

Abstract
Background
Macrophage migration inhibitory factor (MIF) is a key regulator of inflammatory responses, including in the heart. Plasma MIF is elevated early in the course of acute myocardial infarction. In this study, we hypothesized that plasma MIF may also be increased in acute myocardial ischemia.
Methods and Results
Patients undergoing cardiac stress test (stress nuclear myocardial perfusion scan or stress echocardiography) were recruited. Twenty‐two patients had a stress test indicative of myocardial ischemia and were compared with 62 patients who had a negative stress test. Plasma MIF was measured by ELISA before and after the stress test. MIF was also measured in patients with peripheral arterial occlusive disease before and after exercise causing claudication. Gene and protein expression of MIF was measured in mouse cardiac and skeletal muscle tissue by real‐time polymerase chain reaction and western blot, respectively. Plasma MIF was elevated at 5 and 15 minutes after stress (relative to before stress) in patients with a positive test, compared with those with a negative test. In contrast, high‐sensitivity troponin T and C‐reactive protein were not altered after stress in either group. MIF was not altered after exercise in PAOD patients, despite the occurrence of claudication, suggesting that plasma MIF is not a marker for skeletal muscle ischemia. This may be explained by a lower gene and protein expression of MIF in skeletal muscle than the heart.
Conclusions
Our results suggest that plasma MIF is an early marker for acute myocardial ischemia.
Keywords: cytokine, macrophage migration inhibitory factor, myocardial ischemia, peripheral artery disease, stress test
Subject Categories: Ischemia, Basic Science Research, Clinical Studies, Biomarkers
Introduction
Complications of artery disease (eg, acute myocardial infarction [AMI]) are the leading cause of death worldwide. Measurement of circulating biomarkers is of central importance to the evaluation of acute chest pain.1, 2 A number of biomarkers have been used for this evaluation. Currently, the most commonly used biomarker for AMI is cardiac troponin.3 High‐sensitivity troponin assays have significantly improved the sensitivity for early diagnosis of AMI.4, 5, 6 However, the main disadvantage of troponin is that plasma levels only rise several hours after the onset of chest pain patients with AMI.4, 5, 6 None of the other biomarkers, such as creatine kinase (CK) B, C‐reactive protein (CRP), heart‐type fatty acid binding protein, and ischemia modified albumin, rise as rapidly or specifically as troponin.7, 8, 9, 10, 11 Furthermore, troponin and other conventional markers are released by necrotic myocardium, so they cannot be used to detect acute myocardial ischemia.6 Therefore, the detection of myocardial ischemia earlier or in the absence of AMI is challenging, and early and specific markers for acute myocardial ischemia are not currently available.
Macrophage migration inhibitory factor (MIF) is a 12.5‐kDa protein, present in a wide variety of cell types, including cardiomyocytes and immune cells.12, 13, 14, 15 MIF has a range of actions, including metabolic regulation and modulation of inflammatory responses, and plays an important role in cardiovascular disease (CVD).16, 17 MIF is known to play a significant role in AMI and ischemia/reperfusion injury.18, 19, 20 Previous clinical studies by other groups reported an increase in plasma MIF levels in patients with AMI 4 to 6 hours after admission.14, 21 Levels remained elevated over the next 2 weeks.22 Our group further demonstrated that plasma MIF was elevated in patients with AMI at the earliest available samples after admission (average 211 minutes for symptom‐sampling time).23 In mice, we detected a rapid increase in plasma MIF within 15 minutes of coronary artery ligation, accompanied by a reciprocal reduction in myocardial MIF content, suggesting the release of MIF from myocardium into the circulation.23 The release at 15 minutes occurred before the development of irreversible cardiac ischemic damage.
So, we hypothesized that acute myocardial ischemia induced by a stress cardiac test in patients with coronary artery disease (CAD), but without AMI, would be sufficient to cause MIF release from the heart into the circulation. Demonstration of such release may also indicate the potential for plasma MIF as an early marker for the presence of myocardial ischemia.
Methods
Study Subjects
We recruited patients who had already been scheduled to undergo stress nuclear myocardial perfusion scan or stress echocardiography at the Alfred Hospital. The stress studies were undertaken to evaluate chest pain or as part of preoperative assessment for evidence of inducible acute myocardial ischemia in patients with known CAD. This study was approved by the Alfred Hospital Ethic Committee in accord with National Health and Medical Research Council's (NHMRC) National Statement on Ethical Conduct in Research Involving Humans. All patients provided written informed consent.
Patients undergoing a 6‐minute walk test for the evaluation of peripheral arterial occlusive disease (PAOD), but who had no known CAD, were recruited from the Department of Cardiovascular Medicine, 1st Affiliated Hospital, Jiaotong University (Xi'an, China). A further series of patients undergoing coronary percutaneous coronary intervention (PCI) were also recruited from the same department. Ethics approval for both studies was obtained from the human ethics committee of 1st Affiliated Hospital, Jiaotong University, and all patients gave informed consent.
Stress Myocardial Perfusion Scan
Continuous erect graded bicycle exercise (2 min/workload) was performed with 12 lead electrocardiogram (ECG) monitoring. Tc‐99m sestamibi (3 MBq/kg) was given intravenously in the last 1.5 minutes of exercise. Supine and prone single‐photon emission computed tomography (SPECT) images were obtained 10 minutes post exercise. Rest images, using the same technique, were performed 4 hours later with a further reinjection of 8 MBq/kg of Tc‐99m sestamibi. Patients with reversible perfusion defects were considered to have a positive nuclear stress test result for inducible myocardial ischemia. ECG criteria were not used to identify positive cases, but cases with ECG‐only changes were not included as controls.
Stress Echocardiography
Patients underwent a symptom‐limited exercise treadmill test, combined with echocardiography, using the standard Bruce protocol. Heart rate, blood pressure, and electrocardiographic measurements were obtained at rest and during each stage. Resting echocardiographic views were recorded before exercise using a standard hospital protocol. Immediately after exercise, echocardiography was repeated to assess the presence of inducible left ventricular regional wall motion abnormalities. ECG criteria were not used to identify positive cases, but cases with ECG‐only changes were not included as controls.
