

Abstract
Background
Cardiomyopathy is a leading cause of mortality among Duchenne muscular dystrophy patients and lacks effective therapies. Phosphodiesterase type 5 is implicated in dystrophic pathology, and the phosphodiesterase type 5 inhibitor tadalafil has recently been studied in a clinical trial for Duchenne muscular dystrophy.
Methods and Results
Tadalafil was evaluated for the prevention of cardiomyopathy in the mdx mouse and golden retriever muscular dystrophy dog models of Duchenne muscular dystrophy. Tadalafil blunted the adrenergic response in mdx hearts during a 30‐minute dobutamine challenge, which coincided with cardioprotective signaling, reduced induction of μ‐calpain levels, and decreased sarcomeric protein proteolysis. Dogs with golden retriever muscular dystrophy began daily tadalafil treatment prior to detectable cardiomyopathy and demonstrated preserved cardiac function, as assessed by echocardiography and magnetic resonance imaging at ages 18, 21, and 25 months. Tadalafil treatment improved golden retriever muscular dystrophy histopathological features, decreased levels of the cation channel TRPC6, increased total threonine phosphorylation status of TRPC6, decreased m‐calpain levels and indicators of calpain target proteolysis, and elevated levels of utrophin. In addition, we showed that Duchenne muscular dystrophy patient myocardium exhibited increased TRPC6, m‐calpain, and calpain cleavage products compared with control human myocardium.
Conclusions
Prophylactic use of tadalafil delays the onset of dystrophic cardiomyopathy, which is likely attributed to modulation of TRPC6 levels and permeability and inhibition of protease content and activity. Consequently, phosphodiesterase type 5 inhibition is a candidate therapy for slowing the development of cardiomyopathy in Duchenne muscular dystrophy patients.
Keywords: dobutamine stress, dystrophin cardiomyopathy, magnetic resonance imaging, phosphodiesterase type 5 inhibition, protease, utrophin
Subject Categories: Translational Studies, Physiology, Fibrosis, Animal Models of Human Disease, Cardiomyopathy
Introduction
Duchenne muscular dystrophy (DMD) is a fatal X‐linked disease that affects ≈1:3500 boys and for which there is no cure and only limited treatment options. DMD is caused by a mutation in the dystrophin gene that results in complete loss of the sarcolemma‐stabilizing protein dystrophin and leads to progressive contraction‐induced degeneration and fibrotic replacement of skeletal and cardiac muscle. With improvements in DMD patient care that have led to prolonged life expectancy, cardiomyopathy is becoming a leading cause of death in DMD patients.1 Effective treatments for DMD‐associated cardiomyopathy is a critical unmet need for these patients.
Standard treatments for cardiomyopathy in DMD patients involves the use of β‐blockers and angiotensin‐converting enzyme inhibitors.1 A potential treatment strategy that showed promise in the treatment of the mild cardiomyopathy seen in mdx mice (the mouse model of human DMD) is inhibition of cyclic guanosine monophosphate (cGMP)–selective phosphodiesterase type 5 (PDE5) using the PDE5 inhibitor sildenafil.2, 3 Inhibition of PDE5 prevents the hydrolysis of cGMP, thereby enhancing nitric oxide signaling effects through activation of cGMP‐dependent protein kinase (PKG).4 In models of heart failure, PDE5 inhibition has improved cardiomyopathy5, 6, 7, 8, 9; however, a recent study of sildenafil in older (aged >15 years) DMD patients with clinical cardiomyopathy (ejection fraction ≤45%) failed to show any significant benefit with treatment for >6 months.10
Sildenafil's lack of effectiveness in the above‐mentioned DMD patient population does not eliminate the possibility that PDE5 inhibition could be prophylactic against cardiomyopathy development and progression if initiated early rather than being able to reverse preexisting pathology. In the current study, we tested this hypothesis using tadalafil, a PDE5 inhibitor that possesses a longer half‐life than sildenafil (t1/2 of 17.5 versus 4 hours for sildenafil). Tadalafil restores limb functional sympatholysis in patients with Becker muscular dystrophy11 and DMD.12 Based on these muscle benefits, a phase III clinical trial for the use of tadalafil in DMD was performed in patients exhibiting normal cardiac function (ClinicalTrials.gov identifier NCT01865084). Consequently, the impact of long‐term PDE5 inhibition on the progression of cardiomyopathy in DMD is a topic of high and immediate clinical significance.
The present study is a cardiocentric evaluation of long‐term tadalafil treatment that utilizes both the mdx mouse and the golden retriever muscular dystrophy (GRMD) models of DMD. We demonstrated that tadalafil reduces myocardial dysfunction and modifies calpain protease expression in mdx mice following acute stress and delays the onset of cardiomyopathy in GRMD dogs. Mechanistically, we provided evidence that tadalafil's effect in GRMD heart involves changes in TRPC6 levels and phosphorylation and m‐calpain abundance and activity, leading to decreased breakdown of utrophin.
Methods
Animals
All animals were handled in compliance with National Institutes of Health and institutional guidelines that were approved by the institutional animal care and use committee of the University of Pennsylvania. Male mdx mice (n=4–6) began either tadalafil treatment (100 mg*L−1 diluted in drinking water) or control treatment (normal water) at 4 weeks of age and continued treatment for either 10 weeks or 8 months.
Five affected GRMD canines were used in the initial arm of this study. Beginning at age 9 months, 2 dogs were orally administered 1 mg*kg−1 tadalafil daily, whereas the other 3 were used as untreated controls. One dog in the control group died suddenly at 20 months from a presumed fatal cardiac event. The remaining 4 dogs were euthanized following their last echocardiogram at age 25 months. In a separate cohort, 2 affected dogs were used, and 1 began tadalafil treatment starting at an age of 2 months. These dogs were euthanized following 6 months of treatment. Quickly following euthanasia, tissue samples were harvested, and samples were identically processed for snap freezing, freezing in OCT, and fixing in 10% formalin.
Human Heart Samples
Snap‐frozen control (aged 20 years) and DMD (aged 17 years) heart samples were obtained from the National Disease Research Interchange (Philadelphia, PA). The cause of death in these persons was not attributed to a cardiac‐related event.
Dobutamine Stress Test
Echocardiography was performed on mice anesthetized with 2% isoflurane using the Vevo 770 system (VisualSonics) by the University of Pennsylvania Small Animal Imaging Facility. After baseline measurements, dobutamine (42 μg*kg−1*min−1; Bedford Laboratories) was continuously infused into the tail vein, and subsequent echocardiography measurements were taken at 15 and 30 minutes. Body temperature was maintained using a heating pad during the course of the experiment. Following completion of the final measurement, mice were euthanized. The hearts were dissected out, snap‐frozen in liquid nitrogen, and stored at −80°C until further analysis.
Dog Echocardiography
Echocardiography was performed, as described previously,13 using a Philips CX‐50 system (Philips) and an 8‐ to 3‐MHz transducer. The same sonographer performed all evaluations, including 2‐dimensional, M‐mode, spectral Doppler, and tissue Doppler scans of the lateral mitral valve annulus.
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging data were acquired, as described previously,13 at ages 18 and 25 months. Briefly, dogs were anesthetized with an intravenous infusion of propofol (induction: 1.0–2.0 mg*kg−1*min−1; maintenance: 0.2 mg*kg−1*min−1) and fentanyl (induction: 0.005 mg*kg−1*min−1; maintenance: 0.7 μg*kg−1*min−1) via the cephalic vein. Respiration, ECG, O2 saturation, and blood pressure were monitored during the magnetic resonance scanning protocol, and magnetic resonance data were acquired in apnea by turning off the ventilator with an infusion of a bolus of cisatracurium (0.1 mg*kg−1).
For image acquisition, dogs were placed in the dorsal position in the bore of a GE 1.5‐T Signa magnetic resonance system (GE Healthcare). A torso array receive‐only coil was positioned over the thoracic region. Initially, sagittal localizers and long‐axis 4‐chamber images of the heart were performed to assist with positioning of the short‐axis images.14 Images were acquired with retrospective ECG gating, and cardiac‐gated tagged images were acquired using a fast spoiled gradient recalled sequence (field of view 24×24 cm2; acquisition matrix 256×128; repetition time 9.2 ms; echo time 5.5 ms; flip angle 20°; grid tag spacing 7 mm) in the short‐axis transventricular view (4–5 contiguous slices, 8‐mm slice thickness). Triggering used a single cardiac‐phase cycle with minimum trigger delay and 16 frames per cardiac cycle.
