

Sudden cardiac death (SCD) claims 340 000 lives per annum, making it the leading cause of cardiovascular‐related deaths in the United States. Most patients die from ischemia‐driven ventricular arrhythmias (VA) secondary to myocardial infarction (MI), but other important causes of SCD include cardiomyopathy (hypertrophic, dilated, and arrhythmogenic), ion channelopathies (Brugada and long QT syndromes [LQTS]), and aortopathies.1, 2 One of the most intriguing and unsolved questions in arrhythmia research is why only some patients succumb to arrhythmia and die while others with similar underlying cardiovascular diseases (CVD) substrates do not. This, along with the abysmal survival rates of 10% following cardiac arrest, makes the search for risk factors, triggers, and maintainers of arrhythmia and SCD crucial public health priorities. Repolarization reserve and arrhythmia remodeling, due in part to long‐term changes in ion channel expression, are constructs that may explain SCD susceptibility in acute MI, cardiomyopathies, and primary ion channelopathies. Obstructive sleep apnea (OSA) may be an independent risk factor for nocturnal MI, SCD, atrial fibrillation (AF), heart failure, and pulmonary and systemic hypertension.3, 4, 5, 6, 7, 8, 9, 10 In this issue of the Journal, Dudley and colleagues elegantly propose a potential mechanistic link between OSA and ion channel remodeling (ICR) by measuring circulating mRNA levels of K+ channels in OSA patients and controls and compare changes in those treated with continuous positive airway pressure (CPAP).11 In this accompanying editorial we provide background on arrhythmia propensity and the role of OSA in SCD.
Propensity for Arrhythmia
The negative cytosolic resting membrane potential (phase 4) of approximately −80 mV is maintained largely by the action of transmembrane cellular pumps (predominantly Na+/K+ ATPase and Na+/Ca2+ exchanger) and closed voltage‐gated ion channels (Figure 1).12, 13 The cardiac action potential (AP) is initiated in the sinoatrial node (SAN) by the slowly leaking inward “funny” current sodium channels (If), leading to the recruitment of Ca2+ channels and causing the positive depolarization current (phase 0). Gap junctions facilitate spread to adjacent cells in the atria, internodal tracts, atrioventricular node (AVN), His bundle, and both right and left bundles, the Purkinje cells, and finally, the ventricular myocardium. Phase 0 in all of the heart cells except for the SAN and AVN is caused by the opening of Na+ channels, flooding the cytoplasm with Na+ ions and causing the positive depolarization of the membrane. As the wave of depolarization spreads, sarcoplasmic Ca2+ release leads to muscular contraction (excitation‐contraction coupling). Immediately after phase 0, repolarization begins with phase 1 (largely due to Cl− ions), followed by phase 2, the plateau stage where the inner membrane potential remains relatively constant due to the influx of Ca2+. Phase 3 is the latter stage of repolarization and the relative refractory period, during which a new stimulus such as a ventricular extrasystole (VE) can trigger arrhythmia. AP duration is determined largely by activation of K+ channels. Thus, inactivation and/or down‐regulation of these K+ channels, which can be primary in genetic LQTS or secondary to other pathological states, can prolong the QTc on the surface ECG. Neurohumoral changes can modify the regionally specific AP further: increased sympathetic tone increases automaticity and conduction velocity and decreases the AP duration, leading to more rapid AP firing and recovery. Conversely, heightened parasympathetic tone decreases automaticity and prolongs the refractory periods.
Figure 1.
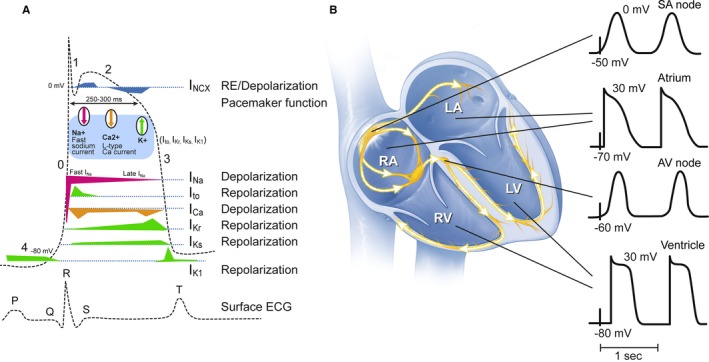
Schematic representation of types of action potential (AP) throughout the heart. The major differences are between the nodal tissues (sinoatrial and atrioventricular nodes) and ventricular Purkinje/myocardium. (A) on the left hand side, the ventricular AP and associated Na+, K+, and Ca2+ channels are shown. Phase 0 corresponds with the QRS wave, phase 3 with the peak of the T wave, phase 4 with the isoelectric line of the surface ECG changes. (B) action potentials from different regions within the heart. IC a indicates inward calcium current L‐type; IK r, delayed inward rectifier K+; IK s, slowly activating K+ channel; IN a, inward voltage‐dependent sodium channel; INCX, sodium‐calcium exchanger; Ito, transient outward; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Adapted from Dumotier, Heart 12 by permission from BMJ Publishing Group Limited, and from Nattel et al, Physiological Reviews,13 with permission by the American Physiological Society.