Blood Sample Collection
Venous blood samples were withdrawn from patients before and 5 and 15 minutes after cardiac stress test or 6‐minute walk test. Venous and coronary blood samples were withdrew in patients undergoing PCI. Plasma was separated from heparinized vacuum tubes (spun at 400g for 15 minutes and then at 3000g for 10 minutes) and stored at −80°C until assayed.
High‐Sensitivity Cardiac Troponin T and High‐Sensitivity CRP
High‐sensitivity troponin T (Roche Diagnostics, Risch‐Rotkreuz, Switzerland) and CRP in plasma were measured at the Department of Chemical Pathology of the Alfred Hospital. The 99th percentile for troponin T for normals has been determined to be ≤13 ng/L. Troponin T was measured at 3 time points (baseline and 5 and 15 minutes after exercise) in all patients. CRP was measured at 3 time points in all positive test patients and the first 31 negative test patients. Given that CRP did not change after exercise, the remaining patients in the negative group only had a baseline measurement.
ELISA Assay
MIF levels in plasma were measured in duplicate using quantikine ELISA human MIF immunoassay according to the manufacturer's instructions (R & D Systems, Minneapolis, MN). The coefficient of variation for intra‐ and interassay variation was 2.2% and 3.6%, respectively.
Western Blot
Frozen tissues of mouse left ventricle or calf skeletal muscle were homogenized in ice‐cold modified RIPA buffer containing (in mmol/L) tris–HCl 50, NaCl 150, β‐glycerophosphate 25, NaF 2, Na3VO4 1, and phenylmethysulfoyl fluoride 1, in the presence of 1% Triton X‐100, protease inhibitors (pH=7.4; Sigma‐Aldrich, St. Louis, MO). Protein concentration was determined using BCA protein assay (Bio‐Rad Laboratories, Hercules, CA). An equal amount of proteins from each sample was separated on 15% SDS‐PAGE and then transferred onto PVDF membranes. The membrane was incubated with anti‐MIF (1:2000; Abcam, Cambridge, UK) or anti‐β‐actin (1:4000; Abcam) overnight and then an anti‐rabbit immunoglobulin G secondary antibody (1:5000; Cell Signaling Technology, Inc., Danvers, MA) for 1 hour. Bands were visualized using enhanced chemiluminenscence (Santa Cruz Biotechnologies, Santa Cruz, CA) and quantified using the software Quantity One (Bio‐Rad). Results were normalized by respective levels of β‐actin.
Real‐Time Polymerase Chain Reaction
Total RNA was isolated from the left ventricle or calf skeletal muscle with Trizol Reagent (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. After DNase treatment (Promega, Madison, WI), RNA was reverse transcribed to first‐strand cDNA using random primers and M‐MLV reverse transcriptase (Invitrogen). Real‐time quantitative polymerase chain reaction (PCR) was performed, in duplicate, using an Invitrogen SYBR green kit on an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). Expression of GAPDH was used as a housekeeping gene for normalization. Transcription abundance was expressed as fold increase over skeletal muscle calculated by the 2−ΔΔCt method.
Cardiac Catheterization Patients
In order to examine the possibility that the elevation of plasma MIF might be attributed to stress‐induced release from atherosclerotic plaque, we recruited CAD patients undergoing PCI of the left anterior descending coronary artery (n=9). Coronary angiography and PCI were conducted in the standard fashion with the additional use of a Terumo Finecross MG (NC‐F 8633A) catheter to allow blood collection from beyond the stenosis. Blood samples were taken from a peripheral vein immediately before the PCI. Further samples were taken from the beyond the left anterior descending (LAD) artery stenosis immediately before and immediately after balloon angioplasty preceding stent deployment. Samples were processed and assayed for MIF as previously described.
Peripheral Obstructive Arterial Disease Patients
In order to determine whether ischemia of skeletal muscle induced MIF release, we further examined whether plasma MIF was also elevated by exercise sufficient to induce claudication in patients with known PAOD but no known history of CAD. Patients who underwent a 6‐minute walk test and who had claudication at the termination of the test were enrolled in the study. Venous blood samples were taken before and 5 and 15 minutes after completion of exercise. Samples were processed and assayed for MIF as previously described.
Statistical Analysis
Continuously variable data are expressed as mean±SEM (laboratory data) or mean±SD (clinical data) for normally distributed data or median (25th, 75th percentiles) for non‐normally distributed data. Continuously variable data were evaluated for normality using the Kolmogorov–Smirnov test and analyzed by Student t test or Mann–Whitney test, depending on whether or not data were normally distributed. Categorical data was presented as percentage and analyzed by χ2 test. Receiver operating characteristic curve analysis was used to calculate the area under the curve (AUC) of percent changes of MIF at 5 and 15 minutes after exercise for diagnosing acute myocardial ischemia. Univariate general linear model was employed to assess whether the association between MIF and the presence of acute myocardial ischemia was influenced by baseline risk factors, such as age, smoking, diabetes mellitus, hypertension, hyperlipidemia, and family history of CVD. All statistical analyses were performed using SPSS software (version 17.0; SPSS, Inc., Chicago, IL) and statistical significance was set at P<0.05 (2‐sided).
Results
Because all patients with a positive stress test were males, we only included male patients with a negative stress test as controls. For balloon angioplasty and PAOD studies, the majority of patients were also males, so we only included male patients throughout this study.