Analysis of the tagged images was performed using harmonic phase analysis (Diagnosoft) for peak circumferential myocardial strain.15, 16 The short‐axis slice in the midpapillary region of the left ventricle (LV) was chosen for analysis. In a middle frame image, epicardial and endocardial traces were drawn and then automatically propagated to create a mesh dividing the myocardium into 6 regions in the short‐axis view of all 16 frames acquired in the cardiac cycle. For consistency, the first segment was placed starting at the anterior region of the ventricular septum. The average circumferential myocardial strain from each segment was calculated for the midwall using Eulerian strain algorithms.17
Histology
Formalin‐fixed samples of LV, right ventricle (RV), interventricular septum (IVS), diaphragm, and quadriceps were embedded in paraffin, sectioned at 5 μm, and stained with Masson's trichrome (Polysciences, Inc) or hemotoxylin and eosin (Sigma‐Aldrich). Slides were viewed using a Leitz DMRBE microscope (Leica) equipped with a Leica DCF480 digital camera.
Immunoprecipitation
Immunoprecipitation experiments were carried out using the Pierce Classic IP Kit (Thermo Scientific). Muscle lysates containing 1 mg total protein were immunoprecipitated with 5 μg of anti‐TRPC3 or TRPC6 (Alomone Labs) overnight at 4°C with end‐over‐end mixing. The immune complex was eluted with reducing sample buffer and boiled at 100°C for 5 minutes before being applied to SDS‐PAGE gel and immunoblotted, as described below.
Immunoblotting
Snap‐frozen mdx mouse whole hearts and GRMD LV samples were finely crushed and homogenized in T‐PER buffer (Thermo Scientific) supplemented with protease and phosphatase inhibitors (Thermo Scientific). The protein concentration of the resulting supernatant was determined using the Bio‐Rad Protein Assay (Bio‐Rad Laboratories). Protein samples were boiled in 4x sample buffer, subjected to SDS‐PAGE using 4% to 12% SDS polyacrylamide gels (Life Technologies), and transferred to nitrocellulose membranes using the iBlot system (Life Technologies). Membranes were blocked in 5% milk‐TBST and incubated with primary antibody overnight at 4°C. Following TBST washes, membranes were incubated in the appropriate horseradish peroxidase–conjugated anti‐rabbit (Cell Signaling), anti‐mouse (Cell Signaling), or anti‐goat (Santa Cruz Biotechnology) secondary antibody for 1 hour at room temperature, washed, incubated for 5 minutes in ECL reagent (Thermo Scientific), and imaged using the LI‐COR C‐DiGit imaging system (LI‐COR Biosciences). Blots for phosphorylated proteins were stripped and reprobed for total protein. All membranes underwent a final probe for GAPDH (1:2000; Santa Cruz Biotechnology) and/or were stained with Ponceau Red for loading control and normalization. Band signal intensities were measured using Image Studio Lite software (LI‐COR Biosciences), normalized to sample loading, and reported relative to respective control samples. The following primary antibodies were used for this study: PDE5 (1:2000; no. 2395; Cell Signaling), TRPC3 (1:500; no. ACC‐016; Alomone Labs), TRPC6 (1:1000; no. ACC‐017; Alomone Labs), cyclic guanosine monophosphate–dependent kinase 1α (PKG1α; 1:1000; no. 13511; Cell Signaling), phospho–glycogen synthase kinase 3β (phospho‐GSK3β; Ser8/9; 1:1000; no. 9323; Cell Signaling), GSK3β (1:1000; no. 9315; Cell Signaling), phospho‐p42/44 extracellular signal–related kinase (ERK; threonine 202 [Thr202]/Tyr204; 1:1000; no. 9101; Cell Signaling), p42/44 ERK (1:1000; no. 9102; Cell Signaling), sarco/endoplasmic reticulum Ca2+‐ATPase 2 (SERCA2; 1:2000; no. ab3625; Abcam), atrial natriuretic peptide (ANP; 1:500; no. 91250; Abcam); endothelial nitric oxide synthase (1:500; no. ab5589; Abcam), neuronal nitric oxide synthase (1:500; no. 1376; Abcam), utrophin (1:500; no. VP‐U579; Vector Laboratories), KH‐type splicing regulatory protein (KSRP); 1:2000; no. A302‐021A; Bethyl Laboratories), RhoA (1:500; no. 2117; Cell Signaling), phospho–cardiac troponin I (phospho‐cTnI; Ser23/24; 1:1000; no. 4004; Cell Signaling), cTnI (1:500; no. sc‐8117; Santa Cruz Biotechnology), phospholamban (1:1000; no. 21923; Santa Cruz Biotechnology), actin (1:2000; no. A3853; Sigma‐Aldrich), talin (1:1000; no. T3287; Sigma‐Aldrich), μ‐calpain (1:1000; no. C0355; Sigma‐Aldrich), m‐calpain (1:500; no. ab39165; Abcam), dysferlin (1:1000; no. ab139379; Abcam), integrin β1 (1:1000; no. ab179471; Abcam), α‐actinin (1:2000; no. A7732; Sigma‐Aldrich), spectrin (1:1000; no. 11755; Abcam), γ‐sarcoglycan (1:100; no. VP‐G803; Vector Laboratories), dystrophin (1:1000; no. 15277; Abcam), matrix metalloproteinase 2 (MMP2; 1:1000; no. NB200‐193; Novus Biologicals), and MMP9 (1:500; no. ab38898; Abcam).
Immunofluorescence
Frozen OCT‐embedded LV samples were sectioned at 10 μm, fixed in ice‐cold acetone, blocked in 5% BSA‐PBS, and incubated with anti‐utrophin (1:50; Vector Laboratories), anti‐laminin (1:1000; Dako), or anti–γ‐sarcoglycan (1:25; Vector Laboratories) primary antibody overnight at 4°C. Following washes, slides were incubated in donkey anti‐mouse Alexa 488 or donkey anti‐rabbit Alexa 568 conjugated secondary antibody (1:500; Life Technologies), coverslipped with VectaShield (with DAPI; Vector Laboratories), and imaged using either a Leica TSC‐8 confocal microscope or a Leitz DMRBE microscope equipped with a Leica DCF480 digital camera. Comparative images were stained, imaged, and processed simultaneously under identical conditions.
Reverse Transcriptase Polymerase Chain Reaction
RNA was isolated from finely crushed snap‐frozen samples using Trizol reagent (Life Technologies), treated with DNase (Promega), and reverse transcribed using the SuperScript III kit (Life Technologies). Resulting cDNA was subjected to polymerase chain reaction using GoTaq (Promega). The following mouse‐specific primers were used: Pde5A (forward) 5′‐ CGGCCTACCT GGCATTCTG‐3′, (reverse) 5′‐AATCAGGTGTTACTTGACCTTGC‐3′; Gapdh (forward) 5′‐AGC AGGCATCTGAGGGCCCA‐3′, (reverse) 5′‐TCCCAACTCGGCCCCCAACA‐3′. The following canine‐specific primers were used: Pde5A (forward) 5′‐ ATACTTGCCCTGCTGATTGCTGCT C‐3′, (reverse) 5′‐TGTTGAATAGGCCACGGTTTTGTAATG‐3′; Trpc3 (forward) 5′‐CAAGCTGGCCAACAT AGAGAAGGAGT‐3′, (reverse) 5′‐ CAAGCAGGCCCAGGAATATGATGAAAGAAG‐3′; Trpc6 (forward) 5′‐ATGATATGGGCCGAATGTAAAGAA AT‐3′, (reverse) 5′‐TGCGCCAATGTAGTA GGAGTAGAGG ‐3′; utrophin (forward) 5′‐ AAACATCCTCGGCTTGGCTACC‐3′, (reverse) 5′‐ AG GAGTTTGGCTTCTGCGATGAG‐3′; Gapdh (forward) 5′‐ATGGTGAAGGTCGGAGTC AACG‐3′, (reverse) 5′‐GAAGATGGAGATGGACTTCCC‐3′. Polymerase chain reaction products were run on 2% agarose gels and imaged with the G:Box Chemi IR6 imaging system (Syngene). Values were normalized to Gapdh and reported relative to respective control samples.
Statistical Analysis
Statistical analysis in studies involving mdx mice was performed using the Student t test, 1‐way ANOVA (Tukey honest significant difference post hoc tests), or linear regression, as appropriate (α=0.05). Functional data from GRMD studies were analyzed using the Welch t test (unequal variance), with results of P<0.20 reported with Cohen's d and the Pearson correlation coefficient (r) to show effect size of treatment. GRMD biochemical data were reported with Cohen's d. All outcomes reported in this study were expected to have normal distributions.