Established arrhythmia mechanisms include (1) abnormal impulse initiation due to (1a) automaticity and (1b) triggered mechanisms from early and delayed afterdepolarizations such as VEs and (2) abnormal impulse conduction due to anatomical or functional reentry and reflection. Propensity for arrhythmia is a complex interplay between the substrate (anatomical and functional) and wide‐ranging modulators such as (1) autonomic tone, (2) stretch, (3) electrolyte changes, (4) ectopic beats, and (5) drugs or toxins. OSA is an important treatable and reversible modifier of arrhythmia propensity given direct and indirect effects including autonomic tone, stretch, and increased VE (described below).
Arrhythmogenic Remodeling and Repolarization Reserve
CVD modify not only the macroscopic structure of the heart but also the normally rigidly regulated ion channels, pumps, associated regulatory proteins and their expression, such that this promotes arrhythmia in a process termed “arrhythmogenic remodeling.”12, 13, 14 Many of these alterations are primarily adaptive responses for homeostasis but can result in secondary cardiac dysfunction, particularly VA. Classical models of static ion‐channel structure in heathy tissue have been superseded by a dynamic model of ion‐channel structure in response to normal physiology. Recognized mechanisms of ICR include DNA transcriptional regulation, epigenetic factors, mRNA processing and translation, posttranslational modification, processing, assembly, and membrane transport of proteins, macromolecular assembly, and posttranslational regulation.15, 16 This control also has regional differences, reflecting the known differences in AP generation (see a detailed review17). ICR may help explain changes seen in the surface ECG, particularly QTc prolongation. Significant advances in understanding ICR in CVD have been made,13 but ICR in noncardiac disease, such as highly prevalent OSA, are understudied.
“Repolarization reserve” is the ability of cardiac myocytes to tolerate and compensate for the loss of repolarizing currents by recruiting additional time‐dependent outward (repolarizing) currents IKr and Iks, which minimize repolarization deficit.18 Minor changes to ion channel regulation can mean little loss of repolarization reserve, such that no noticeable clinical or physiological effects are seen. However, major changes to control mechanisms, and thus to repolarization reserve, can exaggerate the effects of any arrhythmic triggers that are directly relevant in prolonged QTc and the risk of VA. The presence of OSA with coexisting CVD may be pivotal in reducing repolarization reserve, which, coupled with ICR, may help explain the higher frequency of SCD.
Obstructive Sleep Apnea
Sleep in itself is associated with changes in autonomic activity and can be considered an autonomic stressor for the heart. In healthy states, non–rapid eye movement (nonREM) sleep, composed of stages N1, N2, and N3, occupies 80% of sleep architecture and is associated with relative autonomic stability in which vagal tone dominates, baroreceptor gain is high, and sympathetic activity is both constant and low.10 Transitions from nonREM to REM sleep can result in bursts of vagal activity causing bradycardia and asystole. During REM sleep, which occurs approximately every 90 minutes of each sleep epoch and >5 times for an average 8 hours of sleep, surges of sympathetic activity occur, which can cause tachycardia, increased VEs, and hypertension.4, 8 Vagal activity is suppressed, baroreceptor gain is reduced, and irregular breathing patterns may occur. Loss of airway muscle tone during inspiration, especially during REM, can lead to obstructive apneas, which can result in hypoxemia and hemoglobin desaturation. In patients with OSA and underlying cardiovascular and/or respiratory disease, hypoxemia can be profound and dysrhythmogenic.10, 19
OSA can acutely elicit sympathetic activation, VA, bradyarrhythmias, and SCD. In the longer term, OSA can result in hypertension, endothelial dysfunction, ventricular remodeling, and myocardial scarring, all of which increase the proclivity for arrhythmia. An alternative mechanism is for OSA to “trigger” an MI due to plaque rupture, de novo thrombosis, or hemodynamic disturbance leading to SCD.