Acute Myocardial Ischemia Study
Basic characteristics
Twenty‐two patients had a positive cardiac stress test. Sixty‐two patients with a negative stress test served as controls. Table 1 shows subject characteristics for both groups. Groups were matched by age. There were no significant differences in prevalence of smoking, diabetes mellitus, hypertension, hyperlipidaemia, family history of CVD, and previous history of myocardial infarction (MI) and stroke. More patients with a positive stress test had known CAD. There were no significant differences in medication, except that more patients in the positive group received angiotensin‐converting enzyme inhibitors. Heart rate and systolic and diastolic blood pressure at baseline and peak exercise were comparable in the positive and negative groups. Thirteen percent in the positive group and 1.6% in the negative group developed chest pain during or after cardiac stress. In the positive group, 50% had ST segment depression, and 13.6% of patients had other ECG changes, such as atrial ectopics and supraventricular tachycardia, whereas the remaining patients had normal ECG during or after cardiac stress. None of them had ST elevation. In the negative group, none of the patients had acute ischemic changes on ECG.
Table 1.
Patient Characteristics in the Cardiac Stress Study
| Positive (22) | Negative (62) | |
|---|---|---|
| n | 22 | 62 |
| Age, y | 65±12 | 62±10 |
| Diabetes mellitus, % | 40.9 | 22.6 |
| Hypertension, % | 72.7 | 50.0 |
| Hyperlipidemia, % | 54.5 | 54.8 |
| Current cigarette smoker, % | 9.1 | 16.1 |
| Family history of CVD, % | 40.9 | 43.5 |
| Chest pain, % | 68.2 | 61.3 |
| Existing CAD, % | 54.5a | 24.2 |
| Previous MI, % | 31.8 | 24.2 |
| Previous stroke, % | 9.1 | 6.5 |
| Medication, % | ||
| β‐blockers | 27.3 | 14.5 |
| Calcium‐channel blockers | 22.7 | 14.5 |
| Angiotensin‐converting enzyme inhibitors | 36.4a | 12.9 |
| Angiotensin receptor blockers | 22.7 | 22.6 |
| Statins | 54.5 | 58.1 |
| Aspirin | 59.1 | 45.2 |
| Clopidogrel | 27.3 | 17.7 |
| Type of test, % | ||
| Exercise stress echo | 27 | 25 |
| Stress myocardial perfusion scan | 73 | 75 |
| Hemodynamic responses to stress | ||
| Resting heart beat, beats/min | 78±15 | 79±13 |
| Peak heart beat, beats/min | 138±15 | 143±15 |
| Rest systolic blood pressure, mm Hg | 137±16 | 133±18 |
| Peak systolic blood pressure, mm Hg | 171±21 | 177±24 |
| Rest diastolic blood pressure, mm Hg | 76±10 | 76±8 |
| Peak diastolic blood pressure, mm Hg | 77±6 | 80±7 |
Data are expressed as mean±SD or percentage. CAD indicates coronary artery disease; CVD, cardiovascular disease; MI, myocardial infarction.
P<0.05 vs negative group.
No changes of troponin T and CRP in acute myocardial ischemia
Eleven of 22 patients in the positive group and 52 of 62 patients in the negative group had troponin T levels below the 99th percentile (ie, <13 ng/L) at baseline, indicating that a higher proportion of patients in the positive group had baseline troponin T levels >13 ng/L (P<0.01). The remaining 11 patients in the positive group had median troponin T level of 20 (15–27) ng/L, and the remaining 10 patients in the negative group had median troponin T level of 17 (14–22) ng/L. All subjects in either the positive or negative group who had undetectable troponin T levels at baseline remained undetectable after cardiac stress. Baseline CRP levels did not significantly differ between the 2 groups (2 [1.53–3.45] vs 1.55 [0.8–3.25] mg/L). Percent changes of troponin T (from those with detectable troponin levels; Figure 1A) and CRP (Figure 1B) at 5 or 15 minutes, relative to baseline values after cardiac stress, was not significant between positive and negative groups.
Figure 1.
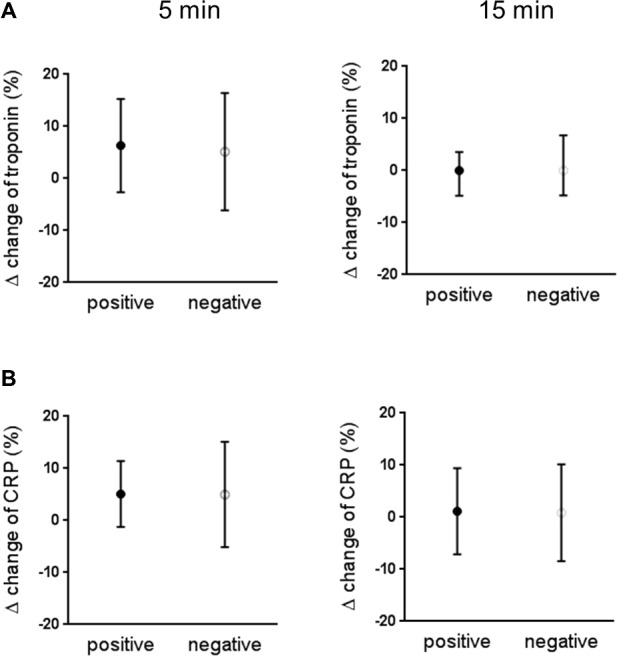
Percent change of troponin T (A) and CRP (B) at 5 and 15 minutes after cardiac stress relative to baseline in the positive and the negative group. Troponin T data were from 11 patients of 22 in the positive group and 10 patients of 62 in the negative group who had troponin T levels above the 99th percentile (ie, >13 ng/L). CRP was measured in all 22 positive cases and 31 negative cases. Data are expressed as median (25th, 75th percentiles). CRP indicates C‐reactive protein.
MIF levels elevated in acute myocardial ischemia
Baseline MIF was comparable between groups (Figure 2A and 2B). MIF levels at 5 and 15 minutes after cardiac stress were significantly elevated compared to baseline values in the positive group (Figure 2A), but not in the negative group (Figure 2B). At 5 minutes after stress, MIF was increased by 39.9% in the positive group, compared to −0.3% in the negative group (Figure 2C; P<0.001). At 15 minutes after stress, MIF was increased by 22.1% in the positive group compared to −8.4% in the negative group (Figure 2D; P<0.001). The association between myocardial ischemia and change of MIF levels analyzed by univariate general linear model was not altered after adjustment for baseline characteristics (age, smoking, diabetes mellitus, hypertension, hyperlipidemia, and family history of CVD).