Results
Tadalafil Modifies β1‐Adrenergic Response in mdx Mouse Hearts
Male mdx mice began tadalafil treatment (0.1 mg*mL−1 supplemented in drinking water) at age 4 weeks. After no adverse effects were observed during 8 months of treatment, we investigated the ability of tadalafil to protect the mdx heart from a challenge with the β1‐adrenergic agonist dobutamine, which causes exacerbated cardiac dysfunction in the absence of dystrophin.18, 19 This experiment, outlined in Figure 1A, revealed that although no difference in ejection fraction or fractional shortening existed at baseline (Figure 1B), tadalafil significantly blunted elevation of both ejection fraction and fractional shortening induced by the dobutamine infusion (Figure 1C). Interestingly, the dobutamine challenge increased tadalafil's target protein, PDE5, which is undetectable by immunoblotting in nonstimulated mdx hearts (Figure 1D). Although PDE5 protein levels were not significantly different between control and tadalafil treatment (Figure 1D and 1E), Pde5 gene expression was elevated by dobutamine administration only in the absence of tadalafil (Figure 1F). This suggests there may be a pool of posttranscriptionally repressed Pde5 that is responsive to acute stress.
Figure 1.

Tadalafil (Tad) protects mdx hearts from stress induced by dobutamine (Dob). Male mdx mice were control or treated with Tad (n=6) for 8 months, underwent a Dob stress test (A), and were evaluated by echocardiography. B, Baseline echocardiograph measures of ejection fraction and fractional shortening in control and Tad‐treated mdx mice. C, Dob induced changes in ejection fraction and fractional shortening (indicated as percentage change from baseline measurement). Representative blots (D) and quantification (E) of phosphodiesterase type 5 (PDE5), cyclic guanosine monophosphate–dependent kinase 1α (PKG1α), phospho–glycogen synthase kinase 3β (p‐GSK3β) and total GSK3β, phospho–extracellular signal–related kinase 1/2 (p‐ERK1/2) and total ERK1/2, and sarco/endoplasmic reticulum Ca2+‐ATPase 2 (SERCA2) protein content, as normalized to Ponceau Red staining and displayed as arbitrary units (AU) in relation to Dob only values.. Values are represented as mean±SEM; *P<0.05 vs Dob‐only treatment. F, Pde5 gene expression in control and Tad‐treated mdx hearts in response to Dob stress, as determined by reverse transcriptase polymerase chain reaction using Gapdh as a normalization control. Values are indicated as mean±SEM; *P<0.05 vs both groups (Tukey honest significant difference post hoc test). G, Preservation of SERCA2 levels shows significant inverse correlation to the Dob‐induced change in fractional shortening during the stress test.
Although there were no changes in the content of PKG1α (the primary intracellular effector kinase of increased cGMP due to PDE5 inhibition), we observed significant increases in phosphorylation of the PKG target GSK1β and depressed phosphorylation of ERK1/2, a mediator of cardiac pathology, in tadalafil‐treated hearts (Figure 1D and 1E). Dobutamine‐induced loss of SERCA2 occurred in control but not in tadalafil‐treated animals, with the change in fractional shortening (depicted in Figure 1C) inversely correlated with SERCA2 content (Figure 1G). This suggests that preservation of SERCA2 may have a role in the reduced dobutamine‐induced inotropy caused by tadalafil treatment. No changes in content of measured proteins were found between untreated and tadalafil‐treated hearts in the absence of dobutamine.
Because cTnI, another direct target of PKG1α kinase activity, also modifies inotropy,20, 21 we evaluated phosphorylated cTnI levels as a protective mechanism. Indeed, dobutamine infusion depressed phosphorylated cTnI levels in nontreated hearts; however, this was due to loss of cTnI rather than to a change in phosphorylation (Figure 2A and 2B). Both SERCA2 and cTnI are targets for intracellular proteases known as calpains,22, 23 thus we looked for loss of other known calpain targets. The sarcomere‐associated proteins phospholamban and actin were both degraded in dobutamine‐stressed controls (Figure 2A); therefore, preservation of contractility‐related proteins by tadalafil could be due to modulation of protease expression and activation. Accordingly, we found the presence of μ‐calpain (also known as calpain 1), a protease that largely targets sarcomere‐associated proteins,24 to be elevated with dobutamine stress, although it was significantly lower in the tadalafil‐treated hearts (Figure 2B). These data presented a model in which transient cardiac stress in the dystrophic heart, such as that induced by adrenergic stimulation, increased PDE5 content and expression, thereby reducing cGMP content and ultimately leading to cardiac dysfunction by increased proteolysis of sarcomeric proteins. The basis for tadalafil's protective effect likely involves the prevention of damage from transient stressors such as adrenergic responses.
Figure 2.

Tadalafil (Tad) decreases μ‐calpain levels and proteolysis of sarcomeric proteins induced by dobutamine (Dob) in mdx hearts. Representative blots (A) and quantification (B) of phospho–cardiac troponin I (p‐cTnI) and total cTnI, phospholamban (PLB), α‐actin, and μ‐calpain from control and Tad‐treated mdx hearts that underwent the Dob stress test. Protein content was normalized to Ponceau Red staining and displayed as arbitrary units (AU) relative to Dob only values. Values are represented as mean±SEM; *P<0.05 vs Dob‐only treatment.
Tadalafil Delays Cardiomyopathy in GRMD Dogs
Although the previous experiment revealed that tadalafil can protect dystrophic cardiomyocytes from stress‐induced dysfunction, we sought to investigate whether prophylactic tadalafil administration can truly delay the onset of dystrophic cardiomyopathy. Because mdx mice display mild, late‐onset, and non–lifespan‐limiting cardiac pathology, we chose to test this hypothesis in GRMD dogs, which exhibit DMD‐like cardiomyopathy that is usually detectable by age 1 year and is often the cause of mortality. Dogs began daily tadalafil treatment at age 9 months and were evaluated by echocardiography at ages 18, 21, and 25 months. Prior to treatment, there were no functional differences between the untreated and tadalafil‐treated groups; however, tadalafil preserved LV systolic function at an age when untreated animals began to lose function in terms of fractional shortening (Figure 3A) and ejection fraction (Figure 3B). Although both measures demonstrated significant effects in the treated versus untreated dogs at 18 and 21 months, the 25‐month measurement appears to show only modest improvement. The treated cohort began exhibiting functional decline at this time point, whereas the apparent decrease in effect was largely due to the sudden death of an untreated dog with substantial cardiomyopathy at age 20 months, potentially from cardiac arrhythmias (but not documented). The difference between the treated and untreated groups at both the 21‐ and 25‐month time points would likely have been greater if not for this death. In addition, aortic peak flow velocity (Figure 3C) was improved compared with untreated controls at 18 months, with strong trends at 21 and 25 months. Importantly, RV systolic function was also preserved with tadalafil; pulmonary arterial peak flow velocity was higher than values for untreated controls at 21 and 25 months (Figure 3C). This finding suggests that a possible additive benefit of PDE5 inhibition is reduced pulmonary hypertension, a cause of RV dysfunction. LV diastolic function (Figure 4A through 4C) was also better preserved with tadalafil treatment, as shown by a better preserved ratio of early passive filling velocity of the LV to the atrial‐dependent active filling velocity (significant at 25 months; Figure 4A). This measure indicates diastolic function by comparing the early passive filling velocity of the LV and the atrial‐dependent active filling velocity, whereas decreased early passive filling velocity can indicate reduced LV compliance. Tadalafil improved early passive filling velocity at 21 and 25 months rather than reduced atrial‐dependent active filling (Figure 4B). Isovolumetric relaxation time (Figure 4C) was also less than that for untreated controls at 21 months; however, it is not likely to entirely account for the improvements in early passive filling velocity. Tadalafil had no effect on body weight, LV wall, or chamber dimensions. Together, these data demonstrate strong positive effects on the maintenance of GRMD cardiac function by tadalafil.
Figure 3.

Tadalafil preserves systolic function in golden retriever muscular dystrophy (GRMD) hearts. Systolic parameters, as measured by echocardiography, of untreated and tadalafil‐treated GRMD dogs (starting at 9 months; n=3 per group, except for 2 for controls at 21 and 25 months due to the death of 1 dog at 20 months) include left ventricular fractional shortening (A) and ejection fraction (calculated from fractional shortening assuming normal ventricular proportions) (B). Tadalafil treatment initiation is indicated by the arrow, and the values for healthy dogs aged 2 years is represented by the bar labeled “Normal”. Aortic and pulmonary arterial peak flow velocity measurements (C) were also performed. All values and individual values are presented, and group values are represented as mean±SD.