OSA is also associated with increased VE activity of 60% when compared to subjects without OSA (4% to 12%), which can be decreased with the use of OSA therapies.10 VEs initiating VA in prolonged QTc, especially during severe hypoxemia, may be key mechanisms for SCD in OSA (Figure 2). Potentiating mechanisms may include hemodynamic instability, increased platelet aggregation, plaque disruption and release of vasoactive substances, inflammation, oxidative stress, endothelial dysfunction, and thrombosis. Direct mechanisms include the effects of a Mueller maneuver (repetitive forced inspiration against a closed upper airway) with substantial negative intrathoracic pressures, which can increase transvalvular gradients, wall stress, and afterload, which can be proarrhythmogenic. This can also increase the risk for aortic dissection and rupture, resulting in SCD. Other possible effects include altering cardiac cellular electrophysiology with prolongation of QT interval, during which the heart is susceptible to VA initiation.
Figure 2.

Schematic outlining potential pathophysiological aspects of OSA, disease mechanisms, and development of associated CVD. Ion channel remodeling may be an important mechanism in associated CVD increasing the propensity for SCD directly and indirectly. Modified from Somers et al, Circulation, 10 with permission from Wolters Kluwer. CVD indicates cardiovascular diseases; OSA, obstructive sleep apnea; SCD, sudden cardiac death; VF, ventricular fibrillation; VT, ventricular tachycardia.
In an elegant prospective observational cohort study in this issue of the Journal, Dudley and colleagues report a possible mechanistic link between OSA and prolonged QT interval and thus SCD.10 Given the inherent procedural risks associated with endomyocardial biopsy of the different regions of the heart, the authors used mRNA in circulating white blood cells as surrogate reflectors of ion channel expression in unaffected controls and patients with OSA before and after 4 weeks of continuous positive airway pressure (CPAP) therapy. The authors found expression of potassium channels KCNQ1, KCNH2, KCNE1, KCNJ2, and KCNA5 to be inversely associated with severity of OSA by the apnea‐hypopnea index. KCNQ1, KCNH2, and KCNE1 were also inversely associated with severity of hypoxemia. Loss‐of‐function mutations in KCNQ1 are associated with LQTS1, KCNH2 with LQTS2, KCNE1 with LQTS5, KCNJ2 with LQTS7 (also known as Andersen‐Tawil syndrome), and KCNA5 with familial AF. Furthermore, the authors found just 4 weeks of CPAP therapy to be sufficient to significantly increase the mRNA levels of KCNQ1 and KCNJ2 in moderate OSA. The dynamic QT interval changes with ventricular rate and during normal sleep, becoming prolonged in response to stressors, including OSA. This novel study suggests a mechanistic association between OSA and altered mRNA expression of K+ channels as a contribution to the association of OSA with SCD. However, the investigators did not assess measured QTc interval on the surface ECG during sleep and wakefulness, which would have provided important proof‐of‐concept insights into this potential link. This study may also be relevant to genetic LQTS, as up to 20% of asymptomatic mutation carriers are phenotype negative with SCD risks comparable to the general population, implicating nongenetic modifiers. Because OSA has a high prevalence in LQTS, this could be an important modifier of phenotype.20
Reducing Arrhythmic Burden and Saving Lives
Although definitive randomized controlled trials remain to be completed, observational data suggest that cardiovascular effects of OSA may be mitigated with safe, established, and effective therapies (including repositioning, oxygen, mandibular devices, and CPAP), making it a potentially modifiable risk factor for MI, hypertension, SCD, and cardiomyopathies.2, 3 Because these therapies have virtually no known major side effects, it is attractive and plausible that the risk of VAs and SCD could be reduced.
Summary
This study is the first to link potential mechanisms between OSA and prolonged QTc and SCD, suggesting an inverse association with mRNA expression of K+ channels with severity of OSA. CPAP therapy diminishing OSA is able to reverse this relationship in 4 weeks for moderate OSA. It remains to be determined whether this effect persists in the long term and what duration of therapy is required to restore circulating levels to those in subjects without OSA. Studies showing reproducibility and an association with measured QTc levels during sleep and wakefulness will strengthen this observation. Also requiring clarification is the nature of the interaction between K+ channel remodeling and hypoxemia and other acute effects of apnea, in the genesis of QT prolongation during apneas.