Figure 2.
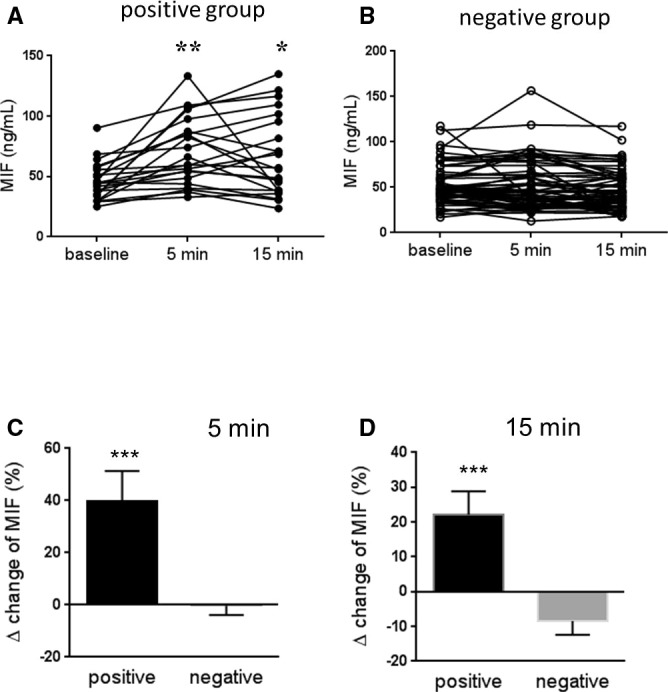
MIF levels at baseline, 5 and 15 minutes after cardiac stress in the positive group (A, n=22) and the negative group (B, n=62). Percent change of MIF at 5 minutes (C) and 15 minutes (D) after cardiac stress relative to baseline in the positive and the negative group. Data are expressed as mean±SEM. *P<0.05; **P<0.01; ***P<0.001 vs baseline or the negative group. MIF indicates macrophage migration inhibitory factor.
The prediction of acute myocardial ischemia by MIF
AUC for the prediction of myocardial ischemia from percent change in MIF at 5 and 15 minutes after exercise for myocardial ischemia is 0.767 (0.692–0.902) and 0.721 (0.610–0.861), respectively. When the values of percent change in MIF at 5 and 15 minutes after exercise were combined, AUC for the prediction of myocardial ischemia was increased to 0.789 (0.673–0.905).
Plasma MIF before and after balloon angioplasty
Basic characteristics of patients are presented in Table 2. Coronary plasma MIF level did not change after balloon angioplasty (P>0.05; Figure 3A).
Table 2.
Patient Characteristics in the Coronary Angioplasty Cohort
| n | 9 |
| Age, y | 62±5 |
| Heart rate, beats/min | 76±9 |
| Systolic blood pressure, mm Hg | 126±6 |
| Diastolic blood pressure, mm Hg | 77±5 |
| Current cigarette smoker (Y/N) | 7/2 |
| Diabetes mellitus (Y/N) | 2/7 |
| Hypertension (Y/N) | 2/7 |
| Hyperlipidemia (Y/N) | 3/6 |
| Medication | |
| Aspirin (Y/N) | 5/4 |
| Clopidogrel (Y/N) | 4/5 |
| Angiotensin‐converting enzyme inhibitors (Y/N) | 5/4 |
| β‐blockers (Y/N) | 4/5 |
| Statins (Y/N) | 5/4 |
Data are expressed as mean±SD or number.
Figure 3.
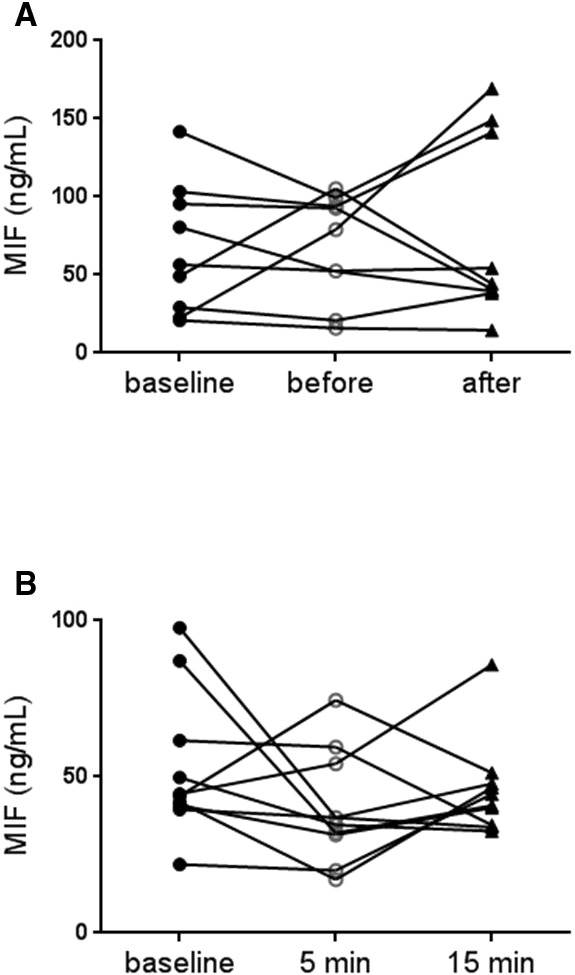
Plasma MIF was measured in peripheral venous blood before and in distal left anterior descending artery before and immediately after balloon angioplasty (A, n=9). Plasma MIF was measured before and 5 and 15 minutes after exercise in patients with peripheral arterial occlusive disease (PAOD; B, n=10). Data are expressed as mean±SEM. MIF indicates macrophage migration inhibitory factor.