Figure 4.

Diastolic function is maintained in tadalafil‐treated golden retriever muscular dystrophy (GRMD) hearts. A, Improved diastolic function, as determined by left ventricular (LV) early/late mitral valve inflow ratio (E/A) and annulus motion (E′/A′). B, This diastolic improvement is due to increased early (E), not reduced late (A), LV filling velocity. C, LV isovolumetric relaxation time (IVRT) improvements were also evident. All values and individual values are presented, and group values are represented as mean±SD.
Magnetic resonance imaging was also performed at ages 18 and 25 months to evaluate the peak circumferential myocardial strain of the LV, a very sensitive measure of myocardial distress.25 For these measurements, the magnetic resonance image of the complete LV wall was partitioned into 6 segments (Figure 5A). At 18 months (Figure 5B), tadalafil‐treated dogs showed improved circumferential myocardial strain values than untreated dogs in the anterior segment of the interventricular septum (segment 2). The majority of the LV free wall, however, did not trend in either direction with tadalafil. At 25 months (Figure 5C), segment 2 remained significantly better in tadalafil‐treated dogs; however, the free‐wall segments trend toward being worse, thus agreeing with echocardiogram data suggesting that tadalafil‐treated hearts were in a state of functional decline at this time point. In addition, untreated dogs showed no signs of improvement at 25 months, although echocardiographic data suggested this may be the case.
Figure 5.

Cardiac magnetic resonance imaging (MRI) segmental peak circumferential myocardial strain (εcc) improved with tadalafil. Segmental MRI of the golden retriever muscular dystrophy myocardium (segmentation depicted in A) reveals differences in εcc between untreated and tadalafil‐treated dog hearts at 18 (B) and 25‐month (C) evaluations. All values and individual values are presented, and group values are represented as mean±SD.
Tadalafil also improved GRMD cardiac histopathology. Masson's trichrome staining of the LV, interventricular septum, and RV (Figure 6) revealed that at age 25 months, untreated GRMD LV (approximately segments 5–6) and interventricular septum (approximately segment 2–3) were largely characterized by disorganized cardiomyocytes, prominent myocardial interstitial and perivascular fibrosis, and large fibrous or fat deposits throughout the myocardium, whereas the RV showed substantial myocardial deterioration and fibrous/fat deposition. Tadalafil reduced most signs of pathology, with comparatively mild perivascular fibrosis being the prominent feature in the LV and interventricular septum and perivascular‐restricted fibrous/fat deposition in the RV, thus agreeing with the functional benefits of tadalafil treatment for dystrophic myocardium. Reductions in fibronectin levels in tadalafil‐treated LVs (Figure 7B) agree with this observed difference in fibrosis.
Figure 6.

Tadalafil reduces histopathology in golden retriever muscular dystrophy (GRMD) hearts. Representative images of Masson's trichrome–stained left ventricle (LV), interventricular septum (IVS), and right ventricle (RV) from untreated and tadalafil‐treated GRMD dogs at 25 months.
Figure 7.
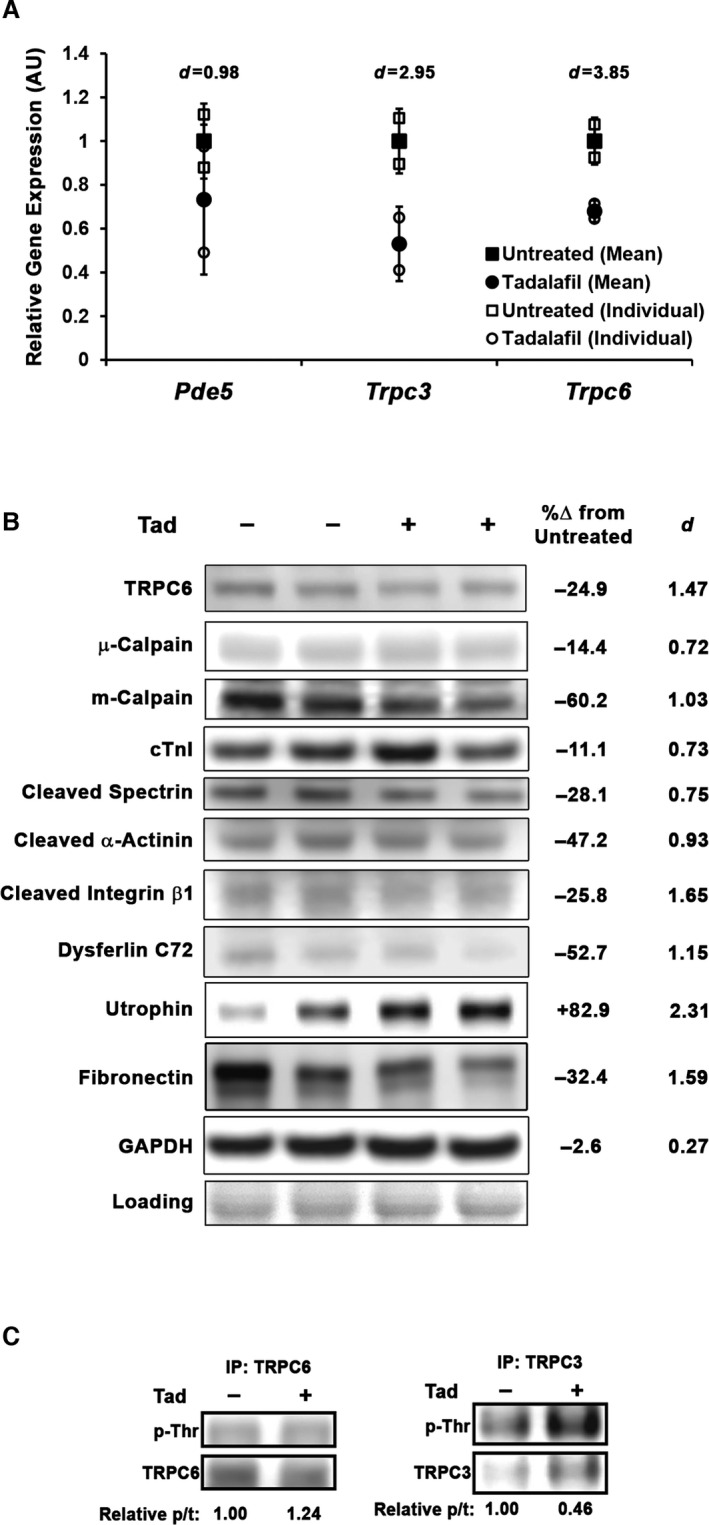
Tadalafil (Tad) modulates TRPC6 and protease levels and activity in golden retriever muscular dystrophy (GRMD) left ventricle (LV). A, Gene expression of Pde5, Trpc3, and Trpc6 in control and Tad‐treated GRMD LV (normalized to Gapdh and displayed as arbitrary units (AU) relative to untreated GRMD values). All values and individual values are presented, and group values are represented as mean±SD. Representative blots of Tad‐induced changes in TRPC6, μ‐calpain, m‐calpain, cardiac troponin I (cTnI), spectrin 150‐kDa cleavage product, α‐actinin 80‐kDa cleavage product, integrin β1 75‐kDa cleavage product, dysferlin C72 fragment, full‐length utrophin, and fibronectin protein content (B), along with percentage difference between untreated and treated values (Cohen's d shown to display effect size). Ponceau Red staining is used as a loading control. C, TRPC6 and TRPC3 immunoprecipitation (IP) results for phosphorylated threonine (p‐Thr) with blot density quantified as phopho‐ to total (p/t) protein ratio.