Sources of Funding
This publication was made possible by Translational Science Award Grant Number UL1 TR000135, supporting the Mayo Clinic Center for Clinical and Translational Science (CCaTS), from the National Center for Advancing Translational Sciences (NCATS), a component of NIH. Somers and Chahal were supported by NIH HL 65176, HL 114676 and HL 114024. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
Disclosures
Somers declares that he has received grant support from a Philips Respironics Foundation gift to Mayo Foundation; is a consultant for GlaxoSmithKline, Price Waterhouse Coopers, ResMed, Respicardia, Rhonda Grey, Dane Garvin, Philips, and U‐Health; and is working with Mayo Health Solutions and their industry partners on intellectual property related to sleep and CVD. Chahal declares no competing interests.
J Am Heart Assoc. 2016;5:e004195 doi: 10.1161/JAHA.116.004195.
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
References
- 1. Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, Mariani R, Gunson K, Jui J. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mansukhani MP, Wang S, Somers VK. Sleep, death, and the heart. Am J Physiol Heart Circ Physiol. 2015;309:H739–H749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chahal CA, Somers VK. Risk factors: sleep apnoea, atrial fibrillation, and heart failure—quo vadis? Nat Rev Cardiol. 2015;12:263–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chahal CA, Somers VK. Secondary hypertension: obstructive sleep apnea. J Am Soc Hypertens. 2015;9:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, Somers VK. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol. 2007;49:565–571. [DOI] [PubMed] [Google Scholar]
- 6. Gami AS, Howard DE, Olson EJ, Somers VK. Day‐night pattern of sudden death in obstructive sleep apnea. N Engl J Med. 2005;352:1206–1214. [DOI] [PubMed] [Google Scholar]
- 7. Gami AS, Olson EJ, Shen WK, Wright RS, Ballman KV, Hodge DO, Herges RM, Howard DE, Somers VK. Obstructive sleep apnea and the risk of sudden cardiac death: a longitudinal study of 10,701 adults. J Am Coll Cardiol. 2013;62:610–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia‐Touchard A, Somers VK, Kara T, Nykodym J, Shamsuzzaman A, Lanfranchi P, Ackerman MJ. Ventricular ectopy during REM sleep: implications for nocturnal sudden cardiac death. Nat Clin Pract Cardiovasc Med. 2007;4:284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuniyoshi FH, Garcia‐Touchard A, Gami AS, Romero‐Corral A, van der Walt C, Pusalavidyasagar S, Kara T, Caples SM, Pressman GS, Vasquez EC, Lopez‐Jimenez F, Somers VK. Day‐night variation of acute myocardial infarction in obstructive sleep apnea. J Am Coll Cardiol. 2008;52:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, Pickering TG, Russell R, Woo M, Young T. Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation. 2008;118:1080–1111. [DOI] [PubMed] [Google Scholar]
- 11. Jiang N, Zhou A, Prasad B, Zhou L, Doumit J, Shi G, Imran H, Kaseer B, Millman R, Dudley SC. Obstructive sleep apnea and circulating potassium channel levels. J Am Heart Assoc. 2016;5:e003666 doi: 10.1161/JAHA.116.003666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dumotier BM. A straightforward guide to the basic science behind arrhythmogenesis. Heart. 2014;100:1907–1915. [DOI] [PubMed] [Google Scholar]
- 13. Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. [DOI] [PubMed] [Google Scholar]
- 14. Michael G, Xiao L, Qi XY, Dobrev D, Nattel S. Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res. 2009;81:491–499. [DOI] [PubMed] [Google Scholar]
- 15. Ohya S, Kito H, Hatano N, Muraki K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol Ther. 2016;160:11–43. [DOI] [PubMed] [Google Scholar]
- 16. Rosati B, McKinnon D. Regulation of ion channel expression. Circ Res. 2004;94:874–883. [DOI] [PubMed] [Google Scholar]
- 17. Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90:939–950. [DOI] [PubMed] [Google Scholar]
- 18. Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. [DOI] [PubMed] [Google Scholar]
- 19. Bradley TD, Floras JS. Sleep apnea and heart failure: part I: obstructive sleep apnea. Circulation. 2003;107:1671–1678. [DOI] [PubMed] [Google Scholar]
- 20. Shamsuzzaman AS, Somers VK, Knilans TK, Ackerman MJ, Wang Y, Amin RS. Obstructive sleep apnea in patients with congenital long QT syndrome: implications for increased risk of sudden cardiac death. Sleep. 2015;38:1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]