Peripheral Ischemia Study
Basic characteristics of patients with PAOD are presented in Table 3. All patients developed claudication during exercise. However, plasma MIF level did not change at 5 or 15 minutes after exercise compared to baseline (P>0.05; Figure 3B).
Table 3.
Patient Characteristics in PAOD Cohort
| n | 10 |
| Age, y | 63±8 |
| Heart rate, beats/min | 75±11 |
| Ankle‐brachial index (lowest) | 0.5±0.3 |
| Six‐minute walk test, m | 267±132 |
| Cigarette smoking (Y/N) | 9/1 |
| Diabetes mellitus (Y/N) | 7/3 |
| Hypertension (Y/N) | 6/4 |
| Hyperlipidemia | 3/7 |
| Medication | |
| Aspirin (Y/N) | 10/0 |
| Angiotensin‐converting enzyme inhibitor (Y/N) | 6/4 |
| Angiotensin receptor blockers (Y/N) | 1/9 |
| Calcium‐channel blockers (Y/N) | 4/6 |
| β‐blockers (Y/N) | 1/9 |
| Statins (Y/N) | 2/8 |
| Cilostazol (Y/N) | 6/4 |
| Sarpogrelate (Y/N) | 4/6 |
Data are expressed as mean±SD or number. PAOD indicates peripheral arterial occlusive disease.
Comparison on MIF Levels Between Heart and Skeletal Muscle in Mice
Given that plasma MIF was increased with myocardial, but not peripheral, muscle ischemia, we compared gene and protein expression of MIF between cardiac and skeletal muscle from mice. Gene expression of MIF was 30‐fold higher in the heart, compared to skeletal muscle (Figure 4B; P<0.01), and protein expression of MIF was 3‐fold higher in the heart compared to skeletal muscle (Figure 4A and 4C; P<0.001).
Figure 4.
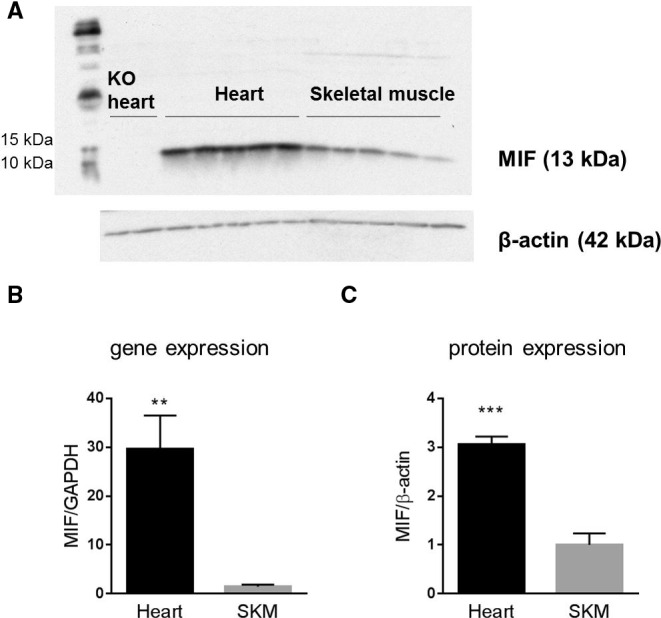
Protein and gene expression of MIF in the heart and skeletal muscle (SKM). A, western blot bands of MIF. First 2 lanes were performed on heart tissue from MIF knockout (KO) mice. B, gene expression of MIF by real‐time PCR (n=5/group). C, Quantification of western blot bands (n=5/group). Data are expressed as mean±SEM. **P<0.01; ***P<0.001 vs skeletal muscle. MIF, macrophage migration inhibitory factor; PCR, polymerase chain reaction.
Discussion
In the present study, we demonstrate that plasma MIF, most likely arising from the heart, is elevated after exercise in subjects who develop myocardial ischemia in comparison to those who do not. In contrast, plasma levels of troponin T and CRP are unchanged after stress. Coronary plasma MIF is not altered after balloon angioplasty of the LAD in patients with LAD stenosis, suggesting that elevated plasma MIF after a positive stress test may be not attributed to release from atherosclerotic plaque. In addition, plasma MIF is not affected by exercise sufficient to induce claudication in patients with PAOD, suggesting that plasma MIF is not a marker for peripheral muscle ischemia. Last, we find that gene and protein expression of MIF in mice is much higher in the heart than in skeletal muscle. Our results suggest that plasma MIF is an early marker for acute myocardial ischemia.
We previously demonstrated that plasma MIF rose earlier than troponin in patients with AMI. In mice, a rapid increase of plasma MIF was induced by 15 minutes occlusion of coronary artery, which is not sufficient to cause cell death, suggesting that acute myocardial ischemia could induce elevation of plasma MIF.23 In contrast, plasma troponin levels were not elevated at this time. In the present study, we further demonstrated that plasma MIF increases shortly after cardiac stress only in patients with evidence of acute myocardial ischemia, but not in those with no evidence of acute myocardial ischemia, suggesting that MIF is an early marker for acute myocardial ischemia. In contrast, troponin T and CRP did not increase in stress‐induced acute myocardial ischemia. Before the stress test, 50% of the patients in the positive group with evidence of acute myocardial ischemia had troponin T levels >13 ng/L, whereas only 16% of the patients in the negative group had troponin T levels >13 ng/L. This finding is consistent with previous studies, which have suggested that troponin predicts cardiovascular events, including incident heart failure and cardiovascular death, even in the general population.24, 25, 26 There have been several previous studies examining whether an increase in plasma troponin occurs in the presence of exercise or pharmacological stress–induced myocardial ischemia. In the majority of these studies, myocardial ischemia was not associated with an increase in plasma troponin. These include studies with troponin T27, 28, 29 and troponin I,30 with 1 study finding a blunting of the rise in troponin T in patients without objective ischemia.31 These studies have used highly sensitive assays. One study32 did report an increase in troponin I in exercise‐induced myocardial ischemia compared with stress test negative controls.