Modulated TRPC6 and Protease Content in Tadalafil‐Treated GRMD Myocardium
In a recent report, Seo et al26 demonstrated that both genetic deletion of the nonspecific cation channel TRPC6 and sildenafil provide nearly equivalent protection against acute stretch‐induced arrhythmias, an effect of calcium dysregulation in isolated mdx cardiomyocytes, suggesting that PDE5 inhibition deactivates TRPC6 permeability, thereby reducing TRPC6‐mediated Ca2+ influx. This effect is likely a result of PKG‐dependent phosphorylation of TRPC6 on Thr69 (in mice; Thr70 in humans and dogs).27, 28 Although LV gene expression of Pde5a did not change with tadalafil treatment (protein levels were undetectable by immunoblot), expression of both Trpc3 and Trpc6 cation channels trended toward a decrease in expression in the LV of treated GRMD dogs (Figure 7A). In agreement, immunoblotting of LV lysates demonstrated that TRPC6 was reduced at the protein level (Figure 7B) and contained ≈25% more total Thr phosphorylation, as determined by immunoprecipitation (Figure 7C), with tadalafil treatment. Although PKG‐mediated modulation of TRPC3 activity via phosphorylation of Thr11 is another potentially beneficial mechanism,29 its protein level could not be detected by direct loading in any sample, and immunoprecipitation experiments revealed that total Thr phosphorylation status of TRPC3 decreased >50% from untreated GRMD LV lysate. These results further strengthen the role of TRPC6 in the dystrophic myocardium phenotype in that reduction of content and increased phosphorylation are concomitant with phenotype improvement.
Protease content was also different between treated and control GRMD LV samples. In these hearts, which represent a basal state (as opposed to the stressed mdx in the previous experiment) of the GRMD heart at 25 months, levels of μ‐calpain were relatively unchanged; in contrast, content of m‐calpain, a predominantly peripheral protease, were strongly reduced with treatment (Figure 7B). Unchanged content of the sarcomeric protein cTnI coincident with reduced cleavage products of the structural/peripheral proteins α‐spectrin (150 kDa), α‐actinin (80 kDa), integrin β1 (75 kDa), and dysferlin (80 kDa)30, 31, 32, 33 in the tadalafil‐treated hearts corroborated the concept of lower levels and activity of peripheral m‐calpain in these nonstimulated dystrophic hearts. We did not, however, note changes in PKG1α content or the phosphorylation status of GSK3β or ERK1/2, although a decrease in ANP and modest increases in endothelial and neuronal nitric oxide synthases were seen (Figure S1A).
In accordance with reduced m‐calpain content and activity in these long‐term tadalafil‐treated hearts, we found an increase >80% in the content of utrophin (Figure 7B), a known calpain target34 that can functionally replace dystrophin in mdx mice.35 Immunofluorescence of frozen LV sections demonstrated that under identical imaging conditions, sarcolemma‐localized utrophin was increased in both untreated and tadalafil‐treated hearts compared with healthy dog levels, with the treated samples demonstrating stronger sarcolemmal staining than the untreated samples (Figure 8). The lack of difference in utrophin gene expression (Figure S1B and S1C) suggested that this enhancement of utrophin protein levels was likely from decreased proteolysis rather than gene upregulation.
Figure 8.

Sarcolemmal localization of utrophin in golden retriever muscular dystrophy (GRMD) left ventricle (LV) is increased by tadalafil. Immunofluorescent detection of utrophin and laminin in the LV of frozen sections from a healthy dog, an untreated GRMD dog, and a tadalafil‐treated GRMD dog demonstrates that elevated utrophin levels found with tadalafil treatment are localized to the sarcolemma with greater homogeneity than in untreated samples. Scale bars represent 50 μm. ROI indicates region of interest.
To address whether these observations at 25 months in tadalafil‐treated GRMD hearts were age‐ or decline‐dependent, we evaluated LV samples from treated and untreated GRMD dogs aged 8 months (1 dog per treatment), which is prior to detectible onset of cardiac functional decline. At 8 months, the tadalafil‐treated LV demonstrated lower TRPC6 and m‐calpain levels concomitant with reduced α‐actinin cleavage product and increased utrophin content (Figure 9A). As in the 25‐month cohort, the tadalafil‐treated LV demonstrated more sarcolemmal utrophin than the control dog (Figure 9B) and reduced signs of myocardial pathology (as shown by hematoxylin and eosin staining). Tadalafil‐induced increases in γ‐sarcoglycan levels and sarcolemmal localization (Figure 9A and 9B) suggested that the observed increases in utrophin coincide with other dystrophin–glycoprotein complex members.
Figure 9.

Effects on TRPC6, m‐calpain, and utrophin that are induced by tadalafil (Tad) occur prior to onset of golden retriever muscular dystrophy (GRMD) cardiomyopathy. A, Immunoblots of TRPC6, μ‐calpain, m‐calpain, α‐actinin 80‐kDa cleavage product, utrophin, and γ‐sarcoglycan at 8 months in untreated and Tad‐treated GRMD left ventricle (LV). Ponceau Red staining was used as a loading control. B, Representative immunofluorescent (IF) images of utrophin and γ‐ sarcoglycan and images of GRMD LVs at 8 months stained with hematoxylin and eosin (H&E). Scale bars represent 100 μm.
We found no trends in content of the RNA binding protein KSRP or the cytoskeleton‐regulating GTPase RhoA (Figure S1D), both posttranscriptional regulators of utrophin,36, 37, 38 that would suggest that they are involved in the tadalafil‐induced elevation of utrophin. The expression pattern of MMP2 and MMP9 (Figure S1F), additional proteases that may be capable of targeting dystrophin–glycoprotein complex members,39, 40 do not support their involvement in this phenomenon, although MMP2 activation appears to be a marker of cardiomyopathy onset in GRMD hearts. Tadalafil does not affect utrophin content of mdx hearts following either short‐term (10 weeks) or long‐term (8 months) treatment (Figure S1E). Together, these data suggest that in addition to protection from intermittent acute stressors (demonstrated earlier), tadalafil delays GRMD cardiomyopathy by constitutive decrease of TRPC6 permeability by both reduction and deactivation of channels, leading to lower m‐calpain content and activity, and results in decreased proteolysis of peripheral and structural proteins, particularly utrophin, in the unstimulated heart, thereby enhancing sarcolemmal stability.
Elevation of TRPC6 and m‐Calpain in DMD Heart
The above‐mentioned data implicate modulation of both TRPC6 and m‐calpain in the beneficial effect of tadalafil on GRMD cardiomyopathy, thus we sought to verify that these targets are increased in DMD patient myocardium, as in GRMD LV (Figure S1G). Indeed, TRPC6, m‐calpain, and the cleavage products of both α‐actinin and integrin β1 were all increased in the DMD heart, whereas μ‐calpain remained unchanged (Figure 10). In addition, we found strong increases in utrophin in the DMD heart, as reported previously41 and consistent with both GRMD and mdx models of DMD (Figure S1H). We cannot, however, speak to the potential variance of these proteins within the DMD population. These data suggest that the beneficial effects of tadalafil by modifying TRPC6 and m‐calpain will likely translate to DMD patient hearts.
Figure 10.
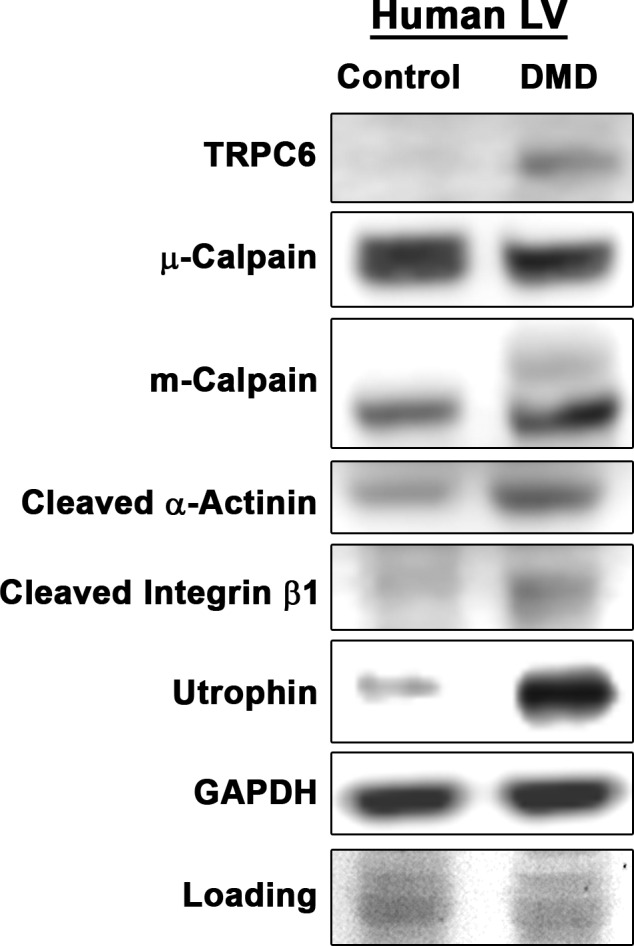
TRPC6 and m‐calpain are elevated in Duchenne muscular dystrophy (DMD) patient myocardium. Immunoblotting of TRPC6, μ‐calpain, m‐calpain, α‐actinin 80‐kDa cleavage product, integrin β1 75‐kDa cleavage product, and utrophin in control and DMD patient myocardium. Ponceau Red staining was used as a loading control.
