characterization of contracting cardiomyocyte colonies in the primary culture of neonatal rat myocardial cells: a model of in vitro cardiomyogenesis. Cell Cycle. 2014;13:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu X, Cao Y, Nie J, Liu H, Lu S, Hu X, Zhu J, Zhao X, Chen J, Chen X, Yang Z, Li X. Genetic and pharmacological inhibition of Rheb1‐mTORC1 signaling exerts cardioprotection against adverse cardiac remodeling in mice. Am J Pathol. 2013;182:2005–2014. [DOI] [PubMed] [Google Scholar]
- 15. Brisco MJ, Morley AA. Quantification of RNA integrity and its use for measurement of transcript number. Nucleic Acids Res. 2012;40:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li M, Sun Y, Tomiya N, Hsu Y, Chai TC. Elevated polyamines in urothelial cells from OAB subjects mediate oxotremorine‐evoked rapid intracellular calcium rise and delayed acetylcholine release. Am J Physiol Renal Physiol. 2013;305:F445–F450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Förster A, Schmitto JD, Gummert J, Schöndube FA, Hasenfuss G, Maier LS, Sossalla S. Ca2+/calmodulin‐dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation. 2013;128:970–981. [DOI] [PubMed] [Google Scholar]
- 18. Giordano E, Hillary RA, Vary TC, Pegg AE, Sumner AD, Caldarera CM, Zhang X, Song J, Wang J, Cheung J, Shantz LM. Overexpression of ornithine decarboxylase decreases ventricular systolic function during induction of cardiac hypertrophy. Amino Acids. 2012;42:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiacdysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sisignano M, Bennett DL, Geisslinger G, Scholich K. TRP‐channels as key integrators of lipid pathways in nociceptive neurons. Prog Lipid Res. 2014;53:93–107. [DOI] [PubMed] [Google Scholar]
- 22. Robbins N, Koch SE, Rubinstein J. Targeting TRPV1 and TRPV2 for potential therapeutic interventions in cardiovascular disease. Transl Res. 2013;161:469–476. [DOI] [PubMed] [Google Scholar]
- 23. Zhong B, Wang DH. N‐oleoyldopamine, a novel endogenous capsaicin‐like lipid, protects the heart against ischemia‐reperfusion injury via activation of TRPV1. Am J Physiol Heart Circ Physiol. 2008;295:H728–H735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhong B, Wang DH. TRPV1 gene knockout impairs preconditioning protection against myocardial injury in isolated perfused hearts in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1791–H1798. [DOI] [PubMed] [Google Scholar]
- 25. Vaughan‐Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. [DOI] [PubMed] [Google Scholar]
- 26. Malinowska B, Baranowska‐Kuczko M, Schlicker E. Triphasic blood pressure responses to cannabinoids: do we understand the mechanism? Br J Pharmacol. 2012;165:2073–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Studer M, McNaughton PA. Modulation of single‐channel properties of TRPV1 by phosphorylation. J Physiol. 2010;588:3743–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamazaki T, Yazaki Y. Molecular basis of cardiac hypertrophy. Z Kardiol. 2000;89:1–6. [DOI] [PubMed] [Google Scholar]
- 29. Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009;123:371–385. [DOI] [PubMed] [Google Scholar]
- 30. Horiuchi‐Hirose M, Kashihara T, Nakada T, Kurebayashi N, Shimojo H, Shibazaki T, Sheng X, Yano S, Hirose M, Hongo M, Sakurai T, Moriizumi T, Ueda H, Yamada M. Decrease in the density of t‐tubular L‐type Ca2+ channel currents in failing ventricular myocytes. Am J Physiol Heart Circ Physiol. 2010;300:H978–H988. [DOI] [PubMed] [Google Scholar]
- 31. Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov‐Yarovoy V, Yamoah EN, Hammond HK, Chiamvimonvat N. Adenylyl cyclase subtype‐specific compartmentalization: differential regulation of L‐type Ca2+ current in ventricular myocytes. Circ Res. 2013;112:1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dries E, Bito V, Lenaerts I, Antoons G, Sipido KR, Macquaide N. Selective modulation of coupled ryanodine receptors during microdomain activation of calcium/calmodulin‐dependent kinase II in the dyadic cleft. Circ Res. 2013;113:1242–1252. [DOI] [PubMed] [Google Scholar]
- 33. Dingar D, Merlen C, Grandy S, Gillis MA, Villeneuve LR, Mamarbachi AM, Fiset C, Allen BG. Effect of pressure overload‐induced hypertrophy on the expression and localization of p38 MAP kinase isoforms in the mouse heart. Cell Signal. 2010;22:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sabri A, Rafiq K, Seqqat R, Kolpakov MA, Dillon R, Dell'italia LJ. Sympathetic activation causes focal adhesion signaling alteration in early compensated volume overload attributable to isolated mitral regurgitation in the dog. Circ Res. 2008;102:1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito N, Ruegg UT, Kudo A, Miyagoe‐Suzuki Y, Takeda S. Capsaicin mimics mechanical load‐induced intracellular signaling events: involvement of TRPV1‐mediated calcium signaling in induction of skeletal muscle hypertrophy. Channels (Austin). 2013;7:221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, Giangaspero F, Santoni G. Capsaicin‐induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem. 2007;102:977–990. [DOI] [PubMed] [Google Scholar]
- 37. Nedeva I, Koripelly G, Caballero D, Chièze L, Guichard B, Romain B, Pencreach E, Lehn JM, Carlier MF, Riveline D. Synthetic polyamines promote rapid lamellipodial growth by regulating actin dynamics. Nat Commun. 2013;4:2165. [DOI] [PubMed] [Google Scholar]
- 38. Bordallo C, Cantabrana B, Velasco L, Secades L, Meana C, Méndez M, Bordallo J, Sánchez M. Putrescine modulation of acute activation of the beta‐adrenergic system in the left atrium of rat. Eur J Pharmacol. 2008;598:68–74. [DOI] [PubMed] [Google Scholar]
- 39. Shantz LM, Feith DJ, Pegg AE. Targeted over expression of ornithine decarboxylase enhances beta‐adrenergic agonist‐induced cardiac hypertrophy. Biochem J. 2001;358:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. López‐Contreras AJ, de la Morena ME, Ramos‐Molina B, Lambertos A, Cremades A, Peñafiel R. The induction of cardiac ornithine decarboxylase by β2 –adrenergic agents is associated with calcium channels and phosphorylation of ERK1/2. J Cell Biochem. 2013;114:1978–1986. [DOI] [PubMed] [Google Scholar]
- 41. Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, Veglia G. Allosteric regulation of SERCA by phosphorylation ‐mediated conformational shift of phospholamban. Proc Natl Acad Sci USA. 2013;110:17338–17343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hughes G, Starling AP, East JM, Lee AG. Mechanism of inhibition of the Ca(2+)‐ATPase by spermine and other polycationic compounds. Biochemistry. 1994;33:4745–4754. [DOI] [PubMed] [Google Scholar]
- 43. Smith H, Brooks JR. Capsaicin‐based therapies for pain control. Prog Drug Res. 2014;68:129–146. [DOI] [PubMed] [Google Scholar]
- 44. Nazıroglu M. TRPV1 channel: a potential drug target for treating epilepsy. Curr Neuropharmacol. 2015;13:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gunthorpe M, Chizh B. Clinical development of TRPV1 antagonists: targeting a pivotal point in the pain pathway. Drug Discovery Today. 2009;14:56–67. [DOI] [PubMed] [Google Scholar]