Although resting plasma high‐sensitivity CRP is known to be elevated in patients with CAD compared with controls,33 there is little data on the short‐term influence of acute myocardial ischemia induced by exercise. In the study of Danzig et al.,34 exercise in patients with CAD was associated with an increase in matrix metalloproteinase 9 and pre‐exercise plasma high‐sensitivity CRP was higher in CAD patients than controls, but no effect of exercise on CRP was reported.
The clinical significance of plasma MIF as an early marker of acute myocardial ischemia is multifold. First, MIF may help to identify myocardial ischemia in patients who present with chest pain. Second, MIF may help to detect the presence of acute myocardial ischemia in patients with existing ischemic heart disease. Third, MIF may help to identify early abnormalities in patients with acute coronary syndromes and AMI, thus limiting disease progression and improving patient outcomes through close monitoring and early diagnosis and treatment.35
MIF is expressed by multiple cell types, including cardiomyocytes14, 36 and immune cells.13, 15 Release of MIF from cardiomyocytes is different from that of troponins. MIF is present in a preformed intracellular storage and can be released by viable and stressed cardiomyocytes.37, 38 In contrast, troponins are structural components of the cardiomyocyte sarcomere, and they enter into the circulation by leakage through the disintegrated cellular membrane of dead cells.6 So, unlike troponins and other biomarkers (CK‐MB and myoglobin), plasma MIF could be an early marker for acute myocardial ischemia. A previous study showed that hypoxia (3 and 6 hours) and H2O2 (within 1 hour) stimulated MIF expression in cultured rat cardiomyocytes.14 Jian et al. further found that the increase of endogenous cardiac MIF expression positively correlated with degree of hypoxia.39 Using isolated perfused mouse heart, Miller et al. observed overflow of MIF into coronary venous drainage after exposure to hypoxia accompanied by decreased heart MIF content.36 In mice, we also detected a rapid increase in plasma MIF within 15 to 60 minutes of coronary artery ligation, accompanied by a reciprocal reduction in myocardial MIF content.23 These results suggest that MIF is released from the myocardium into circulation during acute myocardial ischemia.
It is known that MIF is expressed in atherosclerotic plaque.40, 41 To exclude the possibility that the increase of plasma MIF after ischemia is attributed to release from atherosclerotic plaque, we compared plasma MIF in the coronary artery before and after balloon angioplasty of the LAD in patients with CAD. We found that coronary MIF was not elevated by balloon angioplasty, suggesting that elevation of plasma MIF in patients with stress‐induced myocardial ischemia does not originate from coronary atherosclerotic plaque given that it is unlikely that any pertubation of coronary plaque caused by stress would be as disruptive as that caused by angioplasty. However, we cannot exclude the possibility that patients had ruptured unstable plaque during cardiac stress, whereas the patients had more‐stable plaque during PCI in our study.
The demonstration that myocardial ischemia was sufficient to cause release of MIF raised the possibility that this might also occur in ischemia of other muscles. Leg muscle ischemia was of particular interest and relevance given the prevalence of this condition and the frequent coexistence of both coronary and peripheral arterial disease. In PAOD patients, plasma MIF did not change after exercise sufficient to induce leg pain, suggesting that plasma MIF is not a marker for skeletal muscle ischemia. Thus exercise‐ or stress‐induced elevation in plasma MIF in patients with both coronary and peripheral arterial atherosclerosis is likely to be attributed to myocardial release of MIF, which is an important finding in the possible clinical utility of MIF measurement for the identification of myocardial ischemia. We are not aware of any previous report on circulating MIF and PAOD. It has been shown that MIF is expressed and released by L6 rat myoblasts.42 We compared MIF gene and protein expression levels between cardiac and skeletal muscle in mice. We found that that gene and protein expression of MIF was much higher in the heart than in skeletal muscle. Lower expression of MIF in skeletal muscle may explain the reason why MIF is not elevated in skeletal muscle ischemia.
The study has a number of limitations. Both stress echo and nuclear medicine were used to identify positive cases and the number of positive cases was small. However, only patients with objective evidence of imaging abnormality were included. The small sample size is likely a reflection of the low prevalence of positivity found with such techniques in contemporary practice. The number of study subjects was insufficient to analyze results by method of imaging (stress nuclear myocardial perfusion scan vs stress echocardiography). Of 22 positive cases, 5 patients had minor ischemia and 17 had relatively severe ischemia. Our study was also not powerful enough to assess the relationship between MIF levels and the extent of ischemia attributed to small sample size. A high‐sensitivity troponin T assay was used and levels below the 99th percentile were not reported. We might have missed small changes in patients with low baseline troponin T values, but this has not been clinically utilized. The patients in the PCI and PAOD studies were Chinese, in contrast to the cardiac stress study in which the majority of subjects were Caucasian. However, our previous studies on the release of MIF with AMI were also conducted in both Caucasian and Chinese populations and showed similar responses.23 Ischemia in the PAOD study was defined by the occurrence of typical symptoms, rather than by objective measures of perfusion or muscle function as in the cardiac study. However, all patients had previously had the presence of obstructive disease demonstrated, so it is likely that the appearance of classical claudication was indicative of ischemia.
In conclusion, plasma MIF is elevated in exercise‐induced acute myocardial ischemia, but not in exercise‐induced leg pain in patients with PAOD. The elevation of MIF is likely not attributed to release from atherosclerotic plaque. The demonstration that plasma MIF is elevated specifically in response to myocardial ischemia raises the potential clinical utility of measurement of plasma MIF.
Sources of Funding
The study was supported by a program grant by the National Health and Medical Research Council (NHMRC) to Dart who is an NHMRC Senior Principal Research Fellow (586656) and supported, in part, by the Victorian Government's Operational Infrastructure Support Program. Du is also an NHMRC fellow.
Disclosures
None.
Acknowledgments
We are grateful to the staff in the Department of Nuclear Medicine of the Alfred Hospital for their support with patient recruitment.