Tadalafil Improves Histopathology of GRMD Skeletal Muscle
Because PDE5 inhibition has been effective in improving the phenotype of dystrophic skeletal muscle in numerous mouse studies,42, 43, 44 we evaluated the histopathology of untreated and tadalafil‐treated GRMD diaphragms and quadriceps muscles from the 25‐month cohort of the present study. In agreement with these previous reports, we saw substantial improvements in tissue morphology, including reduced perivascular and endomysial fibrosis, preserved muscle architecture, and more homogeneity of myofiber size and shape (Figure 11). This evidence suggests that tadalafil also protects GRMD skeletal muscle from severe dystrophic pathology.
Figure 11.
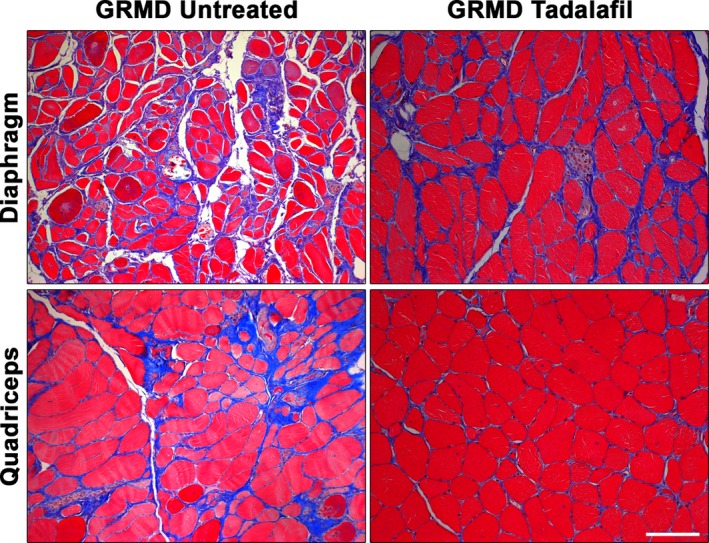
Skeletal muscle histopathology improved by tadalafil treatment. Representative images of Masson's trichrome–stained diaphragm and quadriceps from untreated and tadalafil‐treated golden retriever muscular dystrophy (GRMD) dogs at 25 months. Scale bars represent 100 μm.
Discussion
Cardiomyopathy is emerging as the leading cause of death among DMD patients. Efficacious treatment options to improve both the quality and quantity of life of present‐day DMD patients is a critical clinical need. We reported data demonstrating that daily prophylactic tadalafil administration effectively delays cardiomyopathy onset in a large‐animal model of DMD that recapitulates many aspects of the human disease. We provided evidence that this is achieved by 2 mechanisms: (1) inhibition of stress‐induced elevations in PDE5 that cascade into protease‐mediated dissolution of sarcomeric proteins and (2) inhibition of basal PDE5 activity that reduces abundance and activity of TRPC6 and m‐calpain, resulting in decreased proteolysis of key structural proteins, including utrophin. Because tadalafil is currently approved for other indications and has been studied in a phase III trial for precardiomyopathic DMD patients, it can be evaluated rapidly as a treatment standard to combat DMD‐associated cardiomyopathy.
Clinical manifestations of cardiomyopathy in DMD typically begin in the second decade of life, characterized by prominent LV dysfunction and spontaneous arrhythmias that are often attributed to sudden death.45 That is not to say, however, that cardiac damage begins at this stage. In a real‐life scenario, DMD patients—particularly if still ambulatory—are engaged in physical activity or experience emotional stress or excitement that results in transient bouts of sympathetic stimulation of the heart. Because the dystrophic heart is susceptible to damage during adrenergic stimulation,3, 18, 19, 46 acute damage is possible during each stimulatory event, potentially accumulating toward clinical cardiomyopathy later in life. Tadalafil may be effective at preventing cardiomyopathy in patients by abrogating the damage incurred during these events. This, coupled with improved maintenance of utrophin, would lead to less cardiac damage.
As shown by dobutamine infusion in mdx mice, tadalafil attenuates β1‐adrenergic stimulation of cardiac contractility, thereby reducing energy demands on the stimulated heart; this is particularly important because of the altered energy substrate usage of dystrophic hearts.3 The rapid increase in PDE5 translation during the dobutamine challenge appears to be key in this process and is the first indication of increased PDE5 in the dystrophic heart. Although the increase in Pde5 gene expression is blocked by tadalafil, the increase in PDE5 translation is not significantly affected by tadalafil. This indicates that some degree of contractility escalation is subsequent to PDE5‐dependent hydrolysis of cGMP. In fact, the increase in PDE5 activity appears to lead to a cascade of events resulting in the increase of μ‐calpain and subsequent degradation of a number of proteins involved in contractility, notably SERCA2, which removes Ca2+ from the cytosol. Consequently, the cytosolic Ca2+ load of the cardiomyocytes continues to increase, allowing the heart to progress toward damaging dysfunction and/or arrhythmias.26, 47 We suspect that activation of TRPC6 is a major player in initiating this PDE5‐medated cascade; however, difficulties in the measurement of murine TRPC6 leave us unable to draw that conclusion from this study.
We chose the GRMD model of DMD to test the prophylactic efficacy of tadalafil on delaying cardiomyopathy because this model better recapitulates the severity of the cardiomyopathy seen in DMD compared with mdx mice, which show clinical signs of cardiomyopathy typically after age 12 months.48 We observed that LV dysfunction was prominent in untreated GRMD dogs by age 18 months, with cardiac‐associated mortality occurring as early as 20 months, as was the case for 1 control in this study. Daily administration of tadalafil prior to the onset of cardiac functional decline preserved LV systolic function (ie, ejection fraction and fractional shortening) and diastolic function, especially as noted in the transmitral blood flow pattern. Improvements in GRMD heart histopathology resulted from the treatment, as evaluated with Masson's trichrome staining, and correlated well with the functional improvements detected by echocardiography and magnetic resonance imaging. Because reductions in fibrosis and adipose infiltration were clearly evident, the observed preservation of LV wall compliance likely results from less fibrosis; however, we cannot rule out other potential modifiers of LV stiffness, such as titin modifications,49, 50 which could also contribute to functional improvement. Interestingly, the preserved systolic function and improved histopathology of the RV may indicate a secondary effect of PDE5 inhibition by tadalafil to be a reduction in pulmonary hypertension (another indication intended for sildenafil), resulting in less stress experienced by the RV.
Similar to the unstimulated mdx heart, PDE5 is not readily detectable by immunoblotting in basal GRMD LVs; however, differential content and phosphorylation of TRPC6 and content and activity of m‐calpain between the 2 treatments suggest that basal levels of PDE5 have a functional impact on GRMD hearts. In the absence of stress, PDE5 is primarily localized to the Z‐disk8 and scavenges cGMP from predominantly intrasarcomeric pools of soluble guanylyl cyclase.51 It appears that this basal PDE5 activity is sufficient to modulate both TRPC6 and m‐calpain. These suppositions are based on the report of Seo et al, 26 who showed that the protective effect of sildenafil on stretch‐stimulated Ca2+ overload and arrhythmias of dystrophic mouse heart involved TRPC6 channels. This effect is likely from inhibition of TRPC6 by phosphorylation of Thr69 (Thr70 in humans and dogs) by PKG, resulting in decreased ion permeability.27, 28 Similarly, we found that long‐term tadalafil treatment decreased both gene expression and protein content of TRPC6 and showed an increase in unspecific phosphorylated Thr status of TRPC6. Less expression and activation of these channels in cardiomyocytes leads to reductions in dystrophy‐associated Ca2+ dysregulation, thereby improving the phenotype through what appears to involve m‐calpain in the unstimulated heart. In addition, deactivation and downregulation of TRPC6 in cardiac fibroblasts may prevent their differentiation toward myofibroblasts.52
An unexpected result of tadalafil treatment was the robust increase in utrophin levels in the GRMD LV. Utrophin is a structural ortholog of dystrophin that can replace dystrophin in the dystrophin–glycoprotein complex, restore the mechanical cytoskeleton–extracellular matrix connection, and functionally rescue dystrophin‐deficient skeletal muscle.35 Robust myocardial utrophin upregulation is found in mdx hearts53; however, its lack of increase in GRMD hearts has been attributed to the more severe cardiac phenotype in the canine species.46 We found that tadalafil‐induced increase in GRMD utrophin is localized to the sarcolemma, occurs prior to the onset of cardiac functional decline, and is likely caused by decreased proteolysis by m‐calpain. This myocardial utrophin increase may provide benefit not only by protecting the membrane from damage but also by improving conduction abnormalities by correcting Nav1.5 dysregulation.54 The failure of tadalafil to have the same effect on utrophin in mdx heart could be related to its levels already being stabilized by lack of protease expression or activation in the milder mouse dystrophy. In the severely affected GRMD hearts, tadalafil can spare utrophin from increased degradation. We also demonstrated that that both TRPC6 and m‐calpain were increased in the myocardium of DMD patients, suggesting that the GRMD treatment effects can translate to DMD patients.