(J Am Heart Assoc. 2016;5:e003128 doi: 10.1161/JAHA.115.003128)
References
- 1. Frangogiannis NG. Biomarkers: hopes and challenges in the path from discovery to clinical practice. Transl Res. 2012;159:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dolci A, Panteghini M. The exciting story of cardiac biomarkers: from retrospective detection to gold diagnostic standard for acute myocardial infarction and more. Clin Chim Acta. 2006;369:179–187. [DOI] [PubMed] [Google Scholar]
- 3. Antman EM, Tanasijevic MJ, Thompson B, Schactman M, McCabe CH, Cannon CP, Fischer GA, Fung AY, Thompson C, Wybenga D, Braunwald E. Cardiac‐specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med. 1996;335:1342–1349. [DOI] [PubMed] [Google Scholar]
- 4. Keller T, Zeller T, Peetz D, Tzikas S, Roth A, Czyz E, Bickel C, Baldus S, Warnholtz A, Frohlich M, Sinning CR, Eleftheriadis MS, Wild PS, Schnabel RB, Lubos E, Jachmann N, Genth‐Zotz S, Post F, Nicaud V, Tiret L, Lackner KJ, Munzel TF, Blankenberg S. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med. 2009;361:868–877. [DOI] [PubMed] [Google Scholar]
- 5. Body R, Carley S, McDowell G, Jaffe AS, France M, Cruickshank K, Wibberley C, Nuttall M, Mackway‐Jones K. Rapid exclusion of acute myocardial infarction in patients with undetectable troponin using a high‐sensitivity assay. J Am Coll Cardiol. 2011;58:1332–1339. [DOI] [PubMed] [Google Scholar]
- 6. Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, Biedert S, Schaub N, Buerge C, Potocki M, Noveanu M, Breidthardt T, Twerenbold R, Winkler K, Bingisser R, Mueller C. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med. 2009;361:858–867. [DOI] [PubMed] [Google Scholar]
- 7. de Winter RJ, Koster RW, Sturk A, Sanders GT. Value of myoglobin, troponin T, and CK‐MBmass in ruling out an acute myocardial infarction in the emergency room. Circulation. 1995;92:3401–3407. [DOI] [PubMed] [Google Scholar]
- 8. Freund Y, Chenevier‐Gobeaux C, Leumani F, Claessens YE, Allo JC, Doumenc B, Cosson C, Bonnet P, Riou B, Ray P. Heart‐type fatty acid binding protein and the diagnosis of acute coronary syndrome in the ED. Am J Emerg Med. 2012;30:1378–1384. [DOI] [PubMed] [Google Scholar]
- 9. James SK, Oldgren J, Lindback J, Johnston N, Siegbahn A, Wallentin L. An acute inflammatory reaction induced by myocardial damage is superimposed on a chronic inflammation in unstable coronary artery disease. Am Heart J. 2005;149:619–626. [DOI] [PubMed] [Google Scholar]
- 10. Peacock F, Morris DL, Anwaruddin S, Christenson RH, Collinson PO, Goodacre SW, Januzzi JL, Jesse RL, Kaski JC, Kontos MC, Lefevre G, Mutrie D, Sinha MK, Uettwiller‐Geiger D, Pollack CV. Meta‐analysis of ischemia‐modified albumin to rule out acute coronary syndromes in the emergency department. Am Heart J. 2006;152:253–262. [DOI] [PubMed] [Google Scholar]
- 11. Orn S, Manhenke C, Ueland T, Damas JK, Mollnes TE, Edvardsen T, Aukrust P, Dickstein K. C‐reactive protein, infarct size, microvascular obstruction, and left‐ventricular remodelling following acute myocardial infarction. Eur Heart J. 2009;30:1180–1186. [DOI] [PubMed] [Google Scholar]
- 12. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle‐Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. [DOI] [PubMed] [Google Scholar]
- 13. Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takahashi M, Nishihira J, Shimpo M, Mizue Y, Ueno S, Mano H, Kobayashi E, Ikeda U, Shimada K. Macrophage migration inhibitory factor as a redox‐sensitive cytokine in cardiac myocytes. Cardiovasc Res. 2001;52:438–445. [DOI] [PubMed] [Google Scholar]
- 15. White DA, Fang L, Chan W, Morand EF, Kiriazis H, Duffy SJ, Taylor AJ, Dart AM, Du XJ, Gao XM. Pro‐inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS One. 2013;8:e76206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asare Y, Schmitt M, Bernhagen J. The vascular biology of macrophage migration inhibitory factor (MIF). Expression and effects in inflammation, atherogenesis and angiogenesis. Thromb Haemost. 2013;109:391–398. [DOI] [PubMed] [Google Scholar]
- 17. Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117:1594–1602. [DOI] [PubMed] [Google Scholar]
- 18. Dayawansa NH, Gao XM, White DA, Dart AM, Du XJ. Role of MIF in myocardial ischaemia and infarction: insight from recent clinical and experimental findings. Clin Sci (Lond). 2014;127:149–161. [DOI] [PubMed] [Google Scholar]
- 19. Gao XM, Liu Y, White D, Su Y, Drew BG, Bruce CR, Kiriazis H, Xu Q, Jennings N, Bobik A, Febbraio MA, Kingwell BA, Bucala R, Fingerle‐Rowson G, Dart AM, Morand EF, Du XJ. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia‐reperfusion injury: a predominant role of anti‐inflammation. J Mol Cell Cardiol. 2011;50:991–999. [DOI] [PubMed] [Google Scholar]
- 20. Rassaf T, Weber C, Bernhagen J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2014;102:321–328. [DOI] [PubMed] [Google Scholar]
- 21. Yu CM, Lau CP, Lai KW, Huang XR, Chen WH, Lan HY. Elevation of plasma level of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am J Cardiol. 2001;88:774–777. [DOI] [PubMed] [Google Scholar]
- 22. Takahashi M, Nishihira J, Katsuki T, Kobayashi E, Ikeda U, Shimada K. Elevation of plasma levels of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am J Cardiol. 2002;89:248–249. [DOI] [PubMed] [Google Scholar]