The published clinical study with the short‐acting PDE5 inhibitor sildenafil showed no efficacy in the DMD patients who were treated10; however, those patients had decreased cardiac function on treatment initiation. The present data strongly support the prophylactic use of tadalafil to counter cardiomyopathy in DMD patients rather than treatment after pathology and functional decline are evident.
The recently completed trial of tadalafil in DMD (ClinicalTrials.gov identifier NCT01865084) was performed in young ambulatory patients who were presymptomatic in terms of cardiomyopathy. Unfortunately, that trial did not demonstrate efficacy for its primary end point, the 6‐minute walk test. This is unfortunate because it would have been of great interest to follow the cardiac status of those patients as they approached the age of onset of cardiac symptoms, had they remained on the drug. The anticipated mechanism in the ambulatory trial, suggested by a combination of animal and human studies,12, 42, 43 was to improve blood flow to exercising leg muscles by amplifying deficient nitric oxide signaling to vascular smooth muscle, thus preventing ischemia and preserving muscle. Although our dog studies did not focus on skeletal muscle function, we examined histology of diaphragm and quadriceps muscles from treated and untreated animals. As shown in Figure 11, histology was improved. Consequently, it is unclear whether the patient population was simply not active enough to have ischemia play a role in disease pathology or whether there was not sufficient PDE5 activity in advanced‐stage limb muscles to provide an adequate drug target.
The significance of this work may go beyond dystrophinopathy. The promising results with PDE5 inhibition in animal models have failed to translate into benefit for human DMD patients10 or for human heart failure.55 In these previous trials, PDE5 inhibition was initiated at late stages of disease progression. For idiopathic cardiomyopathies, treatment initiation during early stage disease could have a similar impact of slowing disease progression, as we observed in our GRMD dogs with early stage disease. In all forms of cardiomyopathy, the improved intracellular calcium status and decreased TRPC6 expression and activation that results will potentially decrease protease expression and activation (m‐calpain in this instance), which potentially has a number of deleterious targets beyond utrophin, such as dystrophin in advanced heart failure.56 PDE5 inhibition may translate broadly as a treatment to slow the progression of a number of forms of cardiomyopathy if initiated early rather than in late‐stage disease.
Sources of Funding
This work was funded by a Wellstone Muscular Dystrophy Cooperative Center grant (U54‐AR‐052646) from the NIH to Sweeney, Leducq Foundation funding (13CVD04) to Sweeney, and from funding from the Parent Project Muscular Dystrophy in support of the UF Preclinical Assessment of Therapeutics lab. We also acknowledge the support from grants from the Muscular Dystrophy Association (175552) and NIH (R01AR056973) to Walter. Hammers was supported on the NIH T32 training grant to the Pennsylvania Muscle Institute (T32‐AR053461) for part of this work.
Disclosures
None.
Supporting information
Figure S1. Protein content of markers in dystrophic hearts treated with tadalafil (Tad). A, Immunoblotting quantifications for cyclic guanosine monophosphate–dependent kinase 1α (PKG1α), glycogen synthase kinase 3β (GSK3β), extracellular signal–related kinase 1/2 (ERK1/2), sarco/endoplasmic reticulum Ca2+‐ATPase 2 (SERCA2), atrial natriuretic peptide (ANP), endothelial nitric oxide synthase (eNOS), and neuronal NOS (nNOS; normalized to Ponceau Red staining). Utrophin gene expression in untreated and Tad‐treated golden retriever muscular dystrophy (GRMD) left ventricle (LV) at 25 months (B and C). Immunoblotting for utrophin posttranscriptional regulators KSRP and RhoA at 25 and 8 months in GRMD LV (D), and matrix metalloproteinase 2 (MMP2) and MMP9 (F) in untreated and Tad‐treated GRMD dog LV. E, Utrophin immunoblotting in short‐term (10 weeks) and long‐term (8 months) Tad‐treated mdx hearts. G, TRPC6 and m‐calpain levels in healthy and GRMD dog LV samples. H, Dystrophin and utrophin content in healthy and GRMD dog LV samples and C57BL/10 and mdx mouse hearts. Values are represented as mean±SD; *P<0.05 vs control values.
Acknowledgments
We acknowledge the technical assistance of Patricia O'Donnell, Tracey Sikora, and Therese Ruanethe of the University of Pennsylvania veterinary staff, the University of Pennsylvania Small Animal Imaging Facility, Jen Pham, Pedro Acosta, Diana Menendez, Alexandra Agathis, Chris Phillips, Adam George, Janelle Spinazzola, Jannik Arbogast, and Cora Coker. Special thanks to Dr Elisabeth R. Barton for critical reading and comments during the preparation of this manuscript.
(J Am Heart Assoc. 2016;5:e003911 10.1161/JAHA.116.003911)
References
- 1. Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th‐9th June 2002, Naarden, the Netherlands. Neuromuscul Disord. 2003;13:166–172. [DOI] [PubMed] [Google Scholar]
- 2. Adamo CM, Dai DF, Percival JM, Minami E, Willis MS, Patrucco E, Froehner SC, Beavo JA. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2010;107:19079–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, Deschepper CF, Petrof BJ, Des Rosiers C. Sildenafil and cardiomyocyte‐specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci USA. 2008;105:7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kass DA. Cardiac role of cyclic‐GMP hydrolyzing phosphodiesterase type 5: from experimental models to clinical trials. Curr Heart Fail Rep. 2012;9:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, dos Remedios C, Pritzker M, Hall JL, Garry DJ, Chen Y. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010;121:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M, Takimoto E, Lee DI, Santos CX, Nakamura T, Hsu S, Jiang A, Nagayama T, Bedja D, Yuan Y, Eaton P, Shah AM, Kass DA. Pathological cardiac hypertrophy alters intracellular targeting of phosphodiesterase type 5 from nitric oxide synthase‐3 to natriuretic peptide signaling. Circulation. 2012;126:942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–314. [DOI] [PubMed] [Google Scholar]
- 8. Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP‐specific) modulates beta‐adrenergic signaling in vivo and is down‐regulated in heart failure. FASEB J. 2001;15:1718–1726. [DOI] [PubMed] [Google Scholar]
- 9. Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, Takimoto E, Kass DA. Pressure‐overload magnitude‐dependence of the anti‐hypertrophic efficacy of PDE5A inhibition. J Mol Cell Cardiol. 2009;46:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leung DG, Herzka DA, Thompson WR, He B, Bibat G, Tennekoon G, Russell SD, Schuleri KH, Lardo AC, Kass DA, Thompson RE, Judge DP, Wagner KR. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann Neurol. 2014;76:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martin EA, Barresi R, Byrne BJ, Tsimerinov EI, Scott BL, Walker AE, Gurudevan SV, Anene F, Elashoff RM, Thomas GD, Victor RG. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci Transl Med. 2012;4:162ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nelson MD, Rader F, Tang X, Tavyev J, Nelson SF, Miceli MC, Elashoff RM, Sweeney HL, Victor RG. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology. 2014;82:2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bish LT, Sleeper MM, Forbes SC, Wang B, Reynolds C, Singletary GE, Trafny D, Morine KJ, Sanmiguel J, Cecchini S, Virag T, Vulin A, Beley C, Bogan J, Wilson JM, Vandenborne K, Kornegay JN, Walter GA, Kotin RM, Garcia L, Sweeney HL. Long‐term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6‐mediated exon skipping. Mol Ther. 2012;20:580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mai W, Weisse C, Sleeper MM. Cardiac magnetic resonance imaging in normal dogs and two dogs with heart base tumor. Vet Radiol Ultrasound. 2010;51:428–435. [DOI] [PubMed] [Google Scholar]
- 15. Hor KN, Gottliebson WM, Carson C, Wash E, Cnota J, Fleck R, Wansapura J, Klimeczek P, Al‐Khalidi HR, Chung ES, Benson DW, Mazur W. Comparison of magnetic resonance feature tracking for strain calculation with harmonic phase imaging analysis. JACC Cardiovasc Imaging. 2010;3:144–151. [DOI] [PubMed] [Google Scholar]