- 23. Chan W, White DA, Wang XY, Bai RF, Liu Y, Yu HY, Zhang YY, Fan F, Schneider HG, Duffy SJ, Taylor AJ, Du XJ, Gao W, Gao XM, Dart AM. Macrophage migration inhibitory factor for the early prediction of infarct size. J Am Heart Assoc. 2013;2:e000226 doi: 10.1161/JAHA.113.000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Lemos JA, Drazner MH, Omland T, Ayers CR, Khera A, Rohatgi A, Hashim I, Berry JD, Das SR, Morrow DA, McGuire DK. Association of troponin T detected with a highly sensitive assay and cardiac structure and mortality risk in the general population. JAMA. 2010;304:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. deFilippi CR, de Lemos JA, Christenson RH, Gottdiener JS, Kop WJ, Zhan M, Seliger SL. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. JAMA. 2010;304:2494–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saunders JT, Nambi V, de Lemos JA, Chambless LE, Virani SS, Boerwinkle E, Hoogeveen RC, Liu X, Astor BC, Mosley TH, Folsom AR, Heiss G, Coresh J, Ballantyne CM. Cardiac troponin T measured by a highly sensitive assay predicts coronary heart disease, heart failure, and mortality in the Atherosclerosis Risk in Communities Study. Circulation. 2011;123:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akdemir I, Aksoy N, Aksoy M, Davutoglu V, Dinckal H. Does exercise‐induced severe ischaemia result in elevation of plasma troponin‐T level in patients with chronic coronary artery disease? Acta Cardiol. 2002;57:13–18. [DOI] [PubMed] [Google Scholar]
- 28. de Silva R, Tsujioka H, Gaze D, Banya WA, Shah BN, Zoppelaro G, Hersey J, Gonzalez AM, Collins P, Collinson PO, Senior R, Fox KM. Serial changes in high‐sensitivity cardiac troponin, N‐terminal pro‐B‐type natriuretic peptide, and heart fatty acid binding protein during exercise echocardiography in patients with suspected angina pectoris and normal resting left ventricular function. Clin Chem. 2015;61:554–556. [DOI] [PubMed] [Google Scholar]
- 29. Kurz K, Giannitsis E, Zehelein J, Katus HA. Highly sensitive cardiac troponin T values remain constant after brief exercise‐ or pharmacologic‐induced reversible myocardial ischemia. Clin Chem. 2008;54:1234–1248. [DOI] [PubMed] [Google Scholar]
- 30. Rosjo H, Kravdal G, Hoiseth AD, Jorgensen M, Badr P, Roysland R, Omland T. Troponin I measured by a high‐sensitivity assay in patients with suspected reversible myocardial ischemia: data from the Akershus Cardiac Examination (ACE) 1 study. Clin Chem. 2012;58:1565–1573. [DOI] [PubMed] [Google Scholar]
- 31. Roysland R, Kravdal G, Hoiseth AD, Nygard S, Badr P, Hagve TA, Omland T, Rosjo H. Cardiac troponin T levels and exercise stress testing in patients with suspected coronary artery disease: the Akershus Cardiac Examination (ACE) 1 study. Clin Sci (Lond). 2012;122:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test‐induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J. 2009;30:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buckley DI, Fu R, Freeman M, Rogers K, Helfand M. C‐reactive protein as a risk factor for coronary heart disease: a systematic review and meta‐analyses for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:483–495. [DOI] [PubMed] [Google Scholar]
- 34. Danzig V, Mikova B, Kuchynka P, Benakova H, Zima T, Kittnar O, Skrha J, Linhart A, Kalousova M. Levels of circulating biomarkers at rest and after exercise in coronary artery disease patients. Physiol Res. 2010;59:385–392. [DOI] [PubMed] [Google Scholar]
- 35. Than M, Cullen L, Reid CM, Lim SH, Aldous S, Ardagh MW, Peacock WF, Parsonage WA, Ho HF, Ko HF, Kasliwal RR, Bansal M, Soerianata S, Hu D, Ding R, Hua Q, Seok‐Min K, Sritara P, Sae‐Lee R, Chiu TF, Tsai KC, Chu FY, Chen WK, Chang WH, Flaws DF, George PM, Richards AM. A 2‐h diagnostic protocol to assess patients with chest pain symptoms in the Asia‐Pacific region (ASPECT): a prospective observational validation study. Lancet. 2011;377:1077–1084. [DOI] [PubMed] [Google Scholar]
- 36. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP‐activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. [DOI] [PubMed] [Google Scholar]
- 37. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu CM, Lai KW, Chen YX, Huang XR, Lan HY. Expression of macrophage migration inhibitory factor in acute ischemic myocardial injury. J Histochem Cytochem. 2003;51:625–631. [DOI] [PubMed] [Google Scholar]
- 39. Jian Z, Li JB, Ma RY, Chen L, Zhong QJ, Wang XF, Wang W, Hong Y, Xiao YB. Increase of macrophage migration inhibitory factor (MIF) expression in cardiomyocytes during chronic hypoxia. Clin Chim Acta. 2009;405:132–138. [DOI] [PubMed] [Google Scholar]
- 40. Burger‐Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105:1561–1566. [DOI] [PubMed] [Google Scholar]
- 41. Schmeisser A, Marquetant R, Illmer T, Graffy C, Garlichs CD, Bockler D, Menschikowski D, Braun‐Dullaeus R, Daniel WG, Strasser RH. The expression of macrophage migration inhibitory factor 1alpha (MIF 1alpha) in human atherosclerotic plaques is induced by different proatherogenic stimuli and associated with plaque instability. Atherosclerosis. 2005;178:83–94. [DOI] [PubMed] [Google Scholar]
- 42. Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, Bucala R. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 2000;106:1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]