- 16. Osman NF, Kerwin WS, McVeigh ER, Prince JL. Cardiac motion tracking using CINE harmonic phase (HARP) magnetic resonance imaging. Magn Reson Med. 1999;42:1048–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osman NF, McVeigh ER, Prince JL. Imaging heart motion using harmonic phase MRI. IEEE Trans Med Imaging. 2000;19:186–202. [DOI] [PubMed] [Google Scholar]
- 18. Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS, Metzger JM. Systemic administration of micro‐dystrophin restores cardiac geometry and prevents dobutamine‐induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. [DOI] [PubMed] [Google Scholar]
- 19. Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. [DOI] [PubMed] [Google Scholar]
- 20. Layland J, Li JM, Shah AM. Role of cyclic GMP‐dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. PDE5A suppression of acute beta‐adrenergic activation requires modulation of myocyte beta‐3 signaling coupled to PKG‐mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sorimachi H, Ono Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc Res. 2012;96:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. French JP, Quindry JC, Falk DJ, Staib JL, Lee Y, Wang KK, Powers SK. Ischemia‐reperfusion‐induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H128–H136. [DOI] [PubMed] [Google Scholar]
- 24. Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up‐tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. 2009;81:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashford MW Jr, Liu W, Lin SJ, Abraszewski P, Caruthers SD, Connolly AM, Yu X, Wickline SA. Occult cardiac contractile dysfunction in dystrophin‐deficient children revealed by cardiac magnetic resonance strain imaging. Circulation. 2005;112:2462–2467. [DOI] [PubMed] [Google Scholar]
- 26. Seo K, Rainer PP, Lee DI, Hao S, Bedja D, Birnbaumer L, Cingolani OH, Kass DA. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP‐protein kinase G modulation. Circ Res. 2014;114:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG‐dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishida M, Watanabe K, Sato Y, Nakaya M, Kitajima N, Ide T, Inoue R, Kurose H. Phosphorylation of TRPC6 channels at Thr69 is required for anti‐hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem. 2010;285:13244–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwan HY, Huang Y, Yao X. Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci USA. 2004;101:2625–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T, Kawashima S, Sobue K. Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by calpain. Circ Res. 1995;77:603–610. [DOI] [PubMed] [Google Scholar]
- 31. Sprague CR, Fraley TS, Jang HS, Lal S, Greenwood JA. Phosphoinositide binding to the substrate regulates susceptibility to proteolysis by calpain. J Biol Chem. 2008;283:9217–9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pfaff M, Du X, Ginsberg MH. Calpain cleavage of integrin beta cytoplasmic domains. FEBS Lett. 1999;460:17–22. [DOI] [PubMed] [Google Scholar]
- 33. Redpath GM, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA, North KN, Cooper ST. Calpain cleavage within dysferlin exon 40a releases a synaptotagmin‐like module for membrane repair. Mol Biol Cell. 2014;25:3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Courdier‐Fruh I, Briguet A. Utrophin is a calpain substrate in muscle cells. Muscle Nerve. 2006;33:753–759. [DOI] [PubMed] [Google Scholar]
- 35. Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. [DOI] [PubMed] [Google Scholar]
- 36. Amirouche A, Tadesse H, Miura P, Belanger G, Lunde JA, Cote J, Jasmin BJ. Converging pathways involving microRNA‐206 and the RNA‐binding protein KSRP control post‐transcriptionally utrophin A expression in skeletal muscle. Nucleic Acids Res. 2014;42:3982–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gauthier‐Rouviere C, Bonet‐Kerrache A. RhoA leads to up‐regulation and relocalization of utrophin in muscle fibers. Biochem Biophys Res Commun. 2009;384:322–328. [DOI] [PubMed] [Google Scholar]
- 38. Bonet‐Kerrache A, Fortier M, Comunale F, Gauthier‐Rouviere C. The GTPase RhoA increases utrophin expression and stability, as well as its localization at the plasma membrane. Biochem J. 2005;391:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buchholz B, Perez V, Siachoque N, Miksztowicz V, Berg G, Rodriguez M, Donato M, Gelpi RJ. Dystrophin proteolysis: a potential target for MMP‐2 and its prevention by ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2014;307:H88–H96. [DOI] [PubMed] [Google Scholar]
- 40. Court FA, Zambroni D, Pavoni E, Colombelli C, Baragli C, Figlia G, Sorokin L, Ching W, Salzer JL, Wrabetz L, Feltri ML. MMP2‐9 cleavage of dystroglycan alters the size and molecular composition of Schwann cell domains. J Neurosci. 2011;31:12208–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pons F, Robert A, Fabbrizio E, Hugon G, Califano JC, Fehrentz JA, Martinez J, Mornet D. Utrophin localization in normal and dystrophin‐deficient heart. Circulation. 1994;90:369–374. [DOI] [PubMed] [Google Scholar]
- 42. Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JA, Yasuhara SE. Primary role of functional ischemia, quantitative evidence for the two‐hit mechanism, and phosphodiesterase‐5 inhibitor therapy in mouse muscular dystrophy. PLoS One. 2007;2:e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA, Campbell KP. Sarcolemma‐localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA, Froehner SC. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol. 2012;228:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99:1–19. [DOI] [PubMed] [Google Scholar]
- 46. Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prosser BL, Ward CW, Lederer WJ. X‐ROS signaling: rapid mechano‐chemo transduction in heart. Science. 2011;333:1440–1445. [DOI] [PubMed] [Google Scholar]
- 48. Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju‐Sanka RK, Tou S, Keene BP, Howard JF Jr, Wang J, Fan Z, Schatzberg SJ, Styner MA, Flanigan KM, Xiao X, Hoffman EP. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome. 2012;23:85–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagueh SF, Shah G, Wu Y, Torre‐Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. [DOI] [PubMed] [Google Scholar]
- 50. Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC Jr, Linke WA, Redfield MM. Sildenafil and B‐type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124:2882–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6‐dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weir AP, Burton EA, Harrod G, Davies KE. A‐ and B‐utrophin have different expression patterns and are differentially up‐regulated in mdx muscle. J Biol Chem. 2002;277:45285–45290. [DOI] [PubMed] [Google Scholar]
- 54. Albesa M, Ogrodnik J, Rougier JS, Abriel H. Regulation of the cardiac sodium channel Nav1.5 by utrophin in dystrophin‐deficient mice. Cardiovasc Res. 2011;89:320–328. [DOI] [PubMed] [Google Scholar]
- 55. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toyo‐Oka T, Kawada T, Nakata J, Xie H, Urabe M, Masui F, Ebisawa T, Tezuka A, Iwasawa K, Nakajima T, Uehara Y, Kumagai H, Kostin S, Schaper J, Nakazawa M, Ozawa K. Translocation and cleavage of myocardial dystrophin as a common pathway to advanced heart failure: a scheme for the progression of cardiac dysfunction. Proc Natl Acad Sci USA. 2004;101:7381–7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein content of markers in dystrophic hearts treated with tadalafil (Tad). A, Immunoblotting quantifications for cyclic guanosine monophosphate–dependent kinase 1α (PKG1α), glycogen synthase kinase 3β (GSK3β), extracellular signal–related kinase 1/2 (ERK1/2), sarco/endoplasmic reticulum Ca2+‐ATPase 2 (SERCA2), atrial natriuretic peptide (ANP), endothelial nitric oxide synthase (eNOS), and neuronal NOS (nNOS; normalized to Ponceau Red staining). Utrophin gene expression in untreated and Tad‐treated golden retriever muscular dystrophy (GRMD) left ventricle (LV) at 25 months (B and C). Immunoblotting for utrophin posttranscriptional regulators KSRP and RhoA at 25 and 8 months in GRMD LV (D), and matrix metalloproteinase 2 (MMP2) and MMP9 (F) in untreated and Tad‐treated GRMD dog LV. E, Utrophin immunoblotting in short‐term (10 weeks) and long‐term (8 months) Tad‐treated mdx hearts. G, TRPC6 and m‐calpain levels in healthy and GRMD dog LV samples. H, Dystrophin and utrophin content in healthy and GRMD dog LV samples and C57BL/10 and mdx mouse hearts. Values are represented as mean±SD; *P<0.05 vs